


Immunological mechanisms are increasingly implicated in the pathogenesis of depressive symptoms.Reference Bhattacharya, Derecki, Lovenberg and Drevets1–Reference Strawbridge, Arnone, Danese, Papadopoulos, Herane Vives and Cleare3 Activation of the peripheral immune system has been consistently associated with major depressive disorder (MDD).Reference Maes, Meltzer, Bosmans, Bergmans, Vandoolaeghe and Ranjan4–Reference Haapakoski, Mathieu, Ebmeier, Alenius and Kivimaki8 However, it has also been anticipated that not all patients with MDD will be peripherally inflamed to the same extent. A deeper understanding of how peripheral immune biomarkers relate to some of the dimensions of clinical heterogeneity encompassed by a diagnosis of MDD could be an important step towards mechanistically stratified treatment of depression in the future.Reference Strawbridge, Arnone, Danese, Papadopoulos, Herane Vives and Cleare3, Reference Young, Silber, Bruno, Galatzer-Levy, Pomara and Marmar9, Reference Domenici, Wille, Tozzi, Prokopenko, Miller and McKeown10
C-reactive protein (CRP) is an acute-phase protein that is widely used in clinical practice and has also been measured in many prior studies of MDD.Reference Haapakoski, Mathieu, Ebmeier, Alenius and Kivimaki8 A high-sensitivity assay for CRP is well-validated and accessible. CRP synthesis is induced in the liver by proinflammatory cytokines – especially interleukin 6 (IL-6) – in response to infection, inflammation and tissue damage. In a meta-analysis of 20 case–control studies,Reference Haapakoski, Mathieu, Ebmeier, Alenius and Kivimaki8 CRP was moderately increased ‘on average’ (Cohen's d = 0.47) in patients with MDD. However, there was significant heterogeneity of effect size between studies that may be attributable to clinical heterogeneity, with higher CRP in severe depression (Cohen's d = 0.50) than in mild/moderate depression (Cohen's d = 0.37), as well as methodological differences between studies.Reference Glaser, Robles, Sheridan, Malarkey and Kiecolt-Glaser11
We were motivated to test the hypothesis that the clinically defined subgroup of patients with treatment-resistant depression would have the most abnormally increased CRP. An association between treatment resistance to monoaminergic antidepressant drugs and increased CRP is hypothetically predictable on clinical and mechanistic grounds. Clinical studies indicate that proinflammatory cytokines that induce CRP synthesis are increased in treatment-resistant MDD.Reference Cattaneo, Gennarelli, Uher, Breen, Farmer and Aitchison12, Reference Cattaneo, Ferrari, Uher, Bocchio-Chiavetto, Riva and Consortium13 Mechanistic studies have shown that proinflammatory cytokines can reduce the extracellular availability of serotonin by biasing expression of genes related to serotonin transport and tryptophan metabolism.Reference Janssen, Caniato, Verster and Baune14, Reference Leday, Vertes, Richardson, Greene, Regan and Khan15 Single studies have also reported that elevated CRP may be associated with other dimensions of clinical heterogeneity, namely atypical depression,Reference Hickman, Khambaty and Stewart16 childhood adversity,Reference Miller and Cole17 higher numbers of previous depressive episodesReference Copeland, Shanahan, Worthman, Angold and Costello18 or anxiety in male patients.Reference Liukkonen, Rasanen, Jokelainen, Leinonen, Jarvelin and Meyer-Rochow19
We measured CRP in four groups of participants: patients with MDD who are currently experiencing depression but are not receiving medication (untreated), patients who are currently depressed and are receiving medication (treatment-resistant), patients who are currently receiving medication but are not depressed (treatment-responsive) and healthy volunteers with no history of MDD or monoaminergic drug treatment. The primary hypothesis, that CRP would be most clearly increased above normal levels in treatment-resistant patients with MDD, was tested by planned analyses of between-group differences in mean CRP. In a secondary analysis, we took a more exploratory approach to the question of what other dimensions of clinical heterogeneity in the sample might be related to variation in CRP. We used the multivariate technique of partial least squares (PLS) to explore the relationships between CRP and multiple (139) clinical phenotypes – ranging from body mass index (BMI) to questionnaire items for depressive symptoms, anxiety states or history of childhood adversity.Reference Abdi and Williams20–Reference Menzies, Achard, Chamberlain, Fineberg, Chen and del Campo23 In this way, we could identify a subset of clinical phenotypes weighted strongly on latent dimensions of clinical heterogeneity that were predictive of higher CRP levels. We also tested the confirmatory hypothesis that scores on these clinical dimensions of peripheral inflammation would be higher in the subgroup of patients with treatment resistance defined a priori.
