


Flavonoids constitute the largest class of dietary phytochemicals, which exhibit several well-documented biological and pharmacological activities including modulation of cell proliferation, differentiation and apoptosis, and modulation of inflammation, angiogenesis and metastasis development( Reference González, Ballester and López-Posadas 1 , Reference Lopez-Posadas, Ballester and Mascaraque 2 ). Because of these activities, flavonoids are considered putative nutraceuticals that may help prevent and/or treat cancer and chronic inflammatory conditions such as inflammatory bowel disease (IBD)( Reference Galvez, Sanchez de Medina, Jiménez and Atta 3 ). Among flavonoids, the flavone apigenin (4′,5,7-trihydroxyflavone) has been shown to inhibit cell growth, sensitise cells to apoptosis, and inhibit the development of metastatic disease in murine cells, as it is a promising molecule for cancer prevention and treatment( Reference Wang, Xu and Yin 4 – Reference Shukla, Bhaskaran and Babcook 7 ).
Although it is found in many fruits, vegetables and herbs, the amount of its presence varies, ranging from 302 mg/100 g in parsley to 0·38 mg/100 g in lettuce. In addition to parsley, the richest natural sources are celery, herbs such as rosemary, oregano, thyme, basil and coriander, chamomile, cloves, lemon balm, artichokes, spinach, peppermint, red wine and liquorice. This flavonoid has also attracted attention, thanks to its anti-inflammatory activities both in vitro ( Reference Wang, Xu and Yin 4 , Reference Shao, Jing and Mahmoud 6 , Reference Wang and Huang 8 , Reference Hoensch and Oertel 9 ) and in vivo ( Reference Mafuvadze, Cook and Xu 5 , Reference Shukla, Bhaskaran and Babcook 7 , Reference Man, Hupe and Sun 10 , Reference Ganjare, Nirmal and Patil 11 ). Nevertheless, little attention has been paid to the anti-inflammatory effects of apigenin on colitis( Reference Ganjare, Nirmal and Patil 11 ).
IBD, including ulcerative colitis and Crohn's disease, is a chronic, relapsing and remitting inflammatory condition of the gut. Crohn's disease can affect any part of the gastrointestinal tract, from the mouth to the anus, although the majority of the cases involve the terminal ileum and proximal colon. In contrast, ulcerative colitis is restricted to the colon and rectum. Both are characterised by a pronounced infiltration of neutrophils into colonic lesions, accompanied by epithelial cell damage and ulceration.
The exact pathogenesis of IBD is poorly understood; however, infection, environmental factors, heredity and immunological abnormalities have often been proposed as possible causes( Reference Abraham and Cho 12 ). Because of the limited understanding of the aetiopathogenesis of IBD, treatment is largely unspecific, i.e. based on targeting the inflammation rather than any precipitating factor.
Most of the current therapies for IBD involve treatment with glucocorticosteroids, 5-aminosalicylic acid and immunosuppressive drugs. Although many types of treatment for IBD have been proposed and clinically applied, additional preventive or therapeutic approaches are needed, because many patients either do not respond to the options currently available or suffer significant side effects, thereby precluding their continued use.
Phytochemical compounds such as flavonoids are considered basically safe, sustainable and practical. Furthermore, a change in dietary habits has been proposed as primary therapy for IBD( Reference Gassull 13 ). Flavonoids are quickly and extensively metabolised after absorption, resulting in limited bioavailability of the untransformed polyphenol, although the metabolites may, in some cases, become reactivated at target sites( Reference Kawai, Nishikawa and Shiba 14 ). Apigenin K has been designed as a form of apigenin, with improved water solubility (1·5 mg/ml in water compared with virtually zero solubility for apigenin; Fig. 1), in an attempt to enhance absorption. Therefore, apigenin K may be particularly suited to dampening intestinal inflammation.
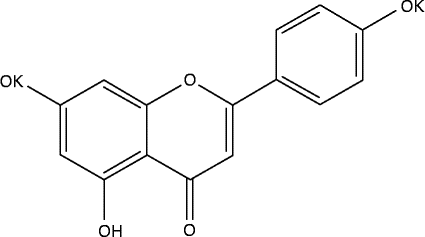
Fig. 1 Structure of apigenin K.
Much of the current knowledge about IBD has been derived from research carried out on animal models of the disease. Ideally, such models should closely resemble Crohn's disease and ulcerative colitis in order to investigate the molecular and cellular mechanisms of inflammation and immunological disorders, and to test potential therapeutic agents. However, there is no such model currently available. Instead, there are a variety of models that not only share a number of the characteristics of ‘human’ IBD, but also exhibit clear differences.
