


Virology and natural history
West Nile virus (WNV) is an enveloped single-stranded, positive-sense RNA virus belonging to the genus Flavivirus in the family Flaviviridae [Reference Büchen-Osmond1]. WNV is antigenically related to other members of the Japanese encephalitis serogroup, which includes St Louis encephalitis virus (SLEV), Japanese encephalitis virus (JEV), Murray Valley encephalitis virus (MVEV), Kunjin virus (KUNV), and Usutu virus (USUV), which have all been shown to cause disease in humans. Culex spp. mosquitoes are the principal vectors of WNV [Reference Hayes2, Reference Petersen and Roehrig3], although the virus has been found in at least 43 other mosquito species [Reference Granwehr4, Reference Higgs, Snow and Gould5]. Virus amplification occurs in susceptible birds, which are believed to be the principal vertebrate reservoir hosts [Reference Hayes2, Reference Granwehr4]. In addition to birds, a very wide range of vertebrate species may present with clinical disease including horses, humans, juvenile squirrels, and even reptilian species such as alligators [Reference Petersen and Roehrig3, Reference Heinz-Taheny6, Reference Miller7]. Prior to its inadvertent introduction into New York in 1999, WNV was usually associated with occasional localized outbreaks of fever and encephalitis in humans, birds and horses in Africa, southern and central Europe and the Volga region of Russia [Reference Hayes2, Reference Cernescu8–Reference Platonov10].
The emergence of epizootic WNV in North America led to intensive investigations of its epidemiology, diagnosis and control. On the basis of phylogenetic studies of geographically representative virus isolates collected from most regions of North America, it seems most likely that a single introduction of the virus occurred, probably in 1999 [Reference Lanciotti11]. The precise reasons are not clearly defined as to why WNV proved to be so successful in dispersing throughout the Americas causing fever and encephalitis in such a wide variety of species. However, the most important contributory factors for dispersal probably include: (i) the availability of competent mosquito vectors and susceptible hosts on which they feed, (ii) highly suitable climatic conditions, and (iii) a reservoir population of flavivirus-naive birds to ensure efficient virus dispersal.
While mosquito-to-human transmission is the predominant means of human infection, the extensive WNV outbreaks in the USA in 2002 revealed four other mechanisms of person-to-person transmission: (1) blood transfusion from an infected viraemic person, (2) transplantation of infected organs, (3) intrauterine infection, and (4) potential passage through breast milk from an infected mother to a nursing child through breastfeeding [12–15].
History and epidemiology of WNV
WNV was first isolated from a febrile patient in Uganda in 1937, making it one of the earliest discovered arthropod-borne viruses [Reference Hayes2]. Prior to 1996, several documented epidemics occurred in the Old World, resulting in numerous cases, but few were reported as having severe neurological disease. Most cases exhibited mild febrile disease or asymptomatic infection. Since the mid-1990s, three epidemiological trends have emerged regarding WNV: (1) increased frequency of outbreaks in humans and horses, (2) increase in reported cases of encephalitis and meningitis (i.e. neuroinvasive disease) in humans, and (3) high case-fatality rates in birds coinciding with human outbreaks, mainly in the USA but also in Israel [Reference Petersen and Roehrig3]. Large outbreaks of WNV in humans were described in densely populated urban areas of Romania (1996), in the Volgograd region of Russia (1999), Northeastern USA (1999), Israel (2000) and the unprecedented expansion of WNV throughout the USA since 2002 (see Table 1) [Reference Hayes2, Reference Petersen and Roehrig3, Reference Cernescu8–Reference Platonov10, Reference Nash16].
