Introduction
Cyathostominae nematodes, also known as small strongyles of horses, belong to the family Strongylidae. Cyathostomins usually present as a mixed infection and produce symptoms such as weight loss, poor coat, lethargy, anaemia and diarrhoea, even with the high mortality caused by larval stage migration (Lichtenfels et al., Reference Lichtenfels, Kharchenko and Dvojnos2008). Cyathostomins can adapt to different climates and have been reported in horses worldwide (Mfitilodze and Hutchinson, Reference Mfitilodze and Hutchinson1990; Smets et al., Reference Smets1999). Their prevalence may be as high as 100%, which can lead to considerable economic damage to horse breeding (Bu et al., Reference Bu2009; Stancampiano et al., Reference Stancampiano2017).
Previous studies on Cyathostominae nematodes have focused mainly on their morphology, life history and epidemiology (Lichtenfels et al., Reference Lichtenfels, Kharchenko and Dvojnos2008; Stancampiano et al., Reference Stancampiano2017), and thus genetic information is limited. Genetic markers, such as the internal transcribed spacer (ITS) of the nuclear ribosomal DNA region and mitochondrial genome sequences, have been used for the genetic classification and evolutionary analysis of cyathostomins (Hung et al., Reference Hung, Chilton and Gasser2000; Gao et al., Reference Gao2017a, Reference Gaob).
Mitochondrial (mt) DNA sequences are used widely as genetic markers, not only for population or ecological genetics studies but also for phylogenetic and evolutionary analyses of many kinds of parasites. Cyathostominae nematodes are a complex nematode group, with 51 reported species. The mt genomes of only six Cyathostominae species (Cylicostephanus goldi, Cylicostephanus minutus, Cylicocyclus nassatus, Cylicocyclus insigne, Cyathostomum catinatum and Poteriostomum imparidentatum) have been sequenced to date (Gao et al., Reference Gao2017a, Reference Gaob). Cyathostomum pateratum is one of the most common cyathostomins in equine species, but there are insufficient mtDNA sequence data available to determine its phylogenetic relationships with other species.
The aims of this study were to determine the mt genome sequence of Cy. pateratum, compare it with the mt genome sequence of Cy. catinatum reported previously, and reconstruct the phylogenetic relationships with other Cyathostominae nematodes using the mtDNA sequence data.
Materials and methods
Parasites and isolation of total genomic DNA
Adult Cy. pateratum were collected from the large intestine of a naturally infected horse from Daqing City, Heilongjiang Province, China. Individual worms were washed with physiological saline and identified based on their morphological characteristics (Lichtenfels et al., Reference Lichtenfels, Kharchenko and Dvojnos2008), then stored at −20°C until used.
Total genomic DNA was extracted from individual worms using a TIANamp Genomic DNA Kit (TIANGEN Biotech, Beijing, China). The species was determined by amplifying the ITS sequence of Cy. pateratum using primers NC5 (5′-GTAGGTGAACCTGCGGAAGGATCATT-3′) and NC2 (5′-TTAGTTTCTTTTCCTCCGCT-3′) (Gao et al., Reference Gao2017a). The ITS sequence of Cy. pateratum obtained in the present study (GenBank: MG727532) matched the previously reported ITS sequence of Cy. pateratum (GenBank: KF850628.1) with 99.9% identity. To better confirm the species, we amplified two genes, nad1 and cox1, and the obtained sequences matched the previously published nad1 (GenBank: KY131816.1) and cox1 (GenBank: AF263484.1) sequences with 96.1% and 92.4% identities, respectively.
PCR amplification and sequencing of the mtDNA genome of Cy. pateratum
The complete mtDNA sequence of Cy. pateratum was amplified using ten specific primers. The primer sequences and polymerase chain reaction (PCR) cycling conditions were the same as those for Cy. catinatum (Gao et al., Reference Gao2017b). The amplicons were separated on 1% agarose gel, purified using a DNA Gel Extraction Kit (AXYGEN, Suzhou, China), ligated into a pMD-18T plasmid vector (Promega, Madison, Wisconsin, USA), and transformed into DH5α competent cells (BioMed, Beijing, China). The positive recombinant plasmids were sequenced by the Life Technology Company (Shanghai, China). The complete mtDNA sequence was assembled manually and aligned against the mtDNA sequences of other Cyathostominae nematodes by Clustal X (v. 1.83) (Thompson et al., Reference Thompson1997). The secondary structures of transfer RNA (tRNA) genes were predicted using the online program tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE) or manually. The boundaries of protein-coding genes and ribosomal RNA (rRNA) genes, and initiation and termination codons were determined by MEGA 5 (Tamura et al., Reference Tamura2011). The identities were calculated using MegAlign 5.01 (Burland, Reference Burland2000). The A + T content was determined using DNAStar (v. 5.0) (Burland, Reference Burland2000). The identities, length and A + T contents of genes of the complete mt genome of Cy. pateratum were compared with the mt genomes of Cy. catinatum and other nematodes.
