


Brain size is decreased and ventricular volume increased in schizophrenia (Reference Lawrie and AbukmeilLawrie & Abukmeil, 1998). The histological correlates of this macroscopic pathology appear to involve alterations in the cortical cytoarchitecture, with changes in the structure and organisation of neurons and their connections (Reference HarrisonHarrison, 1999a ). The synaptic component of the pathology has been investigated by measuring the expression of genes whose protein product is concentrated in synaptic terminals; synaptophysin, a 38-kilodalton synaptic vesicle protein, is the most widely used marker of this kind (Reference Masliah and TerryMasliah & Terry, 1993). In schizophrenia, decreased expression of synaptophysin has been reported in the hippocampus (Eastwood & Harrison, Reference Eastwood and Harrison1995, Reference Harrison and Harrison1999; Reference Harrison, Burnet and HarrisonEastwood et al, 1995; Reference Vawter, Howard and HydeVawter et al, 1999) and prefrontal cortex (Reference Perrone-Bizzozero, Sower and BirdPerrone-Bizzozero et al, 1996; Reference Glantz and LewisGlantz & Lewis, 1997; Reference Karson, Mrak and SchlutermanKarson et al, 1999). However, other studies have shown equivocal (Reference Browning, Dudek and RapierBrowning et al, 1993; Reference Tcherepanov and SokolovTcherepanov & Sokolov, 1997; Reference Honer, Falkai and ChenHoner et al, 1999) or negative (Reference Gabriel, Haroutunian and PowchikGabriel et al, 1997; Reference Rodriguez, Weickert and HermanRodriguez et al, 1998; Reference Young, Arima and XieYoung et al, 1998) results. It is unclear whether the discrepancies reflect anatomical heterogeneity, lack of robustness or methodological differences (e.g. whether protein or messenger RNA (mRNA) has been measured). To address these issues we have examined synaptophysin expression in four cortical areas, using three methods, in two series of brains.
METHOD
Subjects studied
Brain tissue was collected at autopsy, with consent, in two centres, Oxford and London. Demographic details are shown in Table 1. Schizophrenia was diagnosed from the case notes according to ICD-10 (World Health Organization, 1992) and DSM-III-R (American Psychiatric Association, 1987) criteria; most cases were initially of paranoid or disorganised subtype. Nine of the subjects with schizophrenia had been living in the community, the rest were long-term in-patients. All had received antipsychotics and all but two were on medication at death. The mean duration of illness (known for the Oxford series) was 28 years (range 8-57). The control subjects had no neurological or psychiatric history. One case and one control died by suicide. Formal neuropathological assessment of all brains showed no neurodegenerative abnormalities other than minor age-related changes.
Table 1 Demographic details of subjects studied

| Oxford series | London series | |||
|---|---|---|---|---|
| Controls | Schizophrenia cases | Controls | Schizophrenia cases | |
| Number1 | 11 | 11 | 8 | 8 |
| Age (years (s.e.m.)) | 63 (5) | 57 (5) | 61 (8) | 61 (7) |
| (range) | (22-79) | (28-83) | (26-85) | (32-87) |
| Gender (M:F) | 6:5 | 6:5 | 5:3 | 5:3 |
| Post-mortem interval (h (s.e.m.)) | 39 (6) | 45 (5) | 33 (5) | 43 (6) |
| (range) | (19-72) | (23-76) | (10-48) | (22-67) |
| Brain pH2 | 6.48 (0.07) | 6.40 (0.08) | 6.45 (0.14) | 6.37 (0.09) |
| (range) | (6.20-6.76) | (6.06-6.72) | (5.90-6.97) | (6.04-6.80) |
| Brain weight (g (s.e.m.)) | 1253 (49) | 1378 (48) | 1299 (58) | 1171 (43)3 |
Tissue collection and processing
From the Oxford series, blocks were taken from the left dorsolateral prefrontal cortex (Brodmann area (BA) 9/46), primary visual (striate) cortex (BA 17), superior temporal cortex (BA 22), and anterior cingulate cortex (BA 24), and stored at ‒70°C. These regions were chosen because three of them (BA 22, 24 and 9/46) are sites of presumed functional or structural abnormality in schizophrenia, whereas BA 17 is not. Brains in the London series were coronally sliced, snap-frozen and stored at ‒70°C. Blocks from BA 17, 22 and 9/46 were subsequently dissected from the slices at ‒20°C, randomly from either hemisphere, surrounded with embedding compound, and stored at ‒70°C. All material was coded and experiments carried out blind to diagnosis.