Table 1. Human West Nile virus (WNV) cases reported to ArboNET in the USA, 1999–2009 [23]
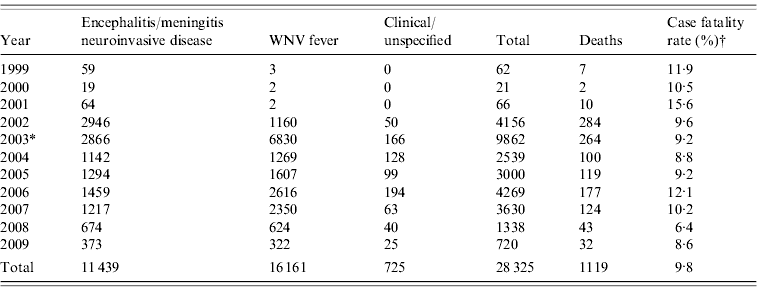
* Case definition changed and CDC requested WNV fever cases be reported to ArboNET.
† Case-fatality rate of neuroinvasive disease cases only.
Introduction of WNV into the USA
In late August of 1999, an unusual cluster of encephalitis cases was reported to the New York City Department of Health [Reference Nash16]. Initially, SLEV was diagnosed as the aetiology [17], but later laboratory sequencing of virus isolates from brain tissue of an encephalitic bird revealed WNV [Reference Jia18, Reference Lanciotti19]. The initial diagnosis of SLEV was a result of serological cross-reaction on laboratory testing with WNV, and retesting of sera from human cases confirmed WNV as the aetiology of this outbreak. This was the first evidence of autochthonous transmission of WNV in the Western Hemisphere.
The WNV strain identified in the 1999 outbreak in New York City showed a close phylogenetic relationship to an isolate obtained from a goose in Israel in 1998, both of which belong to lineage I [Reference Jia18, Reference Lanciotti19]. Very closely related strains of WNV circulate widely in Israel/Egypt/lower central Europe and the Volga Delta. Therefore, while it is unknown specifically how WNV was introduced into the USA, possibilities include introduction of infected mosquitoes or birds carried from one of the aforementioned regions on an incoming flight or ship to New York.
Geographical dispersal of WNV in the USA
Complex ecological factors determine the geographical spread of WNV, causing its distribution to be discontinuous and multi-focal within a given region [Reference Campbell20]. After its introduction in 1999, each transmission season saw WNV expand its range dramatically until 2004 when WNV cases were reported across the entire continental USA [Reference Artsob21–23] (see Table 1). The marked increase in the total number of cases between 2002 and 2003 was mainly due to the result of CDC's request to include West Nile fever (WNF) cases as reportable to ArboNET, which is the national surveillance system for arboviral diseases in the USA, and wider availability of testing for fever cases. The number of WNV neuroinvasive disease cases between 2002 and 2003 decreased slightly (2946 vs. 2866, respectively). After 10 years of virus activity across the USA, more than 27 000 cases of clinical WNV cases have been reported to CDC, including more than 1000 deaths.
WNV also spread beyond the borders of the USA and caused outbreaks of disease in Canada beginning in 2002 [Reference Pepperell24]. Virus activity has now also been detected in Mexico, Central America, South America, and the Caribbean [Reference Komar and Clark25], although reports of human cases are low in those areas. This could possibly be due to ecological differences or the endemic presence of other flaviviruses.
Clinical features and pathology of infection
The incubation period from infection to onset of clinical illness in humans varies between 2 and 14 days [Reference Campbell20]. About 80% of human cases of WNV infection are asymptomatic, while 20–30% experience mild infection classified as ‘WNF’ [Reference Mostashari26, Reference Brown27]. Fewer than 1% of patients develop neuroinvasive disease, characterized by meningitis (WNM) and/or encephalitis (WNE) [Reference Mostashari26, Reference Craven and Roehrig28].
WNF can present clinically with any of the following symptoms: fever, fatigue, malaise, lymphadenopathy, periocular pain, gastrointestinal symptoms, such as nausea, vomiting and abdominal pain, myalgias, headache, and occasionally a maculopapular rash. Younger patients are significantly more likely to present with a rash, and rash is associated with milder illness [Reference Brilla29]. One study found that one-third of those who were diagnosed with WNF were hospitalized and >60% missed school or work [Reference Hayes30]. Although certain aspects of the acute presentation of WNF typically resolve after 1 week, many of the symptoms can persist. WNF patients can experience fatigue and muscle weakness for more than 30 days. Other symptoms such as joint pain, headache and difficulty concentrating persist in 20–40% of WNF patients [Reference Watson31]. Although WNF has generally been characterized as mild and less severe than WNV neuroinvasive disease, it may have a substantial impact on health.