Among these animal models, trinitrobenzenesulfonic acid (TNBS)- and dextran sulphate sodium (DSS)-induced colitis in rodents are the most widely used models for the study of IBD( Reference Wirtz, Neufert and Weigmann 15 ). The TNBS experimental model exhibits many symptoms similar to those seen in human Crohn's disease, such as transmural oedema and inflammation( Reference Morris, Beck and Herridge 16 ), while the DSS model exhibits many symptoms similar to those seen in human ulcerative colitis, including diarrhoea, bloody faeces, body-weight loss or mucosal ulceration( Reference Cooper, Murthy and Shah 17 ). Therefore, these two models are thought to be reliable when studying the pathogenesis of IBD and testing drugs for treatment.
Based on the above-mentioned findings, we set out to test the hypothesis that apigenin exerts a protective effect on intestinal inflammation, using a soluble form of the flavonoid with a well-defined composition, namely apigenin K. Since a single model of colitis does not resemble all the features of human IBD, we used two different models, namely TNBS- and DSS-induced colitis, to strengthen the value of our findings.
Materials and methods
Chemicals
All reagents and primers were obtained from Sigma, unless otherwise indicated. Budesonide was purchased from Molekula Limited. Retrotranscription iScript™ cDNA Synthesis kits and GoTaq qPCR Master Mix were obtained from Bio-Rad and Promega, respectively. The Dynabeads mRNA Purification Kit was obtained from Ambion by Life Technologies. Apigenin K was supplied by Bioiberica.
Animals
Female Wistar rats (175–225 g) obtained from Harlan were housed in makrolon cages and maintained in air-conditioned animal quarters with a 12 h light–12 h dark cycle. They were provided with free access to tap water and a standard chow diet (Panlab A04; Panlab). The present study was carried out in accordance with the European Union's Directive for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (86/609/EEC), and was approved by the Ethical Committee of the University of Granada (reference no. 770).
Induction of colitis and experimental design
A total of three separate experiments were carried out (Fig. 2) . In Expt 1, TNBS colitis was induced( Reference Daddaoua, Puerta and Zarzuelo 18 ). Briefly, the experimental rats were fasted overnight and anaesthetised with isoflurane, and were then given 10 mg TNBS dissolved in 0·25 ml of 50 % ethanol (v/v) by means of a Teflon cannula, inserted 8 cm into the anus. They were kept in a head-down position for an additional 30 s, and returned to their cages. They were then randomly assigned to six different groups, namely control (n 4), TNBS (n 6), budesonide (2 mg/kg, n 4) and apigenin K at three different doses (1, 3 or 10 mg/kg each, in 1 % methylcellulose as the vehicle, n 4 per group) administered orally via a gastroesophageal catheter.

Fig. 2 Experimental design. (a) Expt 1 was a dose-finding pilot study. (b) Expt 2 was intended to explore further the effect of the dose selected. Both experiments were carried out in the rat trinitrobenzenesulfonic acid (TNBS) model. (c) Expt 3 tested the same apigenin K (ApiK) dose (3 mg/kg) in dextran sulphate sodium (DSS) colitis. Treatment for the control and TNBS/DSS groups was vehicle (1 % methylcellulose). DSS was continued from day 0 to day 9. Bude, budesonide. ![]() , Treatment administration;
, Treatment administration; ![]() , TNBS induction;
, TNBS induction; ![]() , DSS induction.
, DSS induction.
All groups received the TNBS challenge except the control group, which was administered a saline enema. All treatments started 2 d before colitis induction, and were maintained until the animals were killed, after 7 d. The budesonide group received 2 mg/kg of the corticoid by the oral route. The control and TNBS groups were administered the vehicle only. Food, water intake and body weight were measured daily.
In Expt 2, TNBS colitis was induced in a fashion similar to the aforementioned approach, but in this case, rats were randomly assigned to four different groups, namely control (n 4), TNBS (n 6), budesonide (2 mg/kg, n 4) and apigenin K at a dose of 3 mg/kg in 1 % methylcellulose (n 4). All groups received the TNBS challenge, except the control group, which was administered a saline enema. Treatments started 2 d before colitis induction, and were maintained until the animals were killed, 7 d after induction. The budesonide group received 2 mg/kg of the corticoid by the oral route. The control and TNBS groups were administered the vehicle only. Food, water intake and body weight were measured daily. Expt 2 was carried out both for confirmation purposes and to obtain samples for PCR analysis.
In Expt 3, DSS colitis was induced as described previously in the literature( Reference Lopez-Posadas, Requena and Gonzalez 19 ). DSS was obtained from MP Biomedicals and was added to drinking-water for 9 d at a concentration of 4 % (w/v). The status of the rats was monitored through a general examination, and specifically by using the disease activity index (DAI), a combined score for weight loss, diarrhoea and haematochezia, which are the three main signs of pathology in this model( Reference Ito, Kita and Shin-Ya 20 ). Rats were randomly assigned to three different groups. The control group (n 6) did not receive DSS, and was administered 1 % methylcellulose daily by means of a gastroesophageal catheter. The remaining rats drank DSS-supplemented water, and received orally either 3 mg/kg of apigenin K (apigenin K group, n 6) or vehicle (1 % methylcellulose, DSS group, n 6). Treatment started 2 d before DSS supplementation, and was maintained until rats were killed by cervical dislocation, 9 d after DSS was started.