From both series, 18-μm cryostat sections were cut for in situ hybridisation (ISH) (to measure synaptophysin mRNA) and for immunoautoradiography (IAR) and immunoblotting (to measure synaptophysin). From BA 9/46 of the Oxford series, autoclaved formalin-fixed wax-embedded sections were also taken, because their better morphology facilitates detailed ‘per cell’ ISH analysis (Reference Harrison and HarrisonEastwood & Harrison, 1999).
Immunoautoradiography and in situ hybridisation
Methods for IAR (Reference Eastwood and HarrisonEastwood & Harrison, 1995) and ISH (Eastwood et al, Reference Harrison, Burnet and McDonald1994, Reference Harrison, Burnet and Harrison1995) have been described. Brief details and modifications are given here.
For IAR, sections were fixed in 4% paraformaldehyde. Non-specific binding sites were blocked by incubating the slides for 30 min with 10% normal sheep serum in phosphate-buffered saline (PBS) containing 0.3% Triton X-100. Sections were incubated overnight at 4°C with mouse antisynaptophysin antibody (SY38; DAKO, High Wycombe, UK) diluted 1:100 with PBS containing 0.3% Triton X-100 and 1% normal sheep serum. After washing in PBS, sections were incubated for 1 h with 0.2 μCi/ml 35S-labelled sheep anti-mouse immunoglobulin (the secondary antibody) in PBS containing 0.3% Triton X-100 and 1% normal sheep serum. Omission of the primary antibody was used as an experimental control. Sections were apposed to film for 10 days alongside 14C microscales.
The ISH probe was a 39-base oligodeoxynucleotide, 3′-labelled with α -[35S]-deoxyadenosine triphosphate (∼ 1200 Ci/mmol). Triplicate sections were incubated overnight at 34°C in 150 μl of hybridisation buffer containing 1 × 106 counts/min of labelled probe and 20 mM dithiothreitol. Post-hybridisation washes were in 150 mmol sodium chloride/15 mmol sodium citrate for 3 × 20 min at 58°C followed by 2 × 60 min washes at room temperature. Slides were apposed to Hyperfilm betamax (Amersham) for 21 days together with 14C microscales. The wax-embedded sections of BA 9/46 were dipped in nuclear track emulsion, exposed for 3 weeks, developed and stained with cresyl violet. ISH using the synaptophysin probe in the sense orientation was used as an experimental control, in addition to previous demonstrations of the probe's specificity under these conditions (Eastwood et al, Reference Harrison, Burnet and McDonald1994, Reference Harrison, Burnet and Harrison1995).