Phylogenetic analyses
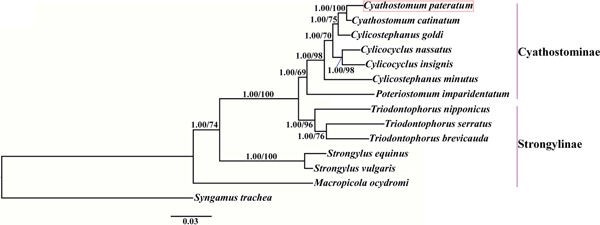
Phylogenetic trees were constructed based on the concatenated amino acid sequences of the 12 protein-coding mt genes of Strongylidae nematodes available in GenBank. The nematodes belonged to two subfamilies as follows: Cyathostominae (Cs. goldi (AP017681), Cc. insigne (GQ888712), Cc. nassatus (KX819273), Cy. catinatum (KY495600), Cs. minutus (KY495601) and P. imparidentatum (KY495602)), and Strongylinae (Strongylus equinus (KM605251), Strongylus vulgaris (AP017698), Triodontophorus brevicauda (KP191043), Triodontophorus nipponicus (KX185155), Triodontophorus serratus (KX185154) and Macropicola ocydromi (KF361320)), with Syngamus trachea (GQ888718) as the outgroup. Bayesian inference and maximum likelihood methods were used to construct the trees as described previously (Gao et al., Reference Gao2017a). Phylograms were drawn using FigTree v. 1.31 (http://tree.bio.ed.ac.uk/software/figtree/).
Results and discussion
Structure of the complete mt genome of Cy. pateratum and comparisons with Cy. catinatum and other nematodes
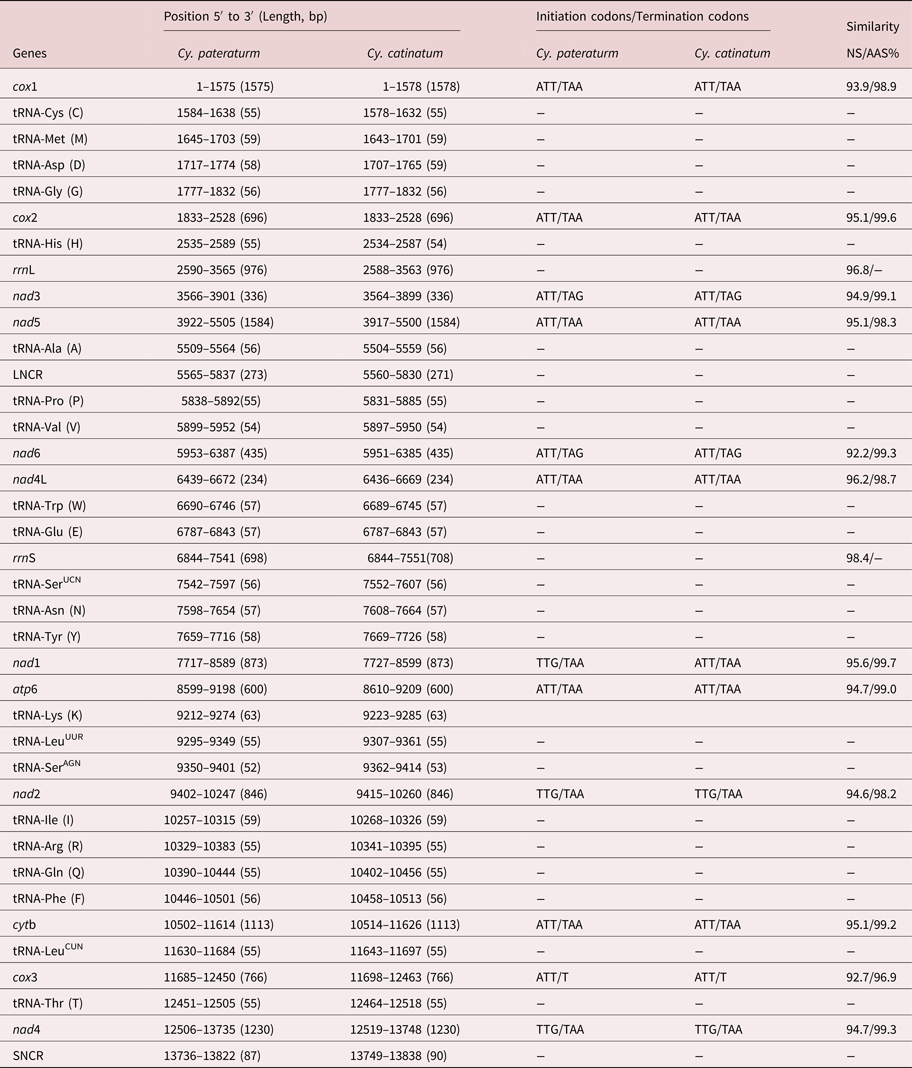
The complete mtDNA sequence of Cy. pateratum was a typical circular DNA molecule 13,822 bp in length (MG727531.1), which is slightly shorter than that of Cy. catinatum (13,838 bp). Both mtDNA sequences contained 36 genes (cox1–3, nad1–6, nad4L, atp6, cytb, rrnL, rrnS and 22 tRNAs) and two non-coding regions (long non-coding region (LNCR) and short non-coding region (SNCR)). All 36 genes were transcribed in the same direction in the two genomes, which is the same as other Chromadorea nematodes, such as Triodontophorus nipponicus and Bunostomum phlebotomum (Gao et al., Reference Gao2014, Reference Gao2017), but different for Enoplea nematodes, such as Trichinella spiralis and Trichuris trichiura (Lavrov and Brown, Reference Lavrov and Brown2001; Liu et al., Reference Liu2012b). The A + T content of the mt genome of Cy. pateratum was 75.68% (A = 30.42%; T = 45.26%), which is similar to that of the majority of Strongylidae nematodes, such as Cy. catinatum (76.12%) and Strongylus vulgaris (76.55%), but higher than that of Enoplea nematodes, such