Method
This was a non-interventional study, conducted as part of the Wellcome Trust Consortium for Neuroimmunology of Mood Disorders and Alzheimer's disease (NIMA). There were five clinical study centres in the UK: Brighton, Cambridge, Glasgow, King's College London and Oxford. All procedures were approved by an independent research ethics committee (National Research Ethics Service East of England, Cambridge Central, UK; approval number 15/EE/0092) and the study was conducted according to the Declaration of Helsinki. All participants provided informed consent in writing and received £100 compensation for taking part.
Sample and eligibility criteria
We recruited four groups of participants, those with treatment-resistant depression, treatment-responsive depression, untreated depression and healthy volunteers.
For all participants, the following inclusion criteria applied: age 25–50 years; able to give informed consent; able to fast for 8 h and abstain from strenuous exercise for 72 h prior to venous blood sampling; and fluent English. The following exclusion criteria applied: pregnancy or breast feeding, alcohol or substance use disorder in the preceding 12 months, participation in an investigational drug study within the preceding 12 months, lifetime history of any medical disorder or current use of any medication (e.g. statins, corticosteroids, antihistamines, anti-inflammatory medications) likely to compromise interpretation of CRP (see Supplementary Material, available at https://doi.org/10.1192/bjp.2018.66).
Adult patients meeting DSM-5 criteria for MDD24 were recruited from National Health Service mental health and primary care services and from the general population by purposive advertising. Lifetime histories of bipolar disorder or non-affective psychosis were additional exclusion criteria. Diagnosis of MDD and other psychiatric disorders was ascertained by the Structured Clinical Interview for DSM-5.Reference Spitzer, Miriam and Williams25 Current depressive symptom severity was defined by total scores from the 17-item Hamilton Rating Scale for Depression (HAM-D),Reference Hamilton26 and lifetime antidepressant medication use was quantified by the Antidepressant Treatment Response Questionnaire (ATRQ).Reference Desseilles, Witte, Chang, Iovieno, Dording and Ashih27 The ATRQ was completed by a member of the study team via an interview with each participant. This structured instrument records all medications received for at least 6 weeks for treatment of depression, for current and past depressive episodes. For each medication ever received, the percentage improvement experienced by the participant during the corresponding episode was documented (<25%, 25–49% improved, 50–75% improved or >75% improved). Treatment response was conservatively defined as >75% improvement in depressive symptoms, as recalled by the participant. The ATRQ provides definitions for the minimum dose for each medication to be considered an adequate treatment course.Reference Desseilles, Witte, Chang, Iovieno, Dording and Ashih27
Patients were assigned to one of three subgroups or strata, per protocol: treatment-resistant (DEP+MED+) patients who had total HAM-D score > 13 and had been medicated with a monoaminergic drug at a therapeutic dose for at least 6 weeks; treatment-responsive (DEP−MED+) patients who had total HAM-D score < 7 and had been medicated with a monoaminergic drug at a therapeutic dose for at least 6 weeks; and untreated (DEP+MED−) patients who had HAM-D score > 17 and had not been medicated with a monoaminergic drug for at least 6 weeks. Cut-offs were defined a priori based on the literature. Total HAM-D score >17 is a standard threshold for entry into placebo-controlled treatment trials of MDD, whereas a lower threshold of total HAM-D score > 13 is typically used to define treatment-resistant depression, because there is usually some modest symptomatic response to treatment even if patients remain depressed.Reference Marcus, McQuade, Carson, Hennicken, Fava and Simon28, Reference Katona, Robertson, Abou-Saleh, Nairac, Edwards and Lock29
A group of healthy volunteers was recruited by advertising with no current or past history of any major psychiatric disorder as defined by DSM-5, and no history of monoaminergic drug treatment for any indication. Healthy volunteers completed the same screening and baseline assessments as patient groups (see below).
Age, gender, medical history, smoking status and family history were documented by semi-structured clinical interviews. Height and weight were measured for calculation of BMI (kg/m2).
Questionnaire assessments
Psychological symptoms and childhood adversity were assessed by administration of the following questionnaires (see Supplementary Material): the Beck Depression Inventory (BDI v2.0Reference Beck, Steer and Brown30), the Spielberger State-Trait Anxiety Rating scale (STAIReference Spielberger, Gorsuch, Lushene, Vagg and Jacobs31), the Chalder Fatigue Score (CFSReference Chalder, Berelowitz, Pawlikowska, Watts, Wessely and Wright32), the Snaith-Hamilton Pleasure Scale (SHAPSReference Snaith, Hamilton, Morley, Humayan, Hargreaves and Trigwell33) and the Childhood Trauma Questionnaire (CTQReference Bernstein, Fink, Handelsman, Foote, Lovejoy and Wenzel34).
