INTRODUCTION
Young onset Parkinson’s disease (YOPD), defined as PD in patients 40 years of age or less, is uncommon (incidence of 0.5/100000)Reference Van Den Eeden, Tanner, Bernstein, Fross, Leimpeter and Bloch 1 and represents 3-5% of all patients with parkinsonism.Reference Schrag, Ben-Shlomo, Brown, Marsden and Quinn 2 Several mutations have been described in association with YOPD but parkin is by far the most common one.Reference Paviour, Surtees and Lees 3 In contrast to late-onset PD, YOPD patients usually have a slower disease progression and less cognitive decline. However, motor fluctuations and dyskinesias appear soon after initiating levodopa treatment.Reference Schrag, Ben-Shlomo, Brown, Marsden and Quinn 2
Wilson’s disease (WD) is an autosomal recessive disorder of copper balance caused by a mutation in the copper-transporting gene ATP7B.Reference Machado, Chien, Deguti, Cancado, Azevedo and Scaff 4 More than 500 ATP7B mutations have now been identified. (http://www.wilsondisease.med.ualberta.ca/database.asp). Worldwide incidence of WD is 30 cases/million and the carrier frequency is 1 in 90.Reference Figus, Angius, Loudianos, Bertini, Dessi and Loi 5 It usually starts with copper liver accumulation during childhood followed, in some cases, by copper storage in the central nervous system, which is responsible for the most common neurological manifestations such as dysarthria, gait disturbances, dystonia, tremor, and parkinsonism. Brain MRI abnormalities are found in almost all individuals with WD and neurological dysfunction.Reference Machado, Chien, Deguti, Cancado, Azevedo and Scaff 4
CASE REPORT
This is a 67-year-old Spanish woman who presented with resting tremor of her left arm at age 38, followed by clumsiness, rigidity and dystonia of the same limb. She was started on levodopa/carbidopa 300 mg/day with a good response. As part of routine screening for YOPD, increased 24 hour urinary copper excretion (90 µg; normal=0-60 μg) was detected, but liver function was normal and Kaysher-Fleischer (KF) rings were absent by slit lamp examination. A liver biopsy detected raised copper in dry tissue (507 μg/g; normal ≤250 μg/g) with no other specific histological findings. She was diagnosed with WD and started on penicillamine 750 mg/day for the next 11 years. Two of her four siblings (a 42 y.o. female and a 33 y.o. male) were completely asymptomatic but also showed abnormal urine copper excretion, and the sister had high copper concentration in liver biopsy (1075 μg/g).