Patients aged >50 years are at highest risk of developing neuroinvasive disease, which clinically presents as meningitis and/or encephalitis [15, Reference Campbell20, Reference Asnis32, Reference Murray33]. Encephalitis can present with altered mental status including confusion, disorientation, and coma. Case-fatality rate in encephalitis cases can be around 15% [Reference Murray33]. In cases of encephalitis and meningoencephalitis, the gross appearance of the brain is normal. Microscopically, microglial nodules are observed as well as neuronal death, necrosis, and mild inflammation, which is mostly mononuclear [Reference Hayes30].
Acute flaccid paralysis (AFP) occurs in 5–15% of patients with neuroinvasive disease [Reference Sejvar, Haddad and Tierney34]. AFP can present as a poliomyelitis-like or Guillain–Barré-like syndrome, with symptoms ranging from single extremity weakness to quadriplegia including paralysis of the respiratory muscles [Reference Ahmed35, 36]. The poliomyelitis-like AFP is present more commonly (84% of WNV AFP cases) and is caused by viral injury to motor neurons, leading to an asymmetrical paralysis which can be permanent [Reference Sejvar, Haddad and Tierney34]. The Guillain–Barré-like AFP occurs less commonly (13% of WNV AFP cases). Nerve conduction studies in these patients suggest a peripheral demyelinating polyneuropathy. Interestingly, AFP may be more common in younger patients [Reference Davis37], but mortality attributable to AFP occurs primarily in the elderly [Reference Saad38]. The prognosis for patients with WNV AFP is poor.
Immune response following infection with WNV
Published studies on the immune response to WNV have focused mostly on acute infections in murine models. Following intradermal deposition of virus by an infected mosquito, the virus is taken up by Langerhans dendritic cells where it initially replicates. These cells migrate to draining lymph nodes and primary viraemia ensues [Reference Johnston, Halliday and King39]. The virus continues to replicate in the spleen, kidney, and epithelial tissues before infecting the brain [Reference Chung40, Reference Daffis41]. In the infected brain, chemokines, monocyte chemoattractant protein (MCP)-5 (CCL12), interferon gamma (IFN-γ)-inducible protein (IP-10), and monokines induced by gamma interferon (MIG) are expressed and trigger expression of IFN-γ and tumour necrosis factor-alpha (TNF-α). These are considered key cytokines in the pathogenesis and immune response during viral infection of the brain. Host antiviral defence relies on interferon regulatory factor (IRF-3) transcription response to promote IFN-α and IFN-β production, along with macrophage expression of host defence molecules RIG-I, MDA-5, ISG54, and ISG56 [Reference Johnston, Halliday and King39].
Kinetics of the humoral immune response
Recent studies have elucidated the kinetics of the humoral response during acute infection in great detail. Analysis of serial samples collected from a large population of blood donors found to be viraemic enabled a very complete portrait of WNV humoral response [Reference Busch42]. The mean duration of the time between first detection of WNV RNA by minipool transcription-mediated amplification and detection of IgM was 3·9 days and 7·7 days for IgG. A subset of the original cohort was followed up long enough to observe IgA and IgM reactivity decline to undetectable levels. The mean time of duration of detectable antibodies was 156 days for IgM (95% CI 70–423 days) and 220 days for IgA (95% CI 48–2100 days) [Reference Busch42].
During the WNV encephalitis epidemic in Bucharest, Romania in 1996, patterns of IgM and IgG reactivity in ELISAs were evaluated in relation to onset of illness [Reference Tardei43]. Anti-WNV IgM antibodies were detectable in serum as early as the second day following onset of illness, followed 2–3 days later by a detectable IgG antibody response. In more than 50% of convalescent sera collected 2 months later, IgM was still present.