Immunoblotting
Immunoblotting (western blotting) was carried out on the BA 9/46 samples of the London series, to complement the IAR measurements. Twenty cryostat sections were collected and homogenised directly in 500 μl of suspension buffer (100 mM sodium chloride, 10 mM Tris-HCl (pH 7.6), 1 mM ethylene diamine tetraacetate (EDTA)) containing 1 μg/ml aprotinin and 100 μg/ml phenylmethyl-sulphonyl fluoride and centrifuged for 5 min at 13 000 g. The resulting pellet was re-suspended in the above buffer containing 1% w/v sodium dodecyl sulphate, boiled for 5 min, spun at 13 000 g for 5 min, and the supernatant collected. The protein concentration of the resulting crude synaptosomal extracts was determined using the Bradford method. Pilot studies found a linear relationship between the amount of protein loaded (2-4.5 μg) and the signal obtained. Hence, 3 μg was loaded for the main analyses. Protein samples were fractionated by electrophoresis on a 10% sodium dodecyl sulphate/polyacrylamide gel for 2 h and transferred onto a polyvinyl difluoride membrane. After blocking of nonspecific binding sites with 2% bovine serum albumin in PBS for 30 min at room temperature, membranes were incubated for 1 h with the SY38 antibody (1:1000) in PBS containing 1% normal sheep serum. Membranes were given three rinses in PBS/0.3% Triton-X100 before incubation for 1 h with 35S-labelled sheep anti-mouse immunoglobulin at 0.2 μCi/ml in PBS. After three washes in PBS with 0.3% Triton-X100, membranes were apposed to film for 48 h alongside 14C microscales. Samples were run in quadruplicate.
Image and data analysis
For ISH and IAR, film autoradiographic measurements were taken through three representative strips of the grey matter of each cortical region; for the immunoblots, the intensity of the 38 kDa band was measured. Values were corrected for background (defined as the sense probe for ISH, and omission of primary antibody for IAR) and converted to nCi/g tissue equivalents by calibrating to the microscales. For analysis of the ISH dipped sections in BA 9/46, grain densities were measured using dark-field microscopy (Reference Harrison, Burnet and HarrisonEastwood et al, 1995) over pyramidal neurons in laminae III, V and VI. The main synaptic targets of these neurons are, respectively, other cortical areas, subcortical nuclei, and mediodorsal thalamus.
Statistical analysis
Brain pH (an index of agonal state), postmortem interval, and age can influence detection of synaptophysin and its mRNA (Eastwood et al, Reference Harrison, Burnet and McDonald1994, Reference Harrison, Burnet and Harrison1995; Reference Harrison and HarrisonEastwood & Harrison, 1999). Their effects were investigated using multiple regression. Analysis of variance, with pH, post-mortem interval and age as covariates, was used to test for effects of diagnosis and diagnosis-by-region interactions. Analyses were carried out for the two series separately, followed by analysis of the combined data set.
RESULTS
The distribution of synaptophysin mRNA (Fig. 1A) and synaptophysin (Fig. 1C) was as anticipated (Reference Harrison, Burnet and McDonaldEastwood et al, 1994). ISH histochemistry images showed enhancement of signal over laminae containing many pyramidal neurons (which express abundant synaptophysin mRNA), whereas the IAR images were more uniform, due to the presence of synaptophysin in synaptic terminals throughout the cortical ribbon. The signal in subcortical white matter was weak for both mRNA and protein, reflecting the small numbers of neurons and synapses therein. The ISH signal was absent or minimal after sense strand hybridisation, as was the IAR signal after omission of the primary antibody (not shown).

Fig. 1 Autoradiographic images of superior temporal cortex (BA 22) showing synaptophysin mRNA (A, B) and synaptophysin (C, D), detected by in situ hybridisation and immunoautoradiography, respectively. A and C: control subject; B and D: subject with schizophrenia.
The regression analyses (Table 2) showed that increasing age, lower pH and longer post-mortem interval were associated with decreased synaptophysin mRNA. In the London series, the abundance of synaptophysin was inversely related to pH and positively related to the post-mortem interval; these correlations were opposite to what had been predicted and are probably chance findings. Nevertheless, the pH and the post-mortem interval were retained as covariates for the IAR analyses.