as T. spiralis (65.2%) and T. trichiura (68.1%), and trematodes, such as Eurytrema pancreaticum (62.5%) (Lavrov and Brown, Reference Lavrov and Brown2001; Liu et al., Reference Liu2012b; Chang et al., Reference Chang2016; Gao et al., Reference Gao2017b). The Cy. pateratum mtDNA sequence contained 20 intergenic sequences 1–51 bp in length. There was no overlap in the complete Cy. pateratum mtDNA sequence, whereas in the complete Cy. catinatum mtDNA sequence there was one overlap of 1 bp between cox1 and tRNA-Cys. Details of the structure of the Cy. pateratum and Cy. catinatum mt genes are in table 1.
Table 1. Locations and similarity of the mitochondrial genes of Cyathostomum pateratum and Cyathostomum catinatum.
Comparative analysis between the mt genomes of Cy. pateratum and Cy. catinatum and those of other nematodes
The complete mtDNA nucleotide sequences of Cy. pateratum and Cy. catinatum shared 94.6% identity. In other congeneric nematode species, such as B. phlebotomum China yak isolate and B. trigonocephalum sheep isolate, the complete mtDNA nucleotide sequences shared only 85.3% identity, whereas for Ascaris lumbricoides and Ascaris suum the identity was 98.81% (Liu et al., Reference Liu2012a; Gao et al., Reference Gao2014). The predicted concatenated amino acid sequences of the 12 protein-coding genes of Cy. pateratum and Cy. catinatum differed by 2.2%, which is similar to what was found in other congeneric species, such as Ancylostoma duodenale and Ancylostoma caninum (4.1%) (Jex et al., Reference Jex2009). The nucleotide and predicted amino acid sequences of the 12 protein-coding genes of Cy. pateratum and Cy. catinatum shared 92.2–96.2% and 96.9–99.6% identity, respectively (table 1). The nucleotide and amino acid sequences of cox3 were the least conserved among the 12 protein-coding genes. The ATT initiation codon and TAA termination codon were the most frequently used codons in Cy. pateratum and Cy. catinatum (table 1), and both had an incomplete codon T at the end of the cox3 gene, which is consistent with cox3 in the S. equinus and Aspiculuris tetraptera mt genomes (Xu et al., Reference Xu2015; Wang et al., Reference Wang2016).