A brain MRI at the age of 45 did not show any abnormality. An 18-Fluorodopa-PET brain scan (1991, Hammersmith Hospital, London, UK) showed an asymmetric putaminal dopaminergic reduction, typical of PD.
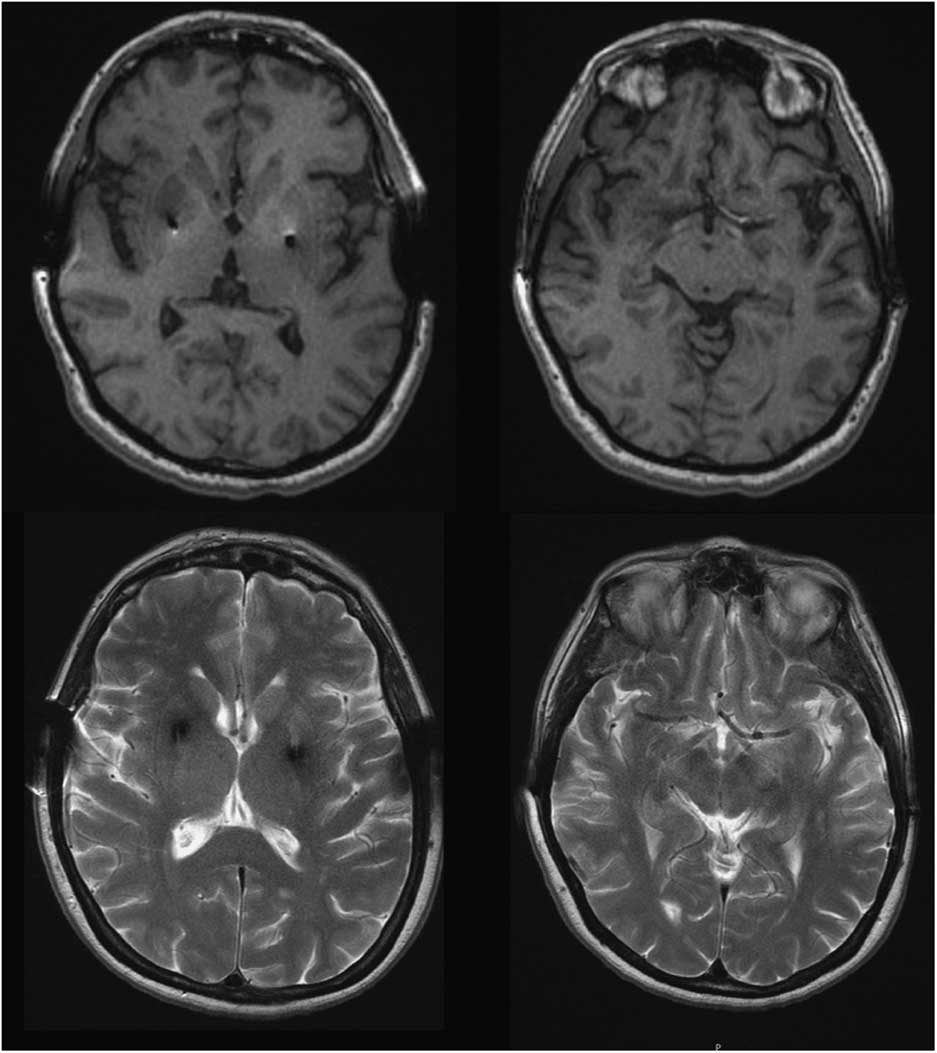
Four years after initiating levodopa she developed motor fluctuations, with very severe “off” dystonia in the lower limbs and peak dose choreic dyskinesias. Several drug regimens were tried, with only gradual and temporary improvement. At age 46, as a consequence of a digestive bleeding due to a stomach ulcer, levodopa treatment was withdrawn and a marked motor deterioration was noted in the form of severe generalized rigidity and bradykinesia (more prominent on the left) and marked hypomimia (UPDRS-III: 58 points). No atypical signs for PD were detected. Amplitude and latency of somatosensory evoked potentials, as well as corticospinal conduction measured by transcranial magnetic stimulation, were normal. Levodopa (1200 mg/day) and pergolide (6 mg/day) led to marked motor improvement (Supplementary Video). Serum copper and ceruloplasmin were low in most follow-up assessments, and urinary copper showed fluctuations. At the age of 49 penicillamine was discontinued because liver function was normal and the KF rings were absent; this was not associated with any change in the clinical status. She underwent bilateral Globus pallidus deep brain stimulation in 1996, with an excellent outcome over the next few years. Subsequent brain MRIs (49, 62, and 65 y.o.) revealed no specific cerebral abnormalities and showed correct placement of the pallidal electrodes (Figure 1). While no significant brain atrophy was observed, longitudinal morphometric analysis could not be performed due to different MRI protocols over time.
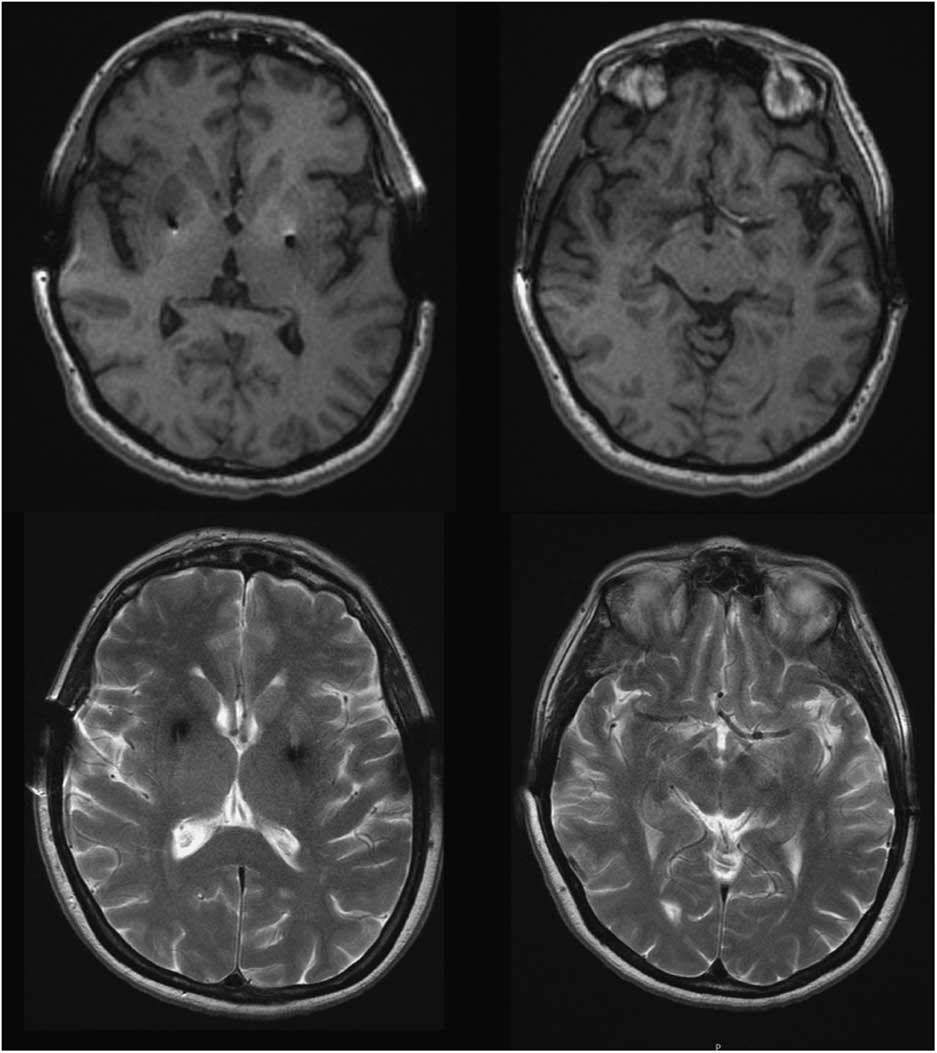
Figure 1 Brain MRI, T1 and T2-weighted images at the age of 65 showed no abnormal findings aside from bilateral GPi electrodes.