Table 2 Effects of age, brain pH and post-mortem interval on synaptophysin expression

| Oxford series | London series | Combined series | ||||
|---|---|---|---|---|---|---|
| β1 | P | β | P | β | P | |
| Synaptophysin mRNA | ||||||
| Age | -0.247 | 0.01 | -0.374 | 0.02 | -0.305 | 0.0001 |
| Brain pH | +0.341 | 0.001 | +0.553 | 0.001 | +0.414 | 0.0001 |
| Post-mortem interval | -0.292 | 0.003 | -0.230 | 0.11 | -0.279 | 0.0001 |
| Synaptophysin | ||||||
| Age | +0.036 | 0.75 | -0.273 | 0.08 | -0.077 | 0.38 |
| Brain pH | -0.179 | 0.13 | -0.298 | 0.05 | -0.229 | 0.01 |
| Post-mortem interval | -0.027 | 0.81 | +0.493 | 0.0001 | +0.137 | 0.12 |
The abundance of synaptophysin mRNA (Table 3) and synaptophysin (Table 5) was significantly higher in the Oxford series than the London series, despite their comparable demographic and storage characteristics (Table 1, and data not shown). Because of this, we computed Z-scores for analysis of the combined series. (The difference may be explained by other perimortem or tissue processing variables. Whatever the reason, it illustrates the potential pitfalls if tissue from cases and controls come from separate sources.)
Table 3 Synaptophysin mRNA in the cerebral cortex in schizophrenia

| Brodmann area | Oxford series | London series | Combined series | |||
|---|---|---|---|---|---|---|
| Controls (n=9-11) | Schizophrenia cases (n=9-11) | Controls (n=6-8) | Schizophrenia cases (n=7-8) | Controls (n=15-18) | Schizophrenia cases (n=16-18) | |
| BA 17 | 140 (15) | 79 (16)** | 112 (21) | 48 (6) | 0.54 (0.21) | -0.57 (0.17)*** |
| BA 22 | 101 (9) | 75 (11)* | 78 (10) | 62 (9) | 0.36 (0.22) | -0.36 (0.23)* |
| BA 24 | 99 (10) | 64 (11) | ND | ND | ND | ND |
| BA 9/46 | 97 (12) | 91 (7) | 79 (9) | 63 (13) | 0.19 (0.25) | -0.18 (0.22) |
| Men only | ||||||
| BA 17 | 150 (24) | 78 (21) | 133 (30) | 44 (40) | 0.80 (0.31) | -0.62 (0.22)** |
| BA 22 | 86 (10) | 85 (20) | 77 (13) | 67 (14) | 0.12 (0.29) | -0.10 (0.37) |
| BA 24 | 103 (14) | 67 (14) | ND | ND | ND | ND |
| BA 9/46 | 86 (19) | 101 (8) | 82 (12) | 66 (21) | 0.07 (0.36) | 0.05 (0.34) |
| Women only | ||||||
| BA 17 | 129 (18) | 81 (26)* | 83 (21) | 55 (18) | 0.21 (0.24) | -0.50 (0.31)** |
| BA 22 | 113 (12) | 66 (11)* | 80 (17) | 53 (8) | 0.63 (0.31) | -0.66 (0.24)** |
| BA 24 | 93 (14) | 60 (18) | ND | ND | ND | ND |
| BA 9/46 | 107 (14) | 81 (10) | 74 (16) | 57 (10) | 0.32 (0.36) | -0.46 (0.2) |
Table 4 Synaptophysin mRNA in BA 9/46 pyramidal neurons

| Controls (n=11) | Schizophrenia cases (n=9) | |
|---|---|---|
| Lamina III | 73 (4) | 66 (5) |
| Lamina V | 94 (4) | 82 (7) |
| Lamina VI | 77 (5) | 68 (8) |
Table 5 Synaptophysin in the cerebral cortex in schizophrenia

| Brodmann area | Oxford series | London series | Combined series | |||
|---|---|---|---|---|---|---|
| Controls (n=10-11) | Schizophrenia cases (n=9-11) | Controls (n=6-8) | Schizophrenia cases (n=7-8) | Controls (n=16-18) | Schizophrenia cases (n=16-18) | |
| BA 17 | 323 (12) | 278 (14)* | 142 (9) | 170 (9) | 0.10 (0.25) | -0.10 (0.22) |