Assessment of colonic damage
In all the three experiments, after the animals were killed, the status of the large intestine was assessed, as described previously( Reference Daddaoua, Puerta and Zarzuelo 18 ). Briefly, the large intestine was opened longitudinally and scored for visible damage on a 0–25 scale by an observer unaware of the treatment, according to the criteria detailed in the aforementioned work, including hyperaemia, fibrosis, thickening and ulceration( Reference Daddaoua, Puerta and Zarzuelo 18 ). Colonic myeloperoxidase (MPO) and alkaline phosphatase (AP) activities were measured spectrophotometrically, as described previously( Reference Sanchez de Medina, Martinez-Augustin and Gonzalez 21 ). In brief, MPO was extracted with hexadecyl trimethyl ammonium bromide and three freeze–thaw cycles with sonication, and measured as a peroxidase enzymatic activity using 0·0005 % H2O2 and 0·168 mg/ml of o-dianisidine as the substrate. AP was quantified using disodium nitrophenylphosphate (5·5 mm) as the substrate in a 50 mm-glycine buffer with 0·5 mm-MgCl2 (pH 10·5). MPO and AP are expressed as mU/mg protein and U/mg protein, respectively (1 U = 1 μmol/min of substrate converted). In addition, the sensitivity of AP to the specific inhibitor levamisole was assessed in vitro.
Analysis of gene expression by quantitative RT-PCR analysis
Total RNA was obtained by the TRIzol method (Invitrogen), 1 μg was retrotranscribed and specific RNA sequences were amplified with a Bio-Rad CFX Connect real-time PCR device using the following primers: AGC TGG GTG CTG GCA TTC TC/TCC TGC AAC AGT TGG GCT TAT C (Def1; defensin 1); TGA TGT GTC TCC TGC TTC TAG CCT/AGC ACC TCA ATG TCG TCT TAT GGC (Il-17c); TGC CCT CAG TTT GTG CAG AA/GCC CAC CCT TAT CAC CAA C (S100a8; S100 calcium binding protein (calgranulin A)); GCT CTG GTC TTC TGG AGT TCC G/TTG GAT GGT CTT GGT CCT TGA CC (Il-6); GTC GTA GCA AAC CAC CAA/GCT GAC TTT CTC CTG GTA TG (Tnf-α); TCG GCT GGA GAA CTA CAA/CTG AAG TCC TTA GGG TTG ATG (Mcp1; monocyte chemotactic protein 1 (Ccl2; chemokine (C–C motif) ligand 2)); GTC AAA CGG GAG ATG AAT GG/TCT TCC TCT GGC GGT AAT AG (Icam1; intercellular adhesion molecule 1); CGA TGT GGA TGC GTT TCA/CAC ACA TTC CTC ACC CTA AC (Cxcl1; C–X–C motif chemokine 1 (growth-regulated oncogene α; GRO-α)); AAT GAC CTG TTC TTT GAG GCT G/CGA GAT GCT GCT GTG AGA TTT (Il-1β); TGG ACT CTG AGC CGC AAT GAG G/GAC GCA TGG CGG ACA ATA GAG G (Il-17); GGT GAC TTG CGA GCC TGT GTG/CCC AGC AGG AAA GCA GAC TCG (Tlr2; Toll-like receptor 2); CGT GGC TTC TAG TGC TGA CG/ACT GGC GAG CCT TAG TTT G (Tgf-β; transforming growth factor-β); CCC AGG AAA GAC AGC AAC CTT/CTG CTT GGC AGT GCT TGA GAA (Foxp3; forkhead box P3); CCA TTG GAG GGC AAG TCT GGT G/CGC CGG TCC AAG AAT TTC ACC (18S; 18S ribosomal RNA); GTC GGT GTG AAC GGA TTT/ATG GGT TTC CCG TTG ATG (GAPDH; glyceraldehyde-3-phosphate dehydrogenase). Because DSS is a potent inhibitor of real-time quantitative PCR amplification, and interferes with tissue-specific gene expression analysis in DSS-exposed rats, we overcame this problem by further purifying DSS-exposed RNA(
Reference Kerr, Ciorba and Matsumoto
22
). Oligodeoxythymidine purification of mRNA was performed using the Invitrogen Dynabeads mRNA Purification Kit. Results are expressed as
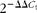 $$2^{ - \Delta \Delta C _{t}} $$
, with 18S as the reference gene in the TNBS-induced experiment and GAPDH in the DSS-induced experiment.
$$2^{ - \Delta \Delta C _{t}} $$
, with 18S as the reference gene in the TNBS-induced experiment and GAPDH in the DSS-induced experiment.