The rrnL genes of Cy. pateratum and Cy. catinatum were located between tRNA-His and nad3 and both were 976 bp in length. The lengths of the rrnS genes differed; 698 bp in Cy. pateratum and 708 bp in Cy. catinatum. The rrnL and rrnS genes of Cy. pateratum and Cy. catinatum varied by 3.2% and 2.6%, respectively. The A + T contents of rrnL and rrnS in Cy. pateratum were 80.64% and 77.65%, and in Cy. catinatum 80.84% and 77.26%, respectively (Gao et al. Reference Gao2017b).
The 22 tRNA genes ranged in length from 52 bp (tRNA-SerAGN) to 63 bp (tRNA-Lys) in the mt genome of Cy. pateratum. The predicted secondary structures of the 22 tRNA genes are shown in supplementary fig. S1. The predicted secondary structure of 20 of the tRNAs (excluding two tRNA-Ser) have a DHU (dihydrouridine) arm and a DHU loop, in which the variable TψC arm and variable loop are replaced by a ‘TV-replacement loop’. Two tRNA-Ser genes of Cy. pateratum mt genome possess a TψC arm and variable loop, but lack the DHU arm and loop, which are common in most other nematode mtDNA sequences (Gao et al., Reference Gao2017a, Reference Gaob). The two non-coding regions (LNCR and SNCR) in the Cy. pateratum mt genome are conserved in the mt genomes of other Cyathostomum nematodes (e.g. Cy. catinatum, Cs. minutus and P. imparidentatum) as well as A. suum, T. serratus and T. nipponicus, but not in other nematodes, such as T. brevicauda and Oxyuris equi, which have one, three or more non-coding regions in their mt genomes (Liu et al., Reference Liu2012a; Duan et al., Reference Duan2015; Zhang et al., Reference Zhang2015; Gao JF et al., Reference Gao2017; Gao Y et al., Reference Gao2017b). The LNCRs of Cy. pateratum and Cy. catinatum were 273 bp and 271 bp in length, and the SNCRs were 87 bp and 90 bp in length, respectively. The non-coding regions in the mt genomes of other nematodes vary greatly; for example, in Syphacia obvelata, a non-coding region is 815 bp in length (Wang et al., Reference Wang2016). The A + T content of the non-coding regions in the Cy. pateratum mtDNA sequences was higher than in other regions, namely 82.75% for the SNCR and 86.35% for the LNCR.
Phylogenetic analysis of Cy. pateratum, Cy. catinatum and other Strongylidae nematodes
We used Bayesian inference and maximum likelihood methods to construct phylogenetic trees of Strongylidae nematodes based on the concatenated amino acid sequences of the 12 protein-coding mt genes. The topologies of the two trees reconstructed using the two different methods were identical. The phylogenetic trees had two large branches (fig. 1); one branch contained only M. ocydromi, a kangaroo parasite, and the other branch contained all the equine parasites. Two Strongylus species (S. equinus, S. vulgaris) clustered together and three Triodontophorus species (T. brevicauda, T. nipponicus, T. serratus) clustered together to form clades separate from the Cyathostominae nematodes. Cyathostomum catinatum and Cy. pateratum formed sister taxa, and congeneric Cs. goldi was closer to the two Cyathostomum species than to Cs. minutus.