High-sensitivity CRP measurement
CRP was measured as one of many immunological markers in a venous blood sample drawn from each participant. Here, we focus on CRP because this has established utility as an immune biomarker of depression, having been widely used in case–control and epidemiological studies, and thus informing our hypothesis that CRP would be increased specifically in treatment-resistant depression. Participants fasted for 8 h and abstained from strenuous exercise for 72 h prior to venous blood sampling between 08:00 and 10:00. Patients taking psychotropic medication(s) continued their usual medication during the assessment day. High-sensitivity CRP was assayed via a central laboratory (see Supplementary Material).
Statistical analysis
For analysis of between-group differences in high-sensitivity CRP and other variables, we first compared all participants with MDD to healthy volunteers, using planned t-tests. We then evaluated pairwise group differences with post hoc t-tests, provided the main effect of group was significant by one-way analysis of variance. When assumptions of normality were violated, data were appropriately transformed and/or non-parametric tests were used for inference. Cohen's d was reported for the effect size of high-sensitivity CRP corrected for BMI in each clinical group compared with healthy volunteers. Additionally, we compared the proportion of participants in each group who had clinically elevated CRP, defined as >3 mg/L.Reference Bassuk, Rifai and Ridker35, Reference Raison, Rutherford, Woolwine, Shuo, Schettler and Drake36 The threshold for statistical significance was defined as two-tailed P < 0.05 throughout.
To identify demographic and clinical phenotypes associated with variation in CRP across all study participants, we utilised the method of PLS, as implemented in JMP Pro software version 13.0.37 PLS is a multivariate technique for modelling relationships between a set of predictor (X) and response (Y) variables in terms of a set of mutually orthogonal latent factors, or PLS components.Reference Wold21, Reference Wold22, Reference Garthwaite38, Reference Höskuldsson39 It requires no distributional assumptions and thus is robust against skewness. This same software was also used for other statistical tests and generation of violin plots for CRP across groups.
Here we modelled high-sensitivity CRP as the response variable, Y. The predictor variables, X, comprised gender, age, BMI, and education level as well as each of the 21 HAM-D, 11 CFS, 21 BDI, 28 CTQ, 40 STAI and 14 SHAPS questionnaire items. Data from all participants were included and missing data were imputed by sample means. Thus, the Y vector was (252 × 1) and the X matrix was (252 × 139). Because of the number of variables and the expectation that many variables would correlate with each other, other statistical approaches (such as linear regression) would not have been valid. In contrast, PLS is an ideal statistical technique under these circumstances.Reference Wold21, Reference Wold22, Reference Garthwaite38, Reference Höskuldsson39 An initial PLS model was fitted including all predictor variables. We then used a two-step approach to identify the subset of predictor variables that significantly contributed to the model: first, we discarded individual X variables with low importance by conventional criteria (variable importance parameter < 0.8 and standardised absolute model coefficient less than the absolute magnitude of 0.05Reference Cox and Gaudard40); second, we utilised a more conservative approach of excluding variables whose standardised model coefficient had a 95% CI (constructed by bootstrapping the data 1000 times) that included zero. PLS models were fitted by leave-one-out cross-validation (non-linear iterative PLS (NIPALS) algorithm), and the optimal number of latent factors was selected by minimising the predictive residual sum of the squares. The statistical significance of the final model was confirmed by comparing the percentage of variation in X and Y accounted for in the experimental data compared with the null distributions of the percentage of X or Y variance sampled by bootstrapping (1000 iterations).
Results
Demographic and clinical data
The size of each group and their demographic and clinical characteristics are summarised in Table 1. The groups did not differ significantly in terms of demographic characteristics. As expected, post hoc tests indicated that each group differed significantly from each other group on HAM-D total score (least significant t = 4.19, d.f. = 248, P < 0.001). The mean number of failed pharmacological treatments for MDD episodes (<75% symptomatic response, defined by ATRQ) is listed for each clinical group in Table 1. The treatment-resistant group had more failed treatments than the untreated group (Wilcoxon Z = 2.843, P = 0.005); both the treatment-resistant group and the untreated group had significantly more failed treatments than the treatment-responsive group (Wilcoxon Z = 5.794, P < 0.001 and Wilcoxon Z = 3.079, P = 0.002, respectively). The majority of treatment-resistant patients were taking selective serotonin reuptake inhibitors (see Table 1 footnote). Summary statistics for questionnaire-based measures and comorbidities are provided in supplementary Tables 1 and 2.