Statistical analyses
Results are expressed as means with their standard errors. Data were analysed by one-way ANOVA and post hoc least significant difference tests, except when the necessary conditions (i.e. normal distribution and equal variance) were not fulfilled, in which case a non-parametric Kruskal–Wallis ANOVA was applied. Logarithmic transformation was applied to the PCR data before statistical analyses. All the analyses were carried out with the SigmaStat program (Jandel), with the exception of DAI data, which were analysed by repeated measurements using NCSS (NCSS-LLC). No significant treatment × time interaction was noted in this case. Statistical significance was set at P< 0·05.
Results
Effects of apigenin K on trinitrobenzenesulfonic acid-induced colitis
To determine the optimal anti-inflammatory dose, we carried out an initial experiment in which the flavonoid was administered to rats at doses of 1, 3 and 10 mg/kg per d, before colitis induction by TNBS. As expected, administration of TNBS induced a severe inflammatory response in the large intestine, characterised by mucosal erosions, epithelial necrosis, submucosal fibrosis and oedema, resulting in a marked increase in the colonic damage score (Table 1). TNBS colitis was also characterised by anorexia and weight loss, augmented colonic damage extension and an increase in the colonic weight:length ratio (Table 1). Macroscopic evaluation of colitic rats treated with apigenin K revealed that the colonic weight:length ratio was not significantly different from that of the control group, with all the three doses assayed. Nor were colonic damage extension data significantly different from those of the control group, except in the case of the lowest dose (Table 1). In both cases, there was no significant reduction compared with the TNBS group, possibly due to the small size of the experimental groups. However, there was no effect on the colonic damage score.
Table 1 Effects of different doses of apigenin K (ApiK) on body-weight gain, food intake, macroscopic and biochemical parameters in rat trinitrobenzenesulfonic acid (TNBS) colitis (Expt 1) (Mean values with their standard errors, n 4–6)
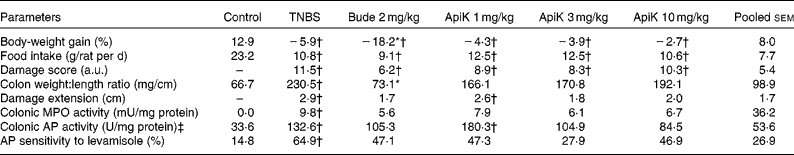
Bude, budesonide; a.u., arbitrary units; MPO, myeloperoxidase; AP, alkaline phosphatase.
* Mean value was significantly different from that of the TNBS group (P< 0·05).
† Mean value was significantly different from that of the control group (P< 0·05).
‡ 1 U = 1 μmol/min.
TNBS colitis was associated with a significant increase in MPO and AP activities, both of which are biochemical parameters of inflammation (Table 1). Furthermore, the sensitivity of AP activity to the specific inhibitor levamisole in vitro was also heightened, consistent with a change in isoform expressed in the inflamed intestine, as described in the literature( Reference Lopez-Posadas, Gonzalez and Ballester 23 ). Similar to the aforementioned data, these parameters showed intermediate values between those of the control group and of the TNBS group, and were not significantly different from either (Table 1). Notably, MPO and AP activities were 33–39 and 21–36 % lower in the TNBS group than in the groups treated with 3 or 10 mg/kg of apigenin K, respectively. There was no effect on body-weight loss or food intake.
In contrast, budesonide – a corticoid with predominantly local action, used as a positive control for colitic therapy in the present study – inhibited colonic thickening significantly and effectively, but was, in other respects, largely comparable with apigenin K (Table 1). Thus, damage extension and score, MPO and AP activities, and sensitivity to levamisole were generally similar to those obtained with apigenin K, especially at the dose of 3 mg/kg. However, budesonide produced significant body wasting compared with the TNBS group (Table 1), without changes in food intake, and a 25 % mortality rate (one-fourth) compared with 0 % in the TNBS group (Fisher's exact test, NS).
The results of Expt 1 suggest that apigenin K at 3 mg/kg may be the most efficient dose for reducing colitis-related inflammation. Therefore, we selected this dose for further testing in Expt 2. This was aimed at confirming therapeutic efficacy, and providing samples for the purpose of undertaking further analyses. As in Expt 1, pre-treatment with apigenin K at 3 mg/kg resulted in a significant reduction of the inflamed area, as well as lower values of score and colonic weight:length ratio (P>0·05; Table 2). Apigenin K pre-treatment was associated with body-weight gain and food intake values close to those of the non-colitic control group. Colonic MPO activity was 30 % lower than that in the TNBS group, although this did not reach statistical significance (Fig. 3). A marginal effect was also apparent on AP activity. Budesonide was markedly less effective in Expt 2 than in Expt 1, especially with regard to the weight:length ratio that was unchanged, while it produced a similar wasting effect in the two experiments (Table 2; Fig. 3).