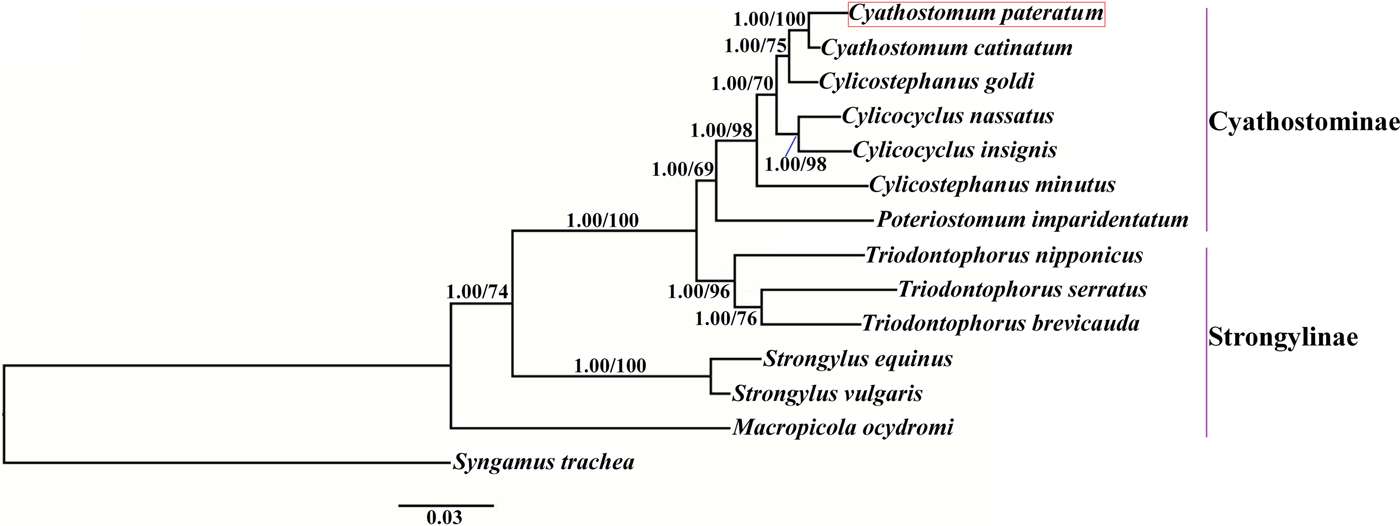
Fig. 1. Phylogenetic relationships among Strongylidae nematodes based on the concatenated amino acid sequences of 12 protein-coding genes by Bayesian inference and maximum likelihood methods. Syngamus trachea was used as the outgroup. The red box marks the species that was sequenced in the present study.
Gao et al. (Reference Gao2017b) discussed the phylogenetic relationships of 20 Strongyloidea nematodes based on the amino acid sequences of 12 protein-coding genes, which indicated that Cs. goldi was closer to Cy. catinatum than to other Cylicostephanus spp. However, this relationship could not be verified because the necessary biological information was lacking. In this study, Cy. catinatum, Cy. pateratum and Cs. goldi formed a cluster that had high statistical support, although Cy. pateratum and Cy. catinatum were closer to each other than to Cs. goldi. This result is consistent with that of McDonnell et al. (Reference McDonnell2000), who analysed the phylogenetic relationships among Strongylinae and Cyathostominae species using partial mitochondrial gene and rRNA ITS sequences.
Cyathostominae nematodes are a complex nematode group, and the genera of equine strongyles with large subglobular buccal capsules were considered to be ancestral to those with small cylindrical capsules (Lichtenfels, Reference Lichtenfels1987). Hung et al. (Reference Hung, Chilton and Gasser2000) used ITS sequences to reconstruct systematic relationships of 30 species of equine strongyles, and their molecular data supported this hypothesis. In the present study, the phylogenetic relationships among Strongylidae nematodes obtained using mtDNA data also support this idea. However, mtDNA data are not available for many Cyathostominae nematodes, so more mt genomes need to be sequenced and evolutionary relationships need to be re-examined to discover some complex species.
In conclusion, the complete mtDNA sequence of Cy. pateratum is reported here for the first time. Comparative and phylogenetic analyses based on the complete mt genome showed that Cy. pateratum is a closely related but distinct species to Cy. catinatum.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X18000688
Acknowledgements
We thank Margaret Biswas, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Financial support
This work was supported by the National Key Research and Development Program of China (2017YFD0501200), the Scientific Research Team and Platform Support Plan of Heilongjiang Bayi Agricultural University and the Natural Science Foundation of Heilongjiang Province of China (C2017048).
Conflict of interest
None.
Ethical standards
This study was performed strictly according to the recommendations of the Guide for the Care and Use of Laboratory Animals of the Ministry of Health, China, and the protocol was reviewed and approved by the Research Ethics Committee of Heilongjiang Bayi Agricultural University, China.