| BA 22 | 312 (20) | 298 (11) | 122 (8) | 154 (9) | -0.18 (0.27) | 0.17 (0.20) |
| BA 24 | 396 (20) | 380 (14) | - | - | - | - |
| BA 9/46 | 318 (14) | 329 (18) | 126 (12) | 150 (17) | -0.19 (0.25) | 0.18 (0.25) |
Synaptophysin mRNA
In the Oxford series there was an overall effect of diagnosis (P <0.001), and a diagnosis-by-region interaction (P=0.047). Synaptophysin mRNA was decreased in BA 17 (P=0.002) and BA 22 (P=0.043), with a trend in BA 24 (P=0.06) (Table 3; compare Figs 1A and 1B). No change was seen in BA 9/46 overall (Table 3), nor in any of the constituent pyramidal neuron populations measured therein using the dipped sections (Table 4). In the London series there was an effect of diagnosis (P=0.019) but no interaction with region (P=0.2), reflecting the smaller sample size and absence of BA 24. The decrease of synaptophysin mRNA from BA 17, though of similar magnitude to that in the Oxford series, was not significant (P=0.056). In the combined sample there was an effect of diagnosis (P <0.001), with less synaptophysin mRNA in schizophrenia in BA 17 (P <0.001) and in BA 22 (P=0.012).
In a post hoc analysis there was a diagnosis-by-gender interaction, significant in the Oxford series (P=0.029) and in the combined sample (P=0.006). In BA 22, synaptophysin mRNA was decreased in the brains of women with schizophrenia (P <0.01) but not in the brains of the men, with a similar trend in BA 9/46 (P=0.058; Table 3). In contrast, the effects of schizophrenia on synaptophysin mRNA in BA 17 and BA 24 were similar in both genders.
There were no significant (P <0.01) correlations of synaptophysin mRNA with duration of illness (after partialling out the effect of age), age at onset, or extent of antipsychotic exposure (rated on a threepoint scale).
Synaptophysin
Synaptophysin measured by IAR did not differ between diagnostic groups in the Oxford (P=0.15) or London (P=0.09) series (Table 5; compare Figs 1C and 1D). Neither were there diagnosis-by-region interactions (Oxford: P=0.3; London: P=0.9). Nevertheless, we examined the data for BA 17 and BA 22 separately because of the loss of the mRNA in those areas. A modest reduction was seen in BA 17 in the Oxford series (P <0.05) but not in the London series nor in the combined sample. There were no changes in BA 22. Immunoblotting corroborated the IAR results, showing synaptophysin to be unchanged in schizophrenia in BA 9/46 (cases: 686 (178) nCi/g; controls: 680 (95) nCi/g).
No diagnosis-by-gender interactions were seen for synaptophysin. Nor did its abundance correlate with duration of illness, age at onset or medication history.
DISCUSSION
Synaptic abnormalities are prominent among the contemporary neuropathological findings in schizophrenia. They provide a plausible anatomical basis for the suggestion that aberrant functional connectivity underlies this disorder (Reference HarrisonHarrison, 1999a ). Nevertheless, the existence of synaptic pathology is by no means firmly established, and its features remain to be characterised.