Table 2 Effects of apigenin K (ApiK) at the dose of 3 mg/kg on body-weight gain, food intake and macroscopic damage parameters in rat trinitrobenzenesulfonic acid (TNBS) colitis (Expt 2) (Mean values with their standard errors, n 4–6)
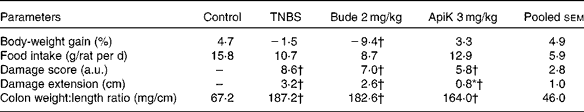
Bude, budesonide; a.u., arbitrary units.
* Mean value was significantly different from that of the TNBS group (P< 0·05).
† Mean value was significantly different from that of the control group (P< 0·05).

Fig. 3 Effects of apigenin K (ApiK) on colonic (a) myeloperoxidase (MPO) and (b) alkaline phosphatase (AP) activities in rat trinitrobenzenesulfonic acid (TNBS) colitis. Rats with TNBS colitis were treated with ApiK or budesonide as described in the ‘Materials and methods’ section. (c) In vitro sensitivity of AP activity to the specific inhibitor levamisole. 1 U = 1 μmol/min. Values are means (n 4–6), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the control group (P< 0·05).
To test the effect of apigenin K at 3 mg/kg on the expression of different genes, colonic mRNA was extracted and studied by quantitative RT-PCR. Since monocytes are attracted to the inflammatory site, CCL2/MCP1 was measured as a marker of monocyte infiltration, together with ICAM1, an intracellular adhesion molecule useful in leucocyte transmigration; IL-1β, a widely known activator of macrophages and a lymphocyte mitogen; IL-6, a pro-inflammatory cytokine produced by these two cell types; and TNF-α, chiefly produced by activated macrophages. The expression of Mcp1, Icam1, Il-1β, Tnf-α and Il-6 was significantly increased after the administration of TNBS (Fig. 4). After treatment with apigenin K, the expression of Mcp1 and Icam1 was found to be significantly down-regulated when compared with the TNBS group (Fig. 4). In accordance with these results, the expression of Il-1β, Tnf-α and Il-6 was also decreased, and was not found to be significantly different from the expression of the control group, indicating a lower degree of macrophage/lymphocyte infiltration/activation (Fig. 4).

Fig. 4 Effects of apigenin K (![]() ) on the expression of colonic inflammatory markers in rat trinitrobenzenesulfonic acid (TNBS,
) on the expression of colonic inflammatory markers in rat trinitrobenzenesulfonic acid (TNBS, ![]() ) colitis. Rats with TNBS colitis were treated with apigenin K as described in the ‘Materials and methods’ section, and inflammatory markers were measured by quantitative RT-PCR. Values are means (n 4–6), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the TNBS group (P< 0·05). † Mean value was significantly different from that of the control (
) colitis. Rats with TNBS colitis were treated with apigenin K as described in the ‘Materials and methods’ section, and inflammatory markers were measured by quantitative RT-PCR. Values are means (n 4–6), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the TNBS group (P< 0·05). † Mean value was significantly different from that of the control (![]() ) group (P< 0·05). Foxp3, forkhead box P3; Tlr2, Toll-like receptor 2; Tgf-β, transforming growth factor-β; S100a8, S100 calcium-binding protein A8; Def1, defensin 1; Ccl2, chemokine (C–C motif) ligand 2; Icam1, intercellular adhesion molecule 1; Cxcl1, chemokine (C–X–C motif) ligand 1.
) group (P< 0·05). Foxp3, forkhead box P3; Tlr2, Toll-like receptor 2; Tgf-β, transforming growth factor-β; S100a8, S100 calcium-binding protein A8; Def1, defensin 1; Ccl2, chemokine (C–C motif) ligand 2; Icam1, intercellular adhesion molecule 1; Cxcl1, chemokine (C–X–C motif) ligand 1.
Because manoeuvres leading to increased differentiation of regulatory T cells have been shown to have anti-inflammatory effects( Reference Park, Jin and Lopez 24 – Reference Furusawa, Obata and Fukuda 26 ), the expression of Tgf-β and Foxp3 was studied. TGF-β is a multifunctional peptide that controls the proliferation and differentiation of multiple cells, and is a necessary factor for the induction of Foxp3 expression in regulatory T cells. The present results show a significant up-regulation of Foxp3 expression in the TNBS group, with no significant change in TGF-β. Pre-treatment with apigenin K resulted in values similar to those in the control group, but did not reach statistical significance, compared with the TNBS group (Fig. 4). The expression of Tlr2, which plays a fundamental role in pathogen recognition and activation of innate immunity, showed a high, but not significant, up-regulation in the TNBS group that was reduced by apigenin K, but, again, not significantly (Fig. 4). The expression of other inflammatory markers such as Il-17, Il-17c, Def1 and Cxcl1 were also analysed, but no significant differences were found with apigenin K pre-treatment (Fig. 4).