Roehrig et al. [Reference Roehrig44] studied the antibody response in 29 patients diagnosed with WNV encephalitis during the New York City outbreak in 1999. At 9 months post-onset, the percent of patients with detectable IgM antibodies appeared to be different with regard to age; 56% of those aged ⩾65 years had detectable IgM compared to 44% of those aged <65 years; however, the sample size was too small to detect a statistical difference. At 500 days post-onset, 7/12 patients who still had evidence of IgM antibodies on previous serial bleeds (9 months and/or 12 months) continued to have detectable IgM. A separate study in Michigan reported persistence of WNV IgM antibodies in the CSF [Reference Kapoor45]. Three patients with WNV neurological disease had detectable IgM in CSF at 110, 141, and 199 days past the acute phase of infection. IgM antibody persistence can hinder diagnosis in successive years in areas affected by large epidemics [Reference Roehrig44]. Overall decline of IgM antibodies is expected; however, extended high titres could represent viral persistence.
Diagnosis of WNV
Clinical disease caused by WNV infection is diagnosed through a combination of findings, including the presence of a clinically compatible illness paired with positive findings through laboratory testing [Reference Granwehr4, Reference Campbell20, Reference Craven and Roehrig28]. Laboratory diagnosis of WNV neuroinvasive disease is typically made by detecting the presence of anti-WNV IgM antibodies in cerebrospinal fluid (CSF) using an IgM-capture enzyme-linked immunosorbent assay (MAC-ELISA) [Reference Granwehr4]. If CSF is not available, then serum can be tested; however, a positive IgM result from a serum sample needs to be interpreted cautiously when diagnosing acute infection since IgM antibodies can persist for extended periods of time [Reference Roehrig44]. Correlation of serological findings with the clinical picture is important, and if necessary, a rise in antibody titre using the plaque reduction neutralization test (PRNT) can be used to confirm the clinical diagnosis of WNV infection. IgG antibodies can also be tested by ELISA; however, this test is not necessary for the diagnosis of an acute clinical case. Processing of clinical samples by the CDC established that MAC-ELISA protocol can take up to 1·5–3 days depending on whether plates are incubated overnight or not. At present there are several commercially available ELISA kits that have comparable sensitivity and specificity that can be completed in a day. Recently, the FDA approved a lateral-flow IgM strip assay [Reference Sambol and Hinrichs46]. This method is comparable in sensitivity and specificity to other immunoassays and takes minutes for completion. There are several additional features of this assay that are particularly relevant to situations outside of a well-equipped diagnostic laboratory. The lateral-flow assay can be performed with the bare minimum of training and it requires no instrumentation. All of these assays listed are labelled for use on serum and can be used to aid in the presumptive diagnosis of WNV in patients presenting with clinically compatible disease symptoms.
PRNT is more specific for WNV, and can be used as a means of confirming an acute case if cross-reaction is occurring with other related flaviviruses, such as SLEV [Reference Craven and Roehrig28]. PRNT is not typically used to confirm all infections since the test requires 3 days for plaque formation if using the wild-type of virus and 5 days if using the Yellow fever/WNV chimera virus [Reference Granwehr4, Reference Komar, Langevan and Monath47]. The IgG ELISA is sensitive and can determine past exposure; however, false positives can result due to cross-reaction with the antibody response to infection caused by other closely related flaviviruses [Reference Niedrig48].
Other laboratory tests can be used to detect infection with WNV; however, these are not commonly used for diagnostic purposes. Reverse transcriptase–polymerase chain reaction (RT–PCR) or RT–PCR with fluorescent probes can be used to detect viral nucleic acid [Reference Granwehr4, Reference Lanciotti49]. RT–PCR with fluorescent probes is primarily suitable for analysis of tissue specimens. In one study, WNV RNA was detected in 6/6 (100%) confirmed positive brain tissue specimens, 16/28 (57%) of CSF specimens, and 0/28 (0%) of serum samples [Reference Lanciotti49]. RT–PCR and immunohistochemistry (IHC) are tests that can be used to determine presence of virus in post-mortem brain tissue. In 2002, the transmission of WNV through blood transfusions was discovered [Reference Busch50]. Following this discovery, a national blood screening programme was implemented. Both in the USA and Canada, blood is now screened using the nucleic acid amplification test (NAAT). NAAT (using nucleic-acid sequence-based amplification and RT–PCR) is a highly sensitive diagnostic tool that can also be used to complement IgM testing in the diagnosis of acute WNV infection if the patient plasma sample is tested within the first few days of clinical illness [Reference Tilley51].