The main positive finding of this study is that synaptophysin mRNA is decreased in some regions of the cerebral cortex (Table 3). The reduction is statistically robust and seen in two series of brains. Antipsychotic treatment is unlikely to have confounded the results, since chronic administration of the drugs to rats leaves the expression of synaptophysin either unaffected or modestly increased (see Reference HarrisonHarrison, 1999b ), and exposure to medication did not correlate with synaptophysin expression here or in previous studies. As such, the basic hypothesis that there is a synaptic abnormality in the cerebral cortex in schizophrenia is supported. Timing and aetiology cannot be determined from a post-mortem study, but the presence of synaptic pathology is consistent with an anomaly in synaptogenesis or synaptic pruning during neurodevelopment (Reference Keshavan, Anderson and PettegrewKeshavan et al, 1994; Reference Harrison, Burnet and HarrisonEastwood et al, 1995). Similarly, despite the relatively large sample size, this present study is underpowered (and limited by the retrospective nature of assessments) to investigate clinicopathological correlations of potential interest, such as between the expression of synaptophysin and the severity of illness or cognitive functioning. The reduced expression of synaptophysin in schizophrenia may well reflect decreased synaptic density, just as in neurodegenerative disorders (Reference Masliah and TerryMasliah & Terry, 1993; Reference Heffernan, Eastwood and NagyHeffernan et al, 1998), but alternative explanations are possible (e.g. the synapses are smaller, or contain fewer vesicles). Either way, the changes seem likely to be associated with impaired synaptic functioning.
Other aspects of the results were unexpected: first, the distribution of changes, which were most striking in BA 17 (visual cortex) and did not occur in BA 9/46; and second, the mRNA decrements were not accompanied by equivalent reductions by synaptophysin (Table 5). These related anatomical and molecular issues are explored below, since they determine the overall interpretation of the findings.
Synaptophysin expression in the cortex in schizophrenia
Decreased synaptophysin has been reported in BA 9/46 and adjacent prefrontal regions in schizophrenia in three studies (Reference Perrone-Bizzozero, Sower and BirdPerrone-Bizzozero et al, 1996; Reference Glantz and LewisGlantz & Lewis, 1997; Reference Karson, Mrak and SchlutermanKarson et al, 1999), but results of a fourth study were negative (Reference Gabriel, Haroutunian and PowchikGabriel et al, 1997), and Honer et al (Reference Honer, Falkai and Chen1999) could only find a reduction when subjects dying by suicide were omitted. We failed to find a change in synaptophysin in BA 9/46 in either brain series, using IAR or immunoblotting, so that the matter remains unresolved. There are no obvious characteristics which distinguish the studies which found reductions from those which did not. The discrepancy is reminiscent of, and might indeed be related to, the continuing controversy regarding hypofrontality in schizophrenia (Reference Weinberger and BermanWeinberger & Berman, 1996). There is more consistency regarding synaptophysin mRNA, since two other studies of prefrontal cortex also found no change (Reference Rodriguez, Weickert and HermanRodriguez et al, 1998; Reference Karson, Mrak and SchlutermanKarson et al, 1999). These measurements did not, however, rule out the possibility of a localised reduction from a sub-population of constituent neurons. The cellular analysis (Table 4) is informative in this respect, showing that synaptophysin mRNA expression is maintained in BA 9/46 in schizophrenia in the three major pyramidal neuron groups.
Synaptophysin and its mRNA were also unchanged in BA 24, in agreement with the findings of Honer et al (Reference Honer, Falkai and Young1997), but not with the finding of a 25% elevation in an elderly sample (Reference Gabriel, Haroutunian and PowchikGabriel et al, 1997). The latter may reflect ongoing age-associated changes in this region in schizophrenia.
Visual symptoms are not a characteristic feature of schizophrenia, and BA 17 was initially included in our studies as an ‘internal control’ region. In the event, the reduction of synaptophysin expression was greatest and most robust in BA 17 (Tables 3 and 5). The implication that this region is not entirely normal, at least histologically, in schizophrenia is supported by neuronal morphometric data (see Reference Selemon and Goldman-RakicSelemon & Goldman-Rakic, 1999) and one previous synaptophysin study (Reference Perrone-Bizzozero, Sower and BirdPerrone-Bizzozero et al, 1996). Moreover, we have found other mRNAs to be reduced in BA 17 (Reference Burnet, Eastwood and HarrisonBurnet et al, 1996; Reference Eastwood and HarrisonEastwood & Harrison, 1998), with a trend for the total mRNA content to be decreased (Reference Harrison, Burnet and FalkaiHarrison et al, 1997), implying that gene expression in this region is non-specifically impaired in schizophrenia. The origin and consequences of this are obscure.