Effects of apigenin K on dextran sulphate sodium-induced colitis
It was demonstrated earlier that apigenin K played a protective role in the TNBS-induced colitis model. For this reason, we decided to confirm the anti-inflammatory effects of apigenin K in a different model of colitis. Supplementation of drinking-water with DSS caused significant colonic damage, as reflected by the increased DAI, beginning day 5 after DSS induction (Fig. 5), and the augmented colonic damage score and weight:length ratio (Table 3). Pre-treatment of rats with 3 mg/kg of apigenin K daily attenuated DSS-induced colitis, as evidenced by a significantly lower DAI from day 6 onwards (Fig. 5), a lower colonic damage score and a higher body-weight gain (Table 3). In addition, DSS-treated rats exhibited anorexia, which was improved in the apigenin K group compared with the control group (Table 3). However, the colonic weight:length ratio was not significantly improved (Table 3).

Fig. 5 Effects of apigenin K (![]() ) on the disease activity index in rat dextran sulphate sodium (DSS,
) on the disease activity index in rat dextran sulphate sodium (DSS, ![]() ) colitis. Rats with DSS colitis were treated with apigenin K as described in the ‘Materials and methods’ section. Data were analysed by repeated-measures ANOVA with Fisher's least significant difference tests. No significant treatment × time interactions were detected. Values are means (n 4–6), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the trinitrobenzenesulfonic acid group (P< 0·05). † Mean value was significantly different from that of the control (
) colitis. Rats with DSS colitis were treated with apigenin K as described in the ‘Materials and methods’ section. Data were analysed by repeated-measures ANOVA with Fisher's least significant difference tests. No significant treatment × time interactions were detected. Values are means (n 4–6), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the trinitrobenzenesulfonic acid group (P< 0·05). † Mean value was significantly different from that of the control (![]() ) group (P< 0·05). a.u., Arbitrary units.
) group (P< 0·05). a.u., Arbitrary units.
Table 3 Effects of apigenin K (ApiK) on body-weight gain and macroscopic damage parameters in rat dextran sulphate sodium (DSS) colitis (Expt 3) (Mean values with their standard errors, n 6)

a.u., Arbitrary units.
* Mean value was significantly different from that of the TNBS group (P< 0·05).
† Mean value was significantly different from that of the control group (P< 0·05).
Colonic MPO and AP activities and sensitivity to levamisole were significantly greater in the DSS group than in the control group (Fig. 6). Pre-treatment with apigenin K at 3 mg/kg reduced MPO activity by 35 % and colonic AP activity by 21 %, with a similar effect on its sensitivity to levamisole, compared with the DSS group (P>0·05; Fig. 6).

Fig. 6 Effects of apigenin K (ApiK) on colonic (a) myeloperoxidase (MPO) and (b) alkaline phosphatase (AP) activities in rat dextran sulphate sodium (DSS) colitis. Rats with DSS colitis were treated with ApiK as described in the ‘Materials and methods’ section. (c) In vitro sensitivity of AP activity to the specific inhibitor levamisole. 1 U = 1 μmol/min. MPO data were analysed by Kruskal–Wallis ANOVA. Values are means (n 4–6), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the control group (P< 0·05).
In the DSS group, there was a general increase in the colonic mRNA levels of several inflammatory parameters, such as Il-17, Il-6, Ccl-2, Tnf-α, Icam1, Il-17c, Cxcl1, Foxp3, Il-1β, Tlr2 and Tgf-β, of which the last three reached statistical significance (Fig. 7). Rats treated with apigenin K showed a trend towards normalisation of a number of these parameters, but a significant effect was observed only for Il-1β (Fig. 7).

Fig. 7 Effects of apigenin K (ApiK, ![]() ) on the colonic expression of inflammatory markers in rat dextran sulphate sodium (DSS,
) on the colonic expression of inflammatory markers in rat dextran sulphate sodium (DSS, ![]() ) colitis. Rats with DSS colitis were treated with ApiK as described in the ‘Materials and methods’ section, and inflammatory markers were measured by quantitative RT-PCR. Values are means (n 4–6), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the trinitrobenzenesulfonic acid group (P< 0·05). † Mean value was significantly different from that of the control (
) colitis. Rats with DSS colitis were treated with ApiK as described in the ‘Materials and methods’ section, and inflammatory markers were measured by quantitative RT-PCR. Values are means (n 4–6), with their standard errors represented by vertical bars. * Mean value was significantly different from that of the trinitrobenzenesulfonic acid group (P< 0·05). † Mean value was significantly different from that of the control (![]() ) group (P< 0·05). Foxp3, forkhead box P3; Tlr2, Toll-like receptor 2; Tgf-β, transforming growth factor-β; S100a8, S100 calcium-binding protein A8; Def1, defensin 1; Ccl2, chemokine (C–C motif) ligand 2; Icam1, intercellular adhesion molecule 1; Cxcl1, chemokine (C–X–C motif) ligand 1.