Table 1 Demographic, clinical and high-sensitivity CRP data

BMI, body mass index; CRP, C-reactive protein; F, one-way analysis of variance; HAM-D, Hamilton Rating Scale for Depression; K, Kruskal–Wallis test; L, likelihood ratio; MDD, major depressive disorder.
a. The majority of treatment-resistant patients were taking selective serotonin reuptake inhibitors (70%) with smaller numbers exposed to noradrenergic and specific serotonergic reuptake inhibitors (15%), mixed reuptake inhibitors (25%), tricyclic antidepressants (4%), mood stabilisers (4%) and dopamine receptor antagonists (3%). Treatment-responsive patients were likewise predominantly treated with selective serotonin reuptake inhibitors (85%), followed by mixed reuptake inhibitors (25%), noradrenergic and specific serotonergic reuptake inhibitors (11%) or tricyclic antidepressants (4%). All treatment-resistant patients were taking at least one conventional antidepressant monoaminergic drug.
CRP
Mean high-sensitivity CRP concentrations (and 95% CIs) are shown in Table 1. Mean CRP was significantly increased in all patients with MDD compared with healthy controls (Wilcoxon Z = 2.7, P = 0.007). Both treatment-resistant and treatment-responsive groups had significantly higher mean high-sensitivity CRP than controls (Wilcoxon Z = 2.9, P = 0.004 and Wilcoxon Z = 2.6, P = 0.010, respectively). Across all patients with MDD, drug treatment class did not significantly affect CRP levels (F = 0.799, P = 0.572), nor did the number of failed treatments in the past (F = 0.245, P = 0.621).
The proportion of participants with high-sensitivity CRP levels exceeding the conventional threshold value of 3 mg/L was also significantly different between the pooled MDD groups and controls (likelihood ratio χ2 = 10.01, P = 0.002). Treatment-resistant, untreated and treatment-responsive MDD groups all had significantly increased proportions of participants with high-sensitivity CRP > 3 mg/L compared with healthy volunteers (likelihood ratio χ2 = 8.4, P = 0.004; likelihood ratio χ2 = 8.6, P = 0.003; and likelihood ratio χ2 = 5.0, P = 0.025, respectively). No other post hoc test was statistically significant; that is, depressed groups did not differ significantly from each other (all P > 0.09).
Log-transformed and BMI-corrected CRP