The reduction in synaptophysin mRNA in BA 22 in brains of schizophrenia cases was limited to women. As this was not predicted, the observation must be replicated before suggesting that there are gender differences in synaptic pathology in schizophrenia. However, we note that diagnosis-by-gender interactions have been reported in other recent morphometric studies of schizophrenia (Reference Highley, Esiri and McDonaldHighley et al, 1998), and this should be investigated further.
Interpreting differential changes in synaptophysin mRNA and protein
Interpreting the significance of a loss of mRNA when the level of encoded protein is preserved is not straightforward. It might be due to an altered balance of transcriptional v. translational gene regulation (see Reference Burnet, Eastwood and HarrisonBurnet et al, 1996). For example, only a small fraction of an mRNA is translationally active and being used for protein synthesis. Perhaps the proportion of ‘active’ synaptophysin mRNA is increased in schizophrenia, compensating for the decreased total amount. Or it may be that the methods used are more sensitive in detecting changes in abundance of an mRNA than of a protein. A further possibility — that the mRNA levels are simply more labile and susceptible to confounding variables (e.g. pH or post-mortem interval) — is not likely, given that these factors were matched between groups and covaried for in the analyses.
For pre-synaptic proteins there is an additional issue, first raised by a similar finding of a greater decrease of mRNA than protein in the hippocampus (Reference Eastwood and HarrisonEastwood & Harrison, 1995; Reference Harrison, Burnet and HarrisonEastwood et al, 1995). An mRNA is found only in the neuronal cell body, whereas the protein is in the axon terminals (Fig. 2). In the case of cortical pyramidal neurons, for example, these are located elsewhere, in other cortical regions, the thalamus, striatum and other subcortical nuclei (Reference Peters and JonesPeters & Jones, 1984), where there may be losses of synaptophysin to accompany the decreases in its mRNA identified here. Equally, neurons projecting into one of the measured areas might be overexpressing synaptophysin, compensating for decreased expression by intrinsic neurons. In other words, our mRNA data suggest that neurons in BA 17 and BA 22 are forming fewer, or otherwise aberrant, synaptic connections, but we have not located the affected synapses. Though important, this can only be a partial explanation, since many cortical synapses do arise from interneurons and from axon collaterals of adjacent pyramidal neurons. Further studies are needed to distinguish molecular from anatomical explanations for the greater loss of synaptophysin mRNA than synaptophysin.
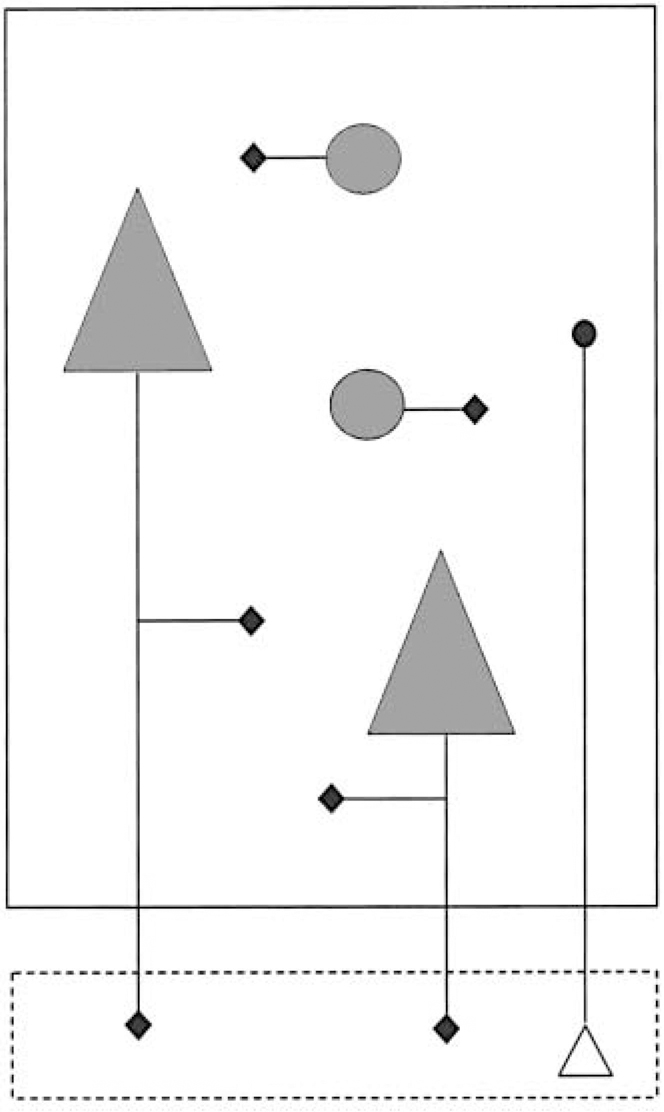