) group (P< 0·05). Foxp3, forkhead box P3; Tlr2, Toll-like receptor 2; Tgf-β, transforming growth factor-β; S100a8, S100 calcium-binding protein A8; Def1, defensin 1; Ccl2, chemokine (C–C motif) ligand 2; Icam1, intercellular adhesion molecule 1; Cxcl1, chemokine (C–X–C motif) ligand 1.
Discussion
Based on the amelioration of various general and biochemical parameters, our data demonstrate that apigenin K, a soluble derivative of apigenin, is efficacious in TNBS and DSS colitis, the two most widely used models of IBD. Apigenin has been shown to exert significant immunomodulatory actions in vitro, notably on monocytes, dendritic cells, lymphocytes, endothelial and intestinal epithelial cells. These actions involve, in part, the inhibition of the NF-κB pathway. One mechanism proposed in the literature is related to changes in p50/p65 phosphorylation and blockade of inhibitor of κB kinase (IKK)-γ( Reference Nicholas, Batra and Vargo 27 ), and inhibition of inhibitor of κB-α phosphorylation( Reference Comalada, Ballester and Bailon 28 ). The inhibition of monocyte activation probably underlies the beneficial effect of apigenin on lipopolysaccharide-induced shock in mice( Reference Nicholas, Batra and Vargo 27 ). In EL4 T lymphoma and primary lymph node cells, apigenin has been found to inhibit nuclear factor of activated T-cells (NFAT) DNA binding, resulting in lower IL-4 release( Reference Park, Kim and Kim 29 ); reduce T-cell proliferation and activation( Reference Yin, Gong and Wu 30 , Reference Liang, Zeng and Huang 31 ); and exhibit cytotoxic/apoptotic effects in rat splenocytes( Reference López-Posadas, Ballester and Abadía 32 ). In Mode-K intestinal epithelial cells, apigenin inhibits IP-10 (CXCL10) secretion via the blockade of Akt( Reference Ruiz and Haller 33 ).
Thus, apigenin appears to exert generally anti-inflammatory effects. However, all in vitro actions documented for apigenin are obviously a direct effect (i.e. exerted by apigenin itself), and it is important not to assume that they do occur necessarily in vivo. Indeed, oral bioavailability of apigenin is very low, as is the case with most flavonoids( Reference Walle 34 – Reference Manach, Williamson and Morand 37 ), so that in vitro actions may not translate easily into clinically relevant anti-inflammatory effects. In this regard, apigenin K was developed as an apigenin derivative with improved aqueous solubility, aiming at an increased bioavailability. In so far as the colonic anti-inflammatory effect of apigenin depends on absorption, the use of apigenin K may present obvious advantages compared with the parent compound. However, it should be noted that the exact tissue targets of apigenin, or other flavonoids, are not clearly defined.
Apigenin, as such, has been shown to have beneficial effects in the acetic acid model of colitis( Reference Ganjare, Nirmal and Patil 11 ). This is an acute model with little involvement of immune mechanisms, which is thought to be produced by alteration of the intracellular pH at the epithelial level, resulting in coagulative necrosis of the epithelium and mucosa. This model is used chiefly for screening purposes, but, otherwise, more relevant IBD models such as TNBS or DSS colitis, among others, are preferred.
Apigenin was found in that study to offer protection at 5 mg/kg by the oral route, evidenced by a lower colonic damage score, MPO activity and malondialdehyde level (an index of oxidative stress). This dose is comparable to that used with other flavonoids, tested for intestinal anti-inflammatory activity in vivo, for instance quercitrin, rutin, diosmin, hesperidin or morin( Reference González, Ballester and López-Posadas 1 , Reference Crespo, Galvez and Cruz 38 , Reference Ocete, Galvez and Crespo 39 ). Therefore, we initially assayed three doses of apigenin K of the same magnitude, namely 1, 3 and 10 mg/kg (semi-logarithmic progression), in the TNBS model. The effect on colonic damage score and extension, weight:length ratio, MPO and AP activities indicates that the 3 mg/kg dose is superior to 1 mg/kg, but the 10 mg/kg dose is equivalent to 3 mg/kg; that is, the dose–response curve flattens out at 3 mg/kg. Therefore, we selected this dose to carry out additional measurements by quantitative RT-PCR, and also for testing in the DSS model. It is important to note that the effects of apigenin K on TNBS colitis were largely comparable with those of budesonide, one of the drugs currently used in the management of IBD, while it did not cause body-weight loss.