The distributions of high-sensitivity CRP were positively skewed (moment skewness: 5.08)Reference Terpening41 and therefore were normalised by base log10 transform (see Fig. 1). Log10 CRP was significantly increased in all patients with MDD compared with controls (t = 2.81, d.f. = 250, P = 0.004). Only treatment-resistant and treatment-responsive patients had significantly higher log10 CRP than controls (t = 3.07, d.f. = 248, P = 0.002 and t = 2.32, d.f. = 248, P = 0.021, respectively).
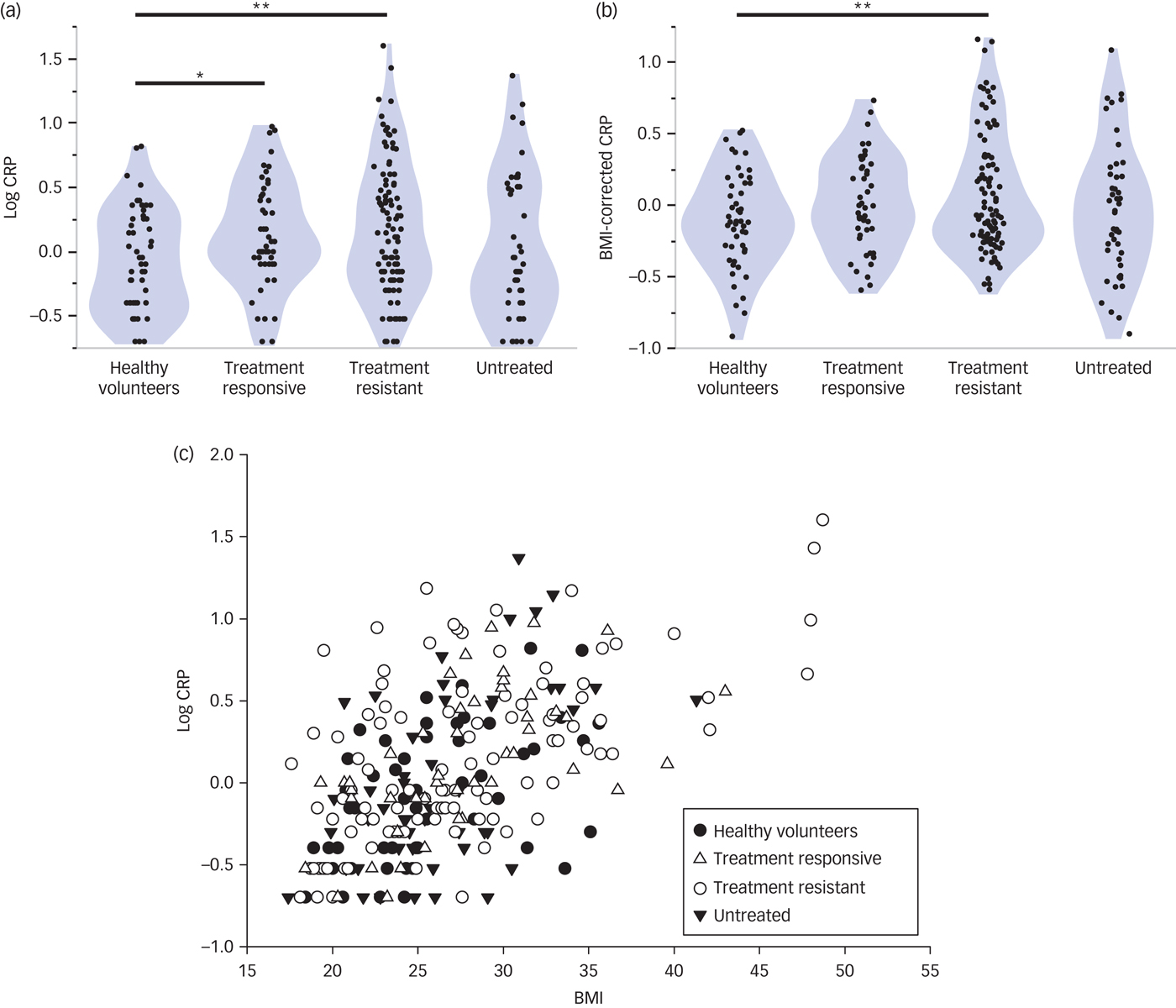
Fig. 1 High-sensitivity CRP and its relationship with BMI.
As anticipated by prior studies,Reference Ridker, Buring, Cook and Rifai42–Reference Janka44 there was a significant positive correlation between BMI and log10 CRP across all study participants (Spearman's rho = 0.56, d.f. = 250, P < 0.001; Fig. 1). Because BMI data were also positively skewed (moment skewness: 1.03),Reference Terpening41 we regressed log10 CRP on log10 BMI and used the residuals as estimates of BMI-corrected CRP (Fig. 1). BMI-corrected CRP was significantly elevated in all patients with MDD compared with controls (t = 2.24, d.f. = 238, P = 0.026). Post hoc t-tests indicated that only the treatment-resistant patients had significantly higher mean BMI-corrected CRP than the controls (t = 2.71, d.f. = 236, P = 0.007; Cohen's d = 0.47).
To assess the possible confounding effect of symptom severity, we identified the subgroup of treatment-resistant patients (n = 48) that had a total HAM-D score > 17, thereby corresponding to the cut-off used to define the untreated group. We confirmed that BMI-corrected CRP was abnormally increased in treatment-resistant patients with HAM-D > 17 (t = 3.0, P = 0.004) with a case–control difference of similar size (Cohen's d = 0.43) to that of treatment-resistant patients with HAM-D > 13.
PLS analysis of the relationship between CRP and clinical variables
A total of 13 out of 139 clinical phenotypes passed criterion for an important effect on CRP levels. Iterative cross-validation of the PLS model including only these important variables yielded an optimal two-factor solution (Fig. 2 and Supplementary Fig. 2), which accounted in total for 34.7% of variation in clinical measures (X), and for 36.1% of variation in CRP (Y). This differed significantly from the proportions of variance expected under the null hypothesis (percentage variance (X) = 12.3%, 95% CI 12.1–12.5%; percentage variance (Y) = 2.7%, 95% CI 1.9–3.0%).