Fig. 2 Cartoon illustrating anatomical aspects of synaptophysin expression. The large rectangle represents a cortical area (e.g. BA 9/46). Synaptophysin mRNA is localised to the cell body of neurons (shown in grey), whereas synaptophysin itself is concentrated in their synaptic terminals (black diamonds). The synapses of interneurons (circles) and some of those arising from pyramidal neurons (triangles) are located in the same area. However the main projection of pyramidal neurons is to other brain areas (represented by the smaller rectangle, below, outlined with broken lines), and this fraction of their synaptophysin will not be detected. Equally, some of the synaptophysin (black circle) in the area being studied is in the terminals of neurons whose cell body (open triangle) lies elsewhere, and whose mRNA will therefore not have been measured.
Further characterisation of synaptic pathology in schizophrenia
Because of the complexities and discrepancies discussed here, it would be premature to conclude that synaptic abnormalities are a feature of schizophrenia, or that the characteristics of any synaptic pathology are known. There is, however, enough evidence to warrant further studies.
Since synaptophysin is present in virtually all synapses, its expression cannot reveal whether subtypes defined by phenotype (e.g. glutamatergic v. GABA-ergic synapses) or by subcellular location (e.g. axo-spinous v. axo-somatic synapses) are differentially affected in a disease. Neither does the expression of unchanged synaptophysin preclude the existence of discrete alterations in a sub-population of synapses. Indeed, there are already suggestions that in schizophrenia the synaptic pathology in the hippocampus (Reference Harrison and EastwoodHarrison & Eastwood, 1998; Reference Young, Arima and XieYoung et al, 1998) differs from that in the prefrontal cortex (Reference Aganova and UranovaAganova & Uranova, 1992; Reference Honer, Falkai and YoungHoner et al, 1997; Reference Thompson, Sower and Perrone-BizzozeroThompson et al, 1998; Reference Woo, Whitehead and MelchitzkyWoo et al, 1998). Molecular dissection as well as regional delineation will, therefore, be needed if the existence and nature of synaptic pathology in the disorder are to be revealed.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ Decreased synaptophysin mRNA implies that some neurons in occipital and superior temporal cortices in schizophrenia form fewer or abnormal synapses.
-
▪ The inferred synaptic pathology is mild and varies from one region to another.
-
▪ The alterations in schizophrenia may be more extensive in women than in men.
LIMITATIONS
-
▪ Synaptic pathology could exist without an alteration in synaptophysin, and vice versa.
-
▪ The diagnostic specificity of the findings is uncertain.
-
▪ The cause and timing of the changes are unknown.
ACKNOWLEDGEMENTS
We thank David Cotter, Margaret Esiri, Ian Everall, Josie Heffernan, Rob Kerwin, Bianca Watts and Padraig Wright for their contributions. The London series was collected courtesy of the MRC Brain Bank at the Institute of Psychiatry.








eLetters
No eLetters have been published for this article.