At the age of 65 a genetic test for Wilson’s disease showed a compound heterozygosity for c.1934T>G (Met645Arg) at exon 6 and c.2303C>T (Pro768Leu) at exon 8 of the ATP7B gene (NM_000053.3). Both mutations were confirmed as being in trans by familial segregation. Parkin and LRRK2 mutations were negative. Her sister showed the same ATP7B genotype. She was started on Zinc (100 mg/day), which has been maintained to date. She remains cognitively normal, but gait and speech have deteriorated in the last five years, as expected for longstanding PD. Otherwise she has remained asymptomatic and liver function continues to be normal. To date several ophthalmological assessments have failed to disclose KF rings. Her siblings show no evidence of neurological disease, but her sister recently showed some evidence of liver steatosis.
DISCUSSION
We report a patient with a clinical presentation and evolution typical of YOPD, and an abnormal copper metabolism with a genetic confirmation for WD. This case highlights a significant question: does the patient have two different diseases? Conversely, does the parkinsonism constitute part of neurological WD? Neurological WD starts as a consequence of copper deposition in the brain, after liver overload typically showing abnormal MRI. The most common findings are areas of high T2 signal in the lentiform and caudate nuclei, thalamus, brainstem, and white matter. Our patient’s brain MRI was normal, as was the KF ring (present in nearly 100% of WD patients with neurological involvement), supporting the absence of neurological involvement of WD.Reference Figus, Angius, Loudianos, Bertini, Dessi and Loi 5 , Reference Lorincz 6
Both c.1934T>G (M645R) and c.2303C>T (P768L) have previously been reported related to WD worldwide,Reference Shah, Chernov, Zhang, Ross, Das and Lutsenko 7 - Reference Ferenci, Członkowska, Merle, Ferenc, Gromadzka, Yurdaydin, Vogel, Bruha, Schmidt and Stremmel 11 and in a remarkably high frequency in the Spanish population.Reference Margarit, Bach, Gomez, Bruguera, Jara and Queralt 12 - Reference Huarte-Muniesa, Lacalle-Fabo, Uriz-Otano, Berisa-Prado, Moreno-Laguna and Burusco-Paternáin 14
The M645R mutation lies between the sixth copper-binding domain and the first transmembrane region of ATP7B. Previous in vitro studies of its biochemical properties found diminished but significant copper transport activity.Reference Huster, Kühne, Bhattacharjee, Raines, Jantsch and Noe 15 Although computational (in silico) models do not clearly predict a deleterious effect of the gene, the multiple independent submissions to the refSNP cluster (rs121907998) (http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp), its extremely low frequency in control general populations (1000 Genomes Project, Exome Aggregation Consortium), and its co-segregation with the disease (even in the homozygous status),Reference Huarte-Muniesa, Lacalle-Fabo, Uriz-Otano, Berisa-Prado, Moreno-Laguna and Burusco-Paternáin 14 meet the criteria required to classify it as “pathogenic” under the guidelines for the interpretation of sequence variants provided by the American College of Medical Genetics and Genomics.Reference Richards, Aziz, Bale, Bick, Das and Gastier-Foster 16 The evidence supporting the pathogenicity of the P768L variant is even stronger: in silico tools predict it to be deleterious, and it has been linked to WD in the homozygous status.Reference Santhosh, Shaji, Eapen, Jayanthi, Malathi and Chandy 17
Patients with ATP7B mutations are typically heterozygous, and unlike homozygote patients, where there is no phenotype-genotype correlation showing entirely different phenotypes in the same mutation,Reference Okada, Shiono, Hayashi, Satoh, Sawada and Suzuki 18 heterozygotes have a hepatic presentation or are presymptomatic, but do not have a neurological presentation.Reference Margarit, Bach, Gomez, Bruguera, Jara and Queralt 12 , Reference Brage, Tome, Garcia, Carracedo and Salas 13 , Reference Garcia-Villarreal, Daniels, Shaw, Cotton, Galvin and Geskes 19 To our knowledge compound M645R/P768L heterozygosity has never been reported in other families before, so the associated described phenotype would probably need to be confirmed in other patients with the same genetic alteration.