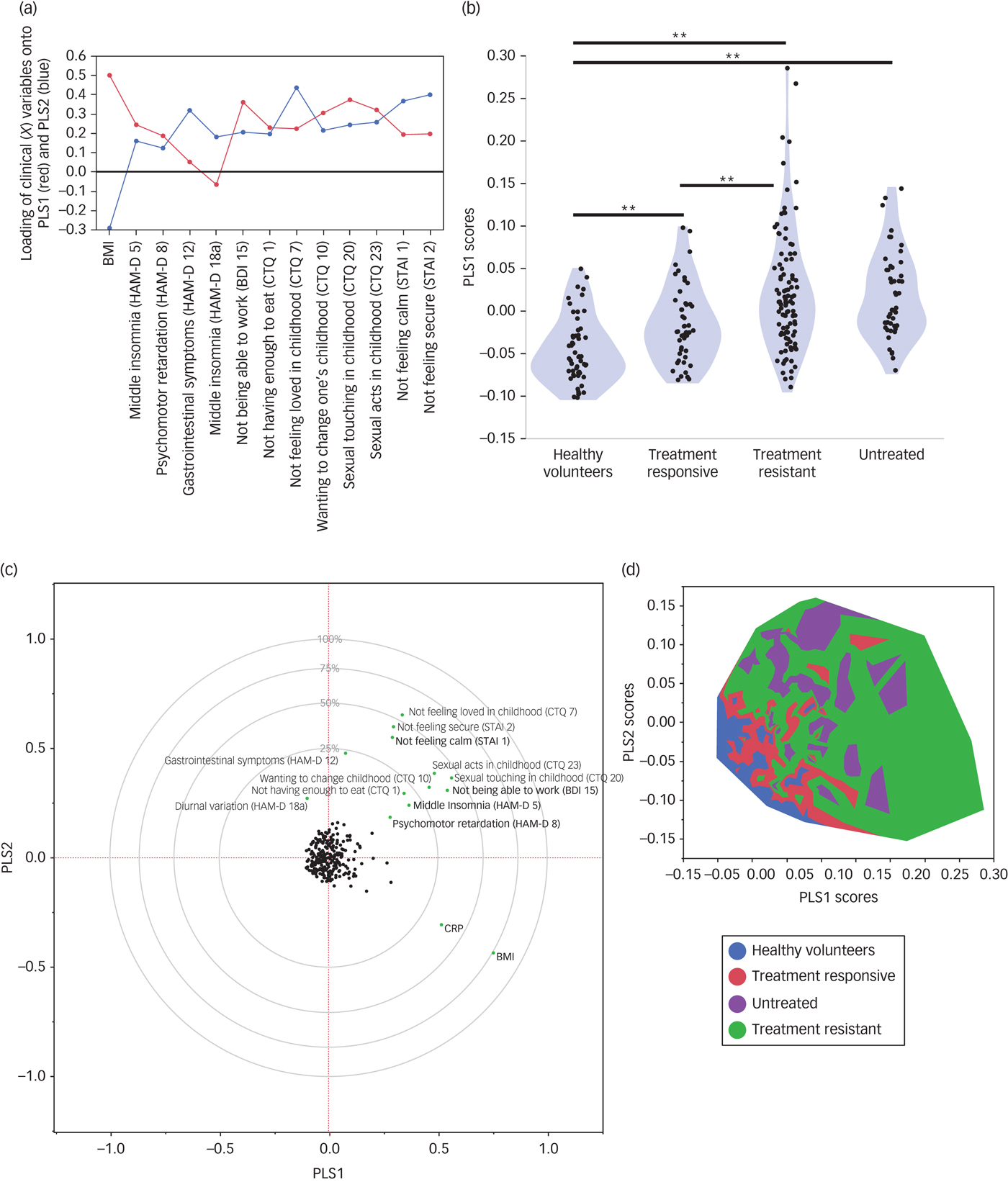
Fig. 2 Partial least squares analysis of the relationships between high-sensitivity CRP and clinical phenotypes.
The first PLS component (PLS1) accounted for 26.7% of the variation in high-sensitivity CRP. Positive scores on PLS1 indicated higher CRP. The clinical phenotypes that were significantly weighted on PLS1 were higher BMI, not feeling loved in childhood (CTQ item seven), not feeling calm (STAI item one), wanting to change one's family in childhood (CTQ item ten), psychomotor retardation (HAM-D item eight), middle insomnia (HAM-D item five) and not being able to work (BDI item 15). The PLS1 scores for individual participants differed significantly between groups (F = 19.88, d.f. = 3248, P < 0.001; Fig. 2). PLS1 scores were highest in the treatment-resistant group, followed by the untreated group, the treatment-responsive group and the healthy volunteers.
The second PLS component (PLS2) orthogonally explained 9.4% of variation in CRP. PLS scores in each group, and variables loading significantly onto PLS2, are summarised in Supplementary Fig. 3. Positive scores on PLS 2 indicated lower CRP. PLS2 scores differed significantly between groups (F = 24.34, d.f. = 3248, P < 0.001). Untreated patients had the highest PLS2 scores, followed by treatment-resistant patients, treatment-responsive patients and healthy volunteers.
Discussion
This is the first study to measure peripheral CRP with the same high-sensitivity assay across a large sample of patients with MDD (n = 198) prospectively stratified in terms of their current and past history of treatment with monoaminergic antidepressant drugs. We replicated the well-established finding that CRP is significantly increased ‘on average’ in patients with MDD, screened for physical comorbidity, and compared with healthy volunteers who did not differ in terms of age, gender, BMI and cigarette smoking status. However, we also found some evidence for our primary hypothesis that CRP was most increased in the subgroup of patients with treatment-resistant depression (n = 102). The standardised size of the case–control difference in CRP between healthy volunteers and treatment-resistant patients (Cohen's d = 0.47) was higher than the case–control difference for treatment-responsive (0.29) or untreated patients (0.18). Controlling for non-normality of the CRP distribution, and for the strong positive correlation between CRP and obesity, we found that the case–control difference in CRP remained significant only for the subgroup of treatment-resistant patients. These results of planned analysis are consistent with the hypothesis that peripheral inflammation is a marker or risk factor for treatment-resistant depression. It should be noted that CRP was somewhat elevated even in treatment-responsive patients, suggesting that a degree of elevated CRP could be trait-related rather than related to current symptoms or treatment status.