Host risk factors for developing severe disease
Elderly patients are at highest risk for encephalitis and death [Reference Nash16, Reference Campbell20, Reference Craven and Roehrig28, Reference Murray52]. Several studies have examined other risk factors for developing severe disease. A study of the WNV outbreak in New York City in 1999 found that advanced age (>75 years) and diabetes were independent risk factors for the seven deaths that were recorded [Reference Nash16]. Unfortunately this study had a small sample size, making it difficult to draw conclusions about important risk factors for developing severe disease from WNV infection.
In a study of hospitalized patients in Colorado, a risk analysis of 65 WNE cases and 53 WNF cases found that alcohol abuse, diabetes, and advanced age were associated with the development of encephalitis [Reference Bode53]. Hypertension and immunosuppression were not significant on univariate analysis and were not retained in the multivariate model. A study of the 2005 WNV cases (n=880) in California found that being older, being male, and having a history of hypertension or diabetes were risk factors for developing neurological disease (including both meningitis and encephalitis) compared to uncomplicated fever cases [Reference Jean54].
In Houston, Texas, a nested case-control study was conducted using medical chart reviews for 172 confirmed WNV cases (113 encephalitis cases and 59 meningitis or fever cases) hospitalized between 2002 and 2004 to determine risk factors for developing encephalitis [Reference Murray55]. A multiple logistic regression model identified advanced age, history of hypertension, and history of cardiovascular disease as independent risk factors for developing encephalitis. Age-adjusted risk factors for death from encephalitis included being black, chronic renal disease, co-infection with hepatitis C, and immunosuppression. A separate but related matched case-control study was conducted in Houston to determine risk factors for developing encephalitis by comparing WNV encephalitis cases to non-WNV-infected age-, gender- and race/ethnicity-matched controls. The multivariable conditional logistic regression analysis identified hypertension, immunosuppression, and cardiovascular disease as independent risk factors associated with being an encephalitis case [Reference Murray55, Reference Mateo56].
Immunosuppression or immune senescence may also play a role by increasing magnitude and duration of viraemia [Reference Campbell20]. In a hamster animal model, cyclophosphamide was used to induce immunosuppression [Reference Kumar57]. When compared to normal controls, the WNV-infected immunosuppressed hamsters had a longer period of viraemia, lacked detectable antibodies in haemagglutination inhibition tests, showed greater disease severity and a higher fatality rate. Immunosuppression as a result of single-organ transplant is also believed to be a risk for neuroinvasive disease. One study in Canada found that organ transplant patients were 40 times more likely to develop neuroinvasive disease from natural infection compared to the general population [Reference Kumar57]. On the contrary, a more recent cross-sectional seroprevalence study found no greater risk for neuroinvasive disease between organ transplant recipients and immunocompetent controls, and it proved that asymptomatic infection can occur in immunocompromised patients [Reference Freifeld58].
Long-term sequelae from WNV
After the 2002 outbreak in St Tammany Parish, Louisiana, 16 patients with confirmed WNV neuroinvasive disease were re-examined 8 months later to assess symptoms, functional status, and neurological sequelae [Reference Sejvar, Haddad and Tierney34]. About 8 months after disease onset, 11 (69%) patients were functioning independently at home, three were at home but were dependent, one patient was in a rehabilitation facility, and one patient had died. Persistent symptoms reported by patients included fatigue, headache and myalgias; six patients continued to experience gait and movement disorders, and the three patients diagnosed with AFP had not recovered their limb strength.