Given that our patient fulfils the diagnostic criteria for WD based on abnormal copper metabolism and positive liver biopsy but her hepatic function is normal,Reference Lee, Kim, Lee, Jin, Kim and Lee 20 we believe our patient has a presymptomatic and benign WD. It must also be noted that up to 28.6% of heterozygous asymptomatic carriers have low ceruloplasmin, and 35% show low serum copper levels,Reference Okada, Shiono, Hayashi, Satoh, Sawada and Suzuki 18 , Reference Gibbs and Walshe 21 just like our patient. However, our patient had a pathological biopsy confirmation, which supports the hypothesis that we are dealing with a presymptomatic WD patient and not with an asymptomatic carrier.
Our patient also showed evidence of nigro-striatal dopaminergic deficit that can also be found in WD subjects with neurological symptoms, but with less asymmetry on the dopamine transporter imaging than in PD. WD patients with parkinsonism usually have atypical signs that enable us to rule out PD diagnosis; also, those WD patients with evidence of abnormal dopamine transporter have additional symptoms, such as cerebellar syndrome or early dysphagia, as well as KF rings and abnormal brain MRI signal.Reference Westermark, Tedroff, Thuomas, Hartvig, Langstrom and Andersson 22 - Reference Barthel, Hermann, Kluge, Hesse, Collingridge and Wagner 24 Some other genetic disorders may cause parkinsonism with evidence of dopaminergic deficit (Spinocerebellar ataxia 2, 3, 6 and 17; Huntington’s disease PLA2G6 (PARK 14) and ATP13A2 (PARK 9); and genetic frontotemporal dementia with parkinsonism, mainly MAPT mutations). The presence of additional atypical signs, however, help to distinguish them from PD. As in typical PD, our patient had a good response to levodopa, and dyskinesias and deterioration after withdrawal. In WD there is typically no levodopa responsiveness to parkinsonian signs, probably due to a post-synaptic deficit of dopaminergic neurotransmission in addition to a pre-synaptic deficit.Reference Barthel, Hermann, Kluge, Hesse, Collingridge and Wagner 24
We cannot totally rule out that copper metabolism abnormalities caused nigro-striatal neurodegeneration or triggered the pathological process of PD, since decreased serum ceruloplasmin levels have been shown to be associated with nigral iron deposition and could be a potential risk factor for PD.Reference Jin, Wang, Zhao, Jin, Fei and Zhang 25 Moreover, on the basis of a single study of three elderly patients with late onset parkinsonism levodopa responsive heterozygotes for a 15bp nucleotide deletion at the 5’UTR region of the ATP7B gene, it has been hypothesized that a single mutated ATP7B allele may act as a risk factor for late-onset parkinsonism. However, these subjects were older than our patient and were not diagnosed with WD based on the normal serum ceruloplasmin and copper levels and urinary copper excretion.Reference Sechi, Antonio Cocco, Errigo, Deiana, Rosati and Agnetti 26 In a prevalence study in a young population, a heterozygous H1069Q mutation of the ATP7B gene was detected in one out of 103 patients with young onset Parkinson’s disease, suggesting that the H1069Q mutation does not play a relevant role in the etiology of PD.Reference Moller, Leinweber, Rissling, Oertel, Bandmann and Schmidt 27 Nevertheless, mutations in the ATP7B gene other than H1069Q may play a role in the etiology of YOPD.
In sum, we believe this patient might have both disorders, YOPD and presymptomatic WD. It is likely that in this patient WD would have only been detected much later if parkinsonian manifestations had not developed prematurely.
DISCLOSURES
Carmen Gasca-Salas has the following disclosure: Movement Disorders Society, Travel Grant, Grant. José A. Obeso has the following disclosures: Lectures and Honoraria from UCB, Zambon, Boehringer Ingelheim, and Lundbeck. Angel Alonso and Rafael González-Redondo do not have anything to disclose.
SUPPLEMENTARY MATERIAL
To view supplementary material for this article, please visit https://doi.org/10.1017/10.1017/cjn.2016.327.