Taking a convergent but more exploratory approach to the data, we used multivariate analysis to identify two dimensions of clinical heterogeneity that were predictive of CRP. We found that a subset of clinically measured phenotypes explained approximately 36% of the variance in CRP. High BMI, high scores on vegetative symptoms of depression, low scores on calmness and a history of childhood adversity were all predictive of increased CRP. As expected from the results of our primary analysis, we confirmed that the group of patients defined a priori in terms of treatment resistance had the highest scores on this clinical profile associated with high-sensitivity CRP.
Treatment-resistant depression and peripheral inflammation
Monoamine reuptake inhibitors and related drugs are evidence-based pharmacological treatments for MDD, but response failure afflicts approximately 30% of patients.Reference Al-Harbi45, Reference Schosser, Serretti, Souery, Mendlewicz, Zohar and Montgomery46 Because of the global burden of disability attributable to MDD,Reference Lepine and Briley47 the search for improved understanding of biomarkers of therapeutic resistance to current first-line treatment options is pressing.Reference Bhattacharya, Derecki, Lovenberg and Drevets1, Reference Bhattacharya and Drevets48 Our results provide fresh evidence that patients with treatment-resistant depression have the most abnormally increased CRP levels compared with both treatment-responsive and currently untreated patients. To our knowledge, a specific relationship between CRP and monoaminergic antidepressant drug treatment resistance has not been demonstrated previously, although there is evidence both for increased proinflammatory cytokine concentrationsReference Strawbridge, Arnone, Danese, Papadopoulos, Herane Vives and Cleare3, Reference Janssen, Caniato, Verster and Baune14, Reference Carvalho, Torre, Papadopoulos, Poon, Juruena and Markopoulou49 and for increased peripheral expression of cytokine related genesReference Cattaneo, Gennarelli, Uher, Breen, Farmer and Aitchison12 in treatment-resistant depression. There is also some evidence that baseline inflammatory markers may be useful predictors of treatment response in MDD.Reference Jha, Minhajuddin, Gadad, Greer, Grannemann and Soyombo50, Reference Uher, Tansey, Dew, Maier, Mors and Hauser51 In a rat model of treatment-resistant depression, elevated CRP at baseline differentiated responders from non-responders to ketamine, an N-methyl-D-aspartate receptor antagonist with anti-inflammatory and antidepressant effects.Reference Walker, Foley, Sutor, McGillivray, Frye and Tye52 At a cellular level, neurons, microglia and macrophages respond to inflammatory challenges by activating metabolic pathways that reduce the synaptic availability of serotonin and catalyse the conversion of tryptophan to kynurenine and its putatively neurotoxic, glutamatergic agonist metabolites.Reference Leday, Vertes, Richardson, Greene, Regan and Khan15, Reference Wichers, Koek, Robaeys, Verkerk, Scharpe and Maes53–Reference Postal and Appenzeller55 These effects of inflammation on serotonin transport and tryptophan metabolism may constitute a mechanism by which peripheral inflammation is associated with lack of therapeutic response to selective serotonin reuptake inhibitors.Reference Haroon, Raison and Miller56
Clinical phenotypes predictive of increased CRP in depression
Obesity and its cardiovascular sequelae have been repeatedly associated with increased CRP.Reference Zhao and Lv43, Reference Berg and Scherer57, Reference Capuron, Su, Miller, Bremner, Goldberg and Vogt58 In this study, which excluded patients with a lifetime history of medical disorders including atherosclerosis and diabetes, we confirmed that higher BMI was strongly associated with higher CRP levels. One mechanistic explanation is that macrophages constitute up to 60% of cells in adipose tissue and can release large amounts of IL-6, which is a key driver of CRP synthesis.Reference Mohamed-Ali, Goodrick, Rawesh, Katz, Miles and Yudkin59 Therefore, it is not surprising that inflammation (CRP) and obesity (BMI) were related herein; however, we do not consider that this association trivially accounts for increased CRP in treatment-resistant depression. The groups did not differ significantly in baseline BMI and the case–control difference remained significant for the treatment-resistant patients even after statistical regression to control for individual differences in BMI.