Following the WNV outbreak in Israel in 2000, 32 patients aged ⩾65 years were examined 3 months following discharge to determine their clinical outcome [Reference Labowitz Klee59]. Of these 32 patients, seven (22%) ultimately died. Twenty-two (69%) patients had returned to baseline function, five (16%) patients required prolonged rehabilitation, three (9%) had residual intellectual impairment, and two (6%) were experiencing functional decline. That study appeared to find that a change in consciousness during the acute phase of clinical illness predicted the outcome.
Other studies have described subjective patient-reported outcomes at 1 year post-infection. One study of patients who were infected with WNV in New York City in 1999 found 63% of patients reporting continued difficulties from their infection, with most reporting muscle weakness, loss of concentration, confusion, and lightheadedness, with younger age at infection being the only significant predictor of recovery [Reference Labowitz Klee59]. In Tennessee, 55% of identified patients were still reporting complications from their illness 1-year post-infection, with fatigue, weakness, difficulty ambulating, and memory difficulties most commonly reported [Reference Gottfried, Quinn and Jones60]. In Canada, researchers found that physical and mental outcome measures mostly normalized within the first year of recovery, and recovery took longer in those who presented with neuroinvasive disease and had pre-existing medical conditions [Reference Loeb61]. In a study from Idaho, researchers found that most participants in their study had persistent symptoms more than 6 months following acute infection, with neuroinvasive disease, diabetes, and hypertension being identified as significant predictors for persistent symptoms [Reference Cook62].
Neuropsychiatric consequences have also been reported following WNV infection. A year following the outbreak of WNV in New York City, 38% of patients subjectively reported depression [Reference Labowitz Klee59]. Another 1-year follow-up in Colorado reported 23% of patients with complaints of anxiety and depression [Reference Sejvar63]. In Houston, a study was conducted to evaluate both the subjective and objective measurements for depression 1 year after clinical disease from WNV [Reference Murray, Resnick and Miller64]. In this study, new onset depression was found to be an important outcome, with 31% of patients reporting depression 1 year following infection. Objective measurements using the Center for Epidemiological Studies – Depression (CES-D) scale found that 75% of those cases reporting new-onset depression had scores indicative of mild to severe clinical depression.
The long-term clinical sequelae, including mental and physical deficits, in WNV patients critically need to be defined. By knowing and possibly predicting the outcomes, and whether or not particular interventions can improve recovery, prognosis can be improved and the potential for resolution of disease may be achieved.
Chronic infection with WNV
Animal studies also support the plausibility of persistent WNV infection in humans. Persistent infection with WNV was first documented in rhesus monkeys in 1983 by Pogodina et al. [Reference Pogodina65, Reference Tesh, Xiao and Diamond66]. Rhesus monkeys were infected with several different WNV strains; 87% of animals that survived the infection were sacrificed at various time-points. WNV was recovered from various tissues, including brain, lymph nodes, spleen, and kidneys up to 167 days after infection. Monkeys were not considered clinically ill at the time of death and many had WNV-neutralizing antibodies detected in serum. Interestingly, the phenotypic characteristics of the virus changed over time. During the first 2 months, WNV could be recovered by intracerebral inoculation of newborn mice or by plaque assay in chick embryo fibroblasts. After that point, the virus recovered from persistently infected monkeys no longer killed newborn mice, and plaques were not regularly produced in chick fibroblasts. Co-cultivation of trypsinized organ tissue with pig embryo kidney cells allowed for virus detection, although viral cytopathic effect was rarely seen. WNV antigen could only be demonstrated through immunofluorescence of the indicator pig embryo kidney cells. Genetic studies were not done at the time since the techniques were not widely available. This study demonstrated that WNV had persistently infected some of the macaques for up to 167 days and that the phenotype of the virus changed over time.