Apigenin K had a broad impact on the expression of inflammatory markers in the colonic mucosa in both models of colitis. This is relevant because there is no ideal model of IBD( Reference Koboziev, Karlsson and Zhang 40 ), and each model can only reproduce certain aspects of the human disease, and not others. Thus, clinical efficacy is expected to correlate best with bioactivity established in more than one model. While the inflammatory parameters affected in TNBS and DSS largely overlap with those of the present study, it is interesting to note the distinct effect of DSS colitis on IL-17C and, to a lesser extent, on CXCL1, both of which are cytokines of predominant or exclusive epithelial origin( Reference Reynolds, Martinez and Nallaparaju 41 , Reference Egesten, Eliasson and Olin 42 ). In terms of differential effects of apigenin K on TNBS colitis, the colonic weight:length ratio and damage (necrosis) extension were major targets, while it had no effect on the damage score or body weight. However, in DSS colitis, apigenin K had a marked effect on body-weight gain/DAI, food intake and colonic damage score, with little change in the colonic weight:length ratio.
Some of these differences may be related to the higher severity of TNBS v. DSS colitis in the present study, although these were more qualitative than quantitative, and, therefore, do not explain the results. Instead, the distinct aetiopathogenic characteristics of the two models used may underlie these differences. TNBS acts as a hapten, reacting with proteins in the intestinal mucosa to yield immunogens that, in turn, elicit a T helper 1 response( Reference Allgayer, Deschryver and Stenson 43 ). This can be reproduced in vivo by directly injecting TNBS into the mucosa( Reference Allgayer, Deschryver and Stenson 43 ), but normally a 50 % ethanol solution is used as a vehicle for TNBS to serve as a barrier breaker( Reference Morris, Beck and Herridge 16 , Reference Martinez-Augustin, Merlos and Zarzuelo 44 ). Although ethanol evokes colonic inflammation by itself, the reaction is short-lived, and there are no immunological mechanisms involved. In addition, TNBS may induce oxidative stress, which can contribute to colitis( Reference Grisham, Volkmer and Tso 45 ).
Conversely, DSS induces inflammation by a mechanism that involves epithelial disruption by a direct action on enterocytes( Reference Wirtz, Neufert and Weigmann 15 ). Altered permeability augments the exposure of mucosal immunocompetent cells to bacterial antigens and viable micro-organisms, resulting in a secondary inflammatory reaction. The fact that damage score is significantly improved by apigenin K in DSS, but not in TNBS, colitis is consistent with an important effect of the flavonoid compound on epithelial cells. This is also consistent with the up-regulation of Il-17c in DSS, but not in TNBS, colitis and its amelioration by apigenin K. Similarly, the markedly higher increase observed in DSS colitis compared with TNBS colitis may reflect a higher impact on the epithelium in the former model. In contrast, subepithelial actions may be more relevant to the beneficial effect on TNBS colitis. The known effects of apigenin on monocytes and lymphocytes probably account for much of the anti-inflammatory effect observed in both the models, and they explain the inhibition of cytokines derived from these cell types such as IL-6 or TNF-α. However, the mechanism may be more complex, as suggested, for instance by the apparent lack of effect on CXCL1, which, indeed, may even be viewed as being up-regulated by apigenin K. In this regard, apigenin K shows promising activity against experimental colitis, and may be clinically helpful in the future as a functional food, but further experimentation is warranted.
Acknowledgements
The authors thank Dr Mercedes González for her help in carrying out the study.
The present study was funded by Fundación Ramón Areces; the Ministerio de Economía y Competitividad (SAF2008-01432, AGL2008-04332, SAF2011-22922 and SAF2011-22812); the SENIFOOD project (featuring Bioiberica); and the Junta de Andalucía (CTS164 and CTS6736). C. M. was funded by the Ministerio de Educación, Spain. R. G. was funded by CIBERehd. CIBERehd was funded by the Instituto de Salud Carlos III. The group is a member of the Network for Cooperative Research on Membrane Transport Proteins (REIT), co-funded by the Ministerio de Educación y Ciencia, Spain, and the European Regional Development Fund (grant no. BFU2007-30688-E/BFI).
The present study was partly funded by Bioiberica that supplied apigenin K.
The authors' contributions are as follows: C. M. and R. G. carried out the experiments; M. D. S., A. Z., F. S. M. and O. M.-A. designed the experiments; F. S. M. and O. M.-A. wrote the manuscript. All authors contributed to the discussion and the overall structure of the manuscript.
The authors declare that there are no conflicts of interest.