Of all the depressive symptoms measured, so-called vegetative symptoms (psychomotor retardation, insomnia, difficulty getting started, difficulty working) were more important in explaining higher CRP. These findings are consistent with prior reports that somatic but not cognitive symptoms of depression were associated with increased CRP.Reference Duivis, Vogelzangs, Kupper, de Jonge and Penninx60 Vegetative symptoms of depression are akin to the illness or sickness behaviour that has been repeatedly demonstrated in animal models and experimental medicine studies of humans exposed to acute proinflammatory challenge.Reference Dantzer, O'Connor, Freund, Johnson and Kelley2, Reference Capuron, Gumnick, Musselman, Lawson, Reemsnyder and Nemeroff61 We also found evidence that state anxiety was related with CRP, which is compatible with prior data linking acute endotoxin exposure to anxious and depressive states in healthy volunteers.Reference Reichenberg, Yirmiya, Schuld, Kraus, Haack and Morag62
It is established that childhood trauma increases risk of later mental health disorders, including depression.Reference Sachs-Ericsson, Kendall-Tackett and Hernandez63 In a meta-analysis, individuals exposed to childhood trauma had significantly elevated levels of CRP in adulthood, albeit with a small effect size (Fisher's Z = 0.10).Reference Baumeister, Akhtar, Ciufolini, Pariante and Mondelli64 In a longitudinal study of female adolescents at risk of depression, childhood adversity was found to promote subsequent clustering of depression and inflammation.Reference Lu, Peng, Wang, Vasish, Zhang and Gao65 These results are compatible with our findings that feeling unloved in childhood and wanting to change one's family in childhood were significantly correlated with higher CRP in adults.
Methodological issues
Because of the case–control design, between-group differences in CRP could theoretically be confounded by other factors influencing peripheral inflammation. However, we excluded patients with inflammatory disorders or anti-inflammatory drug treatment, and the groups did not differ on demographic characteristics. The lack of statistically significant case–control differences in BMI-corrected CRP for the comparisons between healthy volunteers and the treatment-responsive and untreated MDD groups could theoretically reflect the smaller sizes of these groups compared with the treatment-resistant MDD group. However, power calculations indicated that the case–control differences in BMI-corrected CRP would probably not have been significant even if the treatment-responsive group had the same size as the resistant group (Table 1). Although the untreated MDD group had not received antidepressant treatment for at least six weeks, the majority (n = 28, 58.3%) had received at least one such treatment in the past (the average number of historically failed treatments in this group was 1.8). As such, this group had some degree of heterogeneity, including both treatment-naïve individuals and those who would have been expected to be treatment-resistant if currently medicated. Treatment resistance was defined by inadequate response to the current drug treatment whereas some other criteria for treatment resistance stipulate a failed response to at least two drugs of different mechanisms of action. There are multiple definitions of treatment resistance used in the field; our choice is widely used. The study was not planned or powered to test differences in CRP between subgroups defined by dose or type of current antidepressant medication. We did not find that CRP differed significantly as a function of antidepressant medication class, nor as a function of total number of previous failed treatments (as measured by the ATRQ). A limitation in assessing prior medications was the use of retrospective self-report, albeit based on a comprehensive structured instrument completed by an interviewer. The sample was recruited from the UK population, which is known to differ from the USA and other populations in terms of BMI and other factors that can influence the numerical distribution of CRP, and this may mitigate generalisability of our results, as would our exclusion of people with inflammatory disorders. Finally, CRP is only one of many markers of peripheral inflammation that have been or could be linked to depression. Although these data demonstrate that CRP is robustly associated with treatment-resistant depression, we do not claim that CRP is necessarily the best of all possible peripheral blood biomarkers of treatment-resistant depression.
Conclusions
MDD is associated with increased CRP compared with healthy volunteers and the case–control difference appears higher in treatment-resistant depression. Increased CRP and treatment resistance were also associated with other aspects of clinical heterogeneity in depression including obesity, vegetative symptoms of fatigue and sleep disturbance, state anxiety and a history of childhood adversity. We suggest there may be a clinically and immunologically diagnosable subsyndrome of ‘inflamed depression’ comprising the patients with MDD most likely to benefit therapeutically from second-line treatment with anti-inflammatory drugs.Reference Raison, Rutherford, Woolwine, Shuo, Schettler and Drake36
Supplementary material
Supplementary material is available online at https://doi.org/10.1192/bjp.2018.66.
Funding
This work was funded by a Wellcome Trust strategy award to the Neuroimmunology of Mood Disorders and Alzheimer's Disease (NIMA) Consortium, which is also funded by Janssen, GlaxoSmithKline, Lundbeck and Pfizer. Recruitment of patients was supported by the National Institute of Health Research (NIHR) Clinical Research Network: Kent, Surrey and Sussex & Eastern. The immunological work of the NIMA consortium is supported by the Cambridge NIHR Biomedical Research Centre.
Acknowledgements
We thank all study participants, research teams and laboratory staff, without whom this research would not have been possible. Members of the NIMA Consortium are thanked and acknowledged (see Supplementary Material, available at https://doi.org/10.1192/bjp.2018.66).






 Open access
Open access
eLetters
No eLetters have been published for this article.