More recently, persistent WNV infection of both the brain and the kidneys was demonstrated in experimentally infected hamsters [Reference Siddharthan67–Reference Tonry69]. Hamsters developed chronic infection of epithelium of the distal renal tubules and shed 102–104 plaque-forming units of infectious virus/ml in the urine for up to 8 months post-infection [Reference Tesh68, Reference Tonry69]. Immunohistochemical staining of tissues showed no evidence of WNV antigen in brain, liver, spleen, lungs or bladder, but kidney tissue showed moderate to strong antigen staining [Reference Tesh68]. Infectious virus could be recovered using a co-cultivation technique on fresh kidney tissue. As seen with the monkey studies, the virus underwent phenotypic changes [Reference Wu70]. Progeny virus from a urine isolate inoculated into naive hamsters resulted in asymptomatic infection. Sequencing of the genome identified genetic changes at 116 sites, mainly in coding regions.
In immunocompetent mice, infectious WNV was shown to persist for a month in all mice, and WNV RNA could still be detected in 12% of mice up to 6 months post-infection [Reference Appler71]. Persistence of infection was found to be tissue-dependent, with skin, spinal cord, brain, lymphoid tissue, kidney, and heart being affected. Infectious virus could not be recovered after 4 months post-infection. Another important observation from this study was that persistent infection was identified in animals with no outward signs of clinical disease. These experimental data support the possibility that persistent renal and/or CNS infections may occur in humans and raise concern for persistent infection in people who were mildly or subclinically affected.
A longitudinal study of WNV cases in Houston recently identified WN viral RNA in the urine of 20% of the cohort years after their initial illness [Reference Murray72]. Preliminary research in this cohort found associations between persistence of symptoms, sustained detectable IgM antibody response, and altered cytokine expressions, strengthening the hypothesis that some patients might have persistent infection. Cohort participants' urine was tested for WNV viral RNA and 5/25 (20%) were found to be positive up to 7 years following acute infection. Two of these individuals were found to be in renal failure. Sequencing of amplicons found >99% homology to the WNV NY99 strain. Infectious virus could not be isolated from the urine; therefore, further investigation is needed to understand these findings and whether or not there is any clinical or pathological impact related to the presence of WNV viral RNA. Independent evidence supporting the concept of persistent WNV infection in humans was provided by the detection of WNV RNA and antigen in the central nervous system of an encephalitis patient with B cell lymphoma 99 days after symptom onset [Reference Penn73]. Considering that more than 25 000 clinical cases of WNV have been reported to CDC from across the USA, and an estimated 1·8 million have been infected, chronic infection with WNV could have substantial public health implications.
The future of prevention: antiviral therapy and vaccine development
Currently, the only accepted therapy for WNV infection is supportive care [Reference Craven and Roehrig28]. Clinical trials are still needed to determine the efficacy of treatment with antivirals, immunoglobulins, and novel vaccines. There is no specific antiviral therapy to date for WNV encephalitis, although ribavirin has been shown to inhibit virus in neural cell cultures [Reference Craven and Roehrig28, Reference Jordan74, Reference Enserink75]. Ribavirin was administered to 35 patients during the 2000 outbreak in Israel; however, the treatment did not appear to affect the outcome. Ribavirin does not effectively cross the blood–brain barrier which could limit its usefulness as a treatment.
Several interesting strategies have been employed to develop a vaccine to WNV. DNA vaccines have been tested in animals and shown to produce an effective antibody response [Reference Anwar76]. One of the strategies employed is to fuse DNA transcripts of WNV envelope and membrane proteins to lysosyme-associated membrane proteins. The rationale for this method is that it will result in the ultimate loading of the MHCII antigen groove of professional antigen-presenting cells. The theory is that this triggers antigen-specific CD4+ T-cell responses and the generation and maintenance of memory B-cell and CD8+ T-cell responses. Another approach at creating a DNA-based WNV vaccine has shown great promise [Reference Maeda77]. Polymerase chain reaction ‘stitching’ has been used to create an entire recombinant WNV virion. When a glycosylation motif at position 446 was removed, the virus showed no cytopathic effects in Vero cells. A live, attenuated chimeric WNV vaccine has been developed [Reference Monath78]. Phase I clinical trials have demonstrated that the chimeric vaccine is safe, well tolerated, and highly immunogenic [Reference Guy79]. This same technology has been used to produce a successful equine vaccine, which has had widespread use throughout the USA since it was approved by United States Department of Agriculture in 2002. Other effective inactivated/killed and DNA vaccines have also been approved for use in equines.
One study using an animal model has shown that immunization with heterologous flaviviruses, such as Japanese encephalitis and yellow fever vaccines, appear to be protective against clinical encephalitis and death [Reference Tesh80]. In another study, the WNV non-structural glycoprotein NS1 was used to generate NS1-specific monoclonal antibodies, which were strongly protective against lethal WNV infection in mice when administered as a prophylaxis [Reference Chung81]. Should an effective WNV vaccine or monoclonal antibody be developed, it can be used in targeted populations who are at higher risk of developing encephalitis.
Intravenous immunoglobulin (IVIG) therapy has been examined as a potential option for treatment of acute WNV infections. Published case reports and murine animal model studies suggest rapid improvement in the clinical course following administration of IVIG containing high titres of anti-WNV antibodies [Reference Shimoni82–Reference Ben-Nathan84]. These reports suggest that antibody-containing immunoglobulin might be an effective treatment for cases of WNV encephalitis, particularly in immunocompromised patients; however, proper randomized controlled clinical trials in humans have not been completed to prove the efficacy of treatment with IVIG.
Until effective antivirals, vaccines, or other therapeutics become available, prevention of disease in humans must focus on education, control and reduction of mosquito populations, elimination of breeding sites, and prevention of mosquito bites. In the USA, CDC promotes educating the public on the five ‘Ds’ for WNV prevention: staying indoors between Dusk and Dawn when the most important mosquitoes responsible for spreading WNV are most active, eliminating mosquito breeding sites by Draining standing water, Dress to protect by wearing long sleeves and trousers when outdoors, and use a mosquito repellent containing DEET (N,N-diethyl-meta-toluamide) [23]. Picaridin is another conventional repellent recommended by CDC as well as the biopesticide repellents oil of lemon eucalyptus and IR 3535.
Conclusions
WNV is now endemic in the USA and continues to be a significant source of morbidity and mortality. This virus has made an important impact on the population, and financial, physical, and mental costs related to long-term sequelae will continue to be substantial over time. After the widespread surge of virus activity across the USA over the past 10 years, research has flourished. Our knowledge base today is significantly greater than when this virus first became recognized in the Western Hemisphere in New York City in 1999. Future research related to immune markers, genetic susceptibility, vaccines, impact of persistent infection, and development of therapeutics will hopefully grant us the ability to ameliorate disease incidence, severity, and outcomes in humans.
The future of WNV disease burden in the USA and other countries will be interesting to follow in the coming years. Already, we are seeing a reduction in the number of human cases as the disease has established an endemic pattern. As time continues, the seroprevalence of WNV will increase in both the bird and human population, accumulating immunity and potentially protection from clinical illness during subsequent exposures. However, it is doubtful that herd immunity in the reservoir vertebrate host populations could ever be established considering the natural turnover in these populations, leading to new immunologically naive and susceptible animals. Over the past 10 years in the USA, more than 11 000 cases of encephalitis and meningitis were reported to ArboNET [23]. Considering neurological disease occurs in 1 of every 150 cases of infection [Reference Mostashari26], we can roughly estimate that 1 650 000 people in the USA have been infected with WNV. All infections, regardless of clinical presentation, result in the production of antibodies and establishment of immunity. Currently, the risk for and pathological implications of persistent infection are unknown.
During the past 10 years, arbovirology has effectively been re-born as a result of the appearance of WNV in North America. Armed with our new experience, we now await the introduction of the next arbovirus pathogen, with the most obvious candidates being Chikungunya and Dengue viruses. Over the next 10 years, it will be interesting to speculate research and development outcomes and the future of the expanded distribution of arboviruses around the world.
ACKNOWLEDGEMENTS
The authors thank Dr Liliana F. Rodriguez for assistance with the literature review. This work was funded in part by a grant from the United States Department of Defense, Telemedicine and Advanced Technology Research Center (TexSHIELD W81XWH-07-2-0031).
DECLARATION OF INTEREST
None.

