


Quetiapine is a widely prescribed antipsychotic. 1 First manufactured by AstraZeneca, it was initially introduced for the treatment of schizophrenia and non-affective psychosis in the late 1990s, but is now licensed in several countries for the treatment of bipolar disorder and other conditions. The original immediate-release (IR) version was the third most frequently prescribed antipsychotic in the UK from 2004 to 2007, and by 2008 had been taken by over 25 million people worldwide. 2 The patent for quetiapine IR expired in March 2012 and the generic version is now comparable in cost to haloperidol, leading to considerable cost savings. Although older reviews of comparative and placebo-controlled trials concluded it was an effective treatment for schizophrenia and non-affective psychosis, Reference Srisurapanont, Maneeton and Maneeton3,Reference Leucht, Arbter, Engel, Kissling and Davis4 these were based on a limited number of trials which suffered from severe attrition. The Cochrane review, for example, noted that most of the original placebo-controlled studies were severely compromised by missing outcome data, and that their results were therefore ‘impossible to interpret with confidence’. Reference Srisurapanont, Maneeton and Maneeton3 Three of the four included studies were missing more than half of their 6-week outcome data, and the remaining study had only 12 participants. In each case the outcomes of those leaving early were estimated by the method of carrying their last available observation forward, an imputation strategy now regarded as unreliable. Reference Hamer and Simpson5 In their 2009 review Leucht et al found that, overall, second-generation antipsychotics had a moderate effect on symptoms (Hedges’ g= 0.51). Reference Leucht, Arbter, Engel, Kissling and Davis4 However, they suggested that the high withdrawal rate in these studies might have attenuated the drug-placebo difference. Indeed, in most placebo-controlled trials more than a quarter of participants leave the study, and in a significant number of trials more than half do so. Reference Hutton, Morrison, Yung, Taylor, French and Dunn6 Such high rates of missing data cannot be safely ignored. A recent survey by the Cochrane Schizophrenia Group found consultant psychiatrists, patients, carers and Cochrane researchers in agreement that trials with over 25% missing data lack credibility, Reference Xia, Adams, Bhagat, Bhagat, Bhoopathi and El-Sayeh7 and there is largely a consensus that no statistical approach can produce reliable results when assumptions about the outcomes of participants carry more weight than actual observations. Reference Xia, Adams, Bhagat, Bhagat, Bhoopathi and El-Sayeh7,Reference Leucht, Heres, Hamann and Kane8 Understanding the impact of missing data is particularly challenging if it is missing for non-random reasons that are related to outcome, as may be the case for antipsychotic trials. Reference Rabinowitz and Davidov9 The development of a sustained release version of quetiapine (quetiapine XR) has led to new randomised controlled trials (RCTs) comparing the immediate release version with placebo, some of which had relatively low rates of missing data. Owing to the uncertainty introduced by high attrition in the older studies of quetiapine IR, we set out to perform a new systematic review and meta-analysis.
We had two main objectives. The first was to provide a comprehensive assessment of the efficacy and adverse effects of quetiapine IR for schizophrenia when compared with placebo, with consideration of both outcome quality and the clinical meaningfulness of the results, as informed by recent advances in our understanding of what constitutes a minimum clinically important difference in Positive and Negative Syndrome Scale (PANSS) total scores. Reference Leucht, Kane, Etschel, Kissling, Hamann and Engel10–Reference Hermes, Sokoloff, Stroup and Rosenheck12 Our second objective was to examine the potential impact of missing data on the primary outcomes. More specifically, we examined whether trials with high rates of missing data had smaller effect sizes, Reference Leucht, Arbter, Engel, Kissling and Davis4 and we used a recently published approach to examine the impact on our efficacy estimates of changing assumptions about the likely outcomes of the large numbers of people who leave these trials early. Reference Ebrahim, Akl, Mustafa, Sun, Walter and Heels-Ansdell13
Method
Our search strategy and protocol detailing our inclusion and exclusion criteria are provided in an online data supplement. Two researchers independently searched publication databases, clinical trial registries and previous reviews for randomised controlled trials in which participants with a diagnosis of schizophrenia or early psychosis were randomly allocated to receive double-blind treatment with either placebo or quetiapine IR. No pre-specified limits were placed on study duration.
Data extraction and outcomes
Two reviewers independently extracted data from each study using data extraction forms. We attempted to trace missing summary data by contacting first authors or the study sponsor. Our primary outcomes were the average reduction at study end-point in total score on the PANSS or, if that were not available, on the Brief Psychiatric Rating Scale (BPRS), and the numbers of people achieving an important clinical response. We defined the latter as a greater than 50% reduction in PANSS or BPRS score. Reference Leucht, Kane, Kissling, Hamann, Etschel and Engel14 When these were not reported or provided, we imputed them from means and standard deviations using the validated method of Furukawa et al. Reference Furukawa, Cipriani, Barbui, Brambilla and Watanabe15 (See ‘Changes from protocol’ section in the online supplement.) Our secondary efficacy outcomes included relapse, positive symptoms, negative symptoms, depression, quality of life and need for additional antipsychotic medication or sedatives. We also examined the numbers of participants leaving the study early for any reason, need for hospital care and functioning. For adverse effects, we looked at use of anti-Parkinsonian medication, extrapyramidal side-effects, withdrawal due to adverse events, sedation, total number of drug-attributable adverse events, insomnia, weight gain and weight loss.
We used a strict intention-to-treat (ITT) analysis for dichotomous outcomes, using the total numbers randomised to each group as the denominator in each case. Where possible, we assumed those leaving early or otherwise unaccounted for had an unchanged outcome from randomisation, but carried out sensitivity analyses to test this. Data incorporating last observation carried forward (LOCF) assumptions were used only when there was no alternative. We also wished to use a strict ITT analysis for continuous data, but expected to be limited to summary data derived from smaller samples excluding participants leaving the study early or those without at least one post-baseline assessment. For all outcomes we intended to use summary data based on the mixed-model repeated measures (MMRM) imputation method, followed by LOCF or observed case data if not available. Missing standard deviations were, where possible, calculated from t-test values, P-values, standard errors or confidence intervals. Reference Higgins and Green16 If no variance parameters were reported for a particular study, we imputed standard deviations using the medians of the other studies. Similarly to previous studies, Reference Leucht, Arbter, Engel, Kissling and Davis4,Reference Leucht, Corves, Arbter, Engel, Li and Davis17 we planned to use data from study arms where participants received an optimal drug dose of more than 250 mg. However, we carried out a sensitivity analysis excluding doses of more than 400 mg, as per the recent International Consensus on Antipsychotic Dosing and recent Leucht group analysis. Reference Gardner, Murphy, O'Donnell, Centorrino and Baldessarini18,Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter19
Meta-analytic calculations
For continuous data we calculated the Hedges’ g standardised mean difference (SMD) using Comprehensive Meta-Analysis version 2 for Windows 7. For the primary analysis of the 2–12 week study end-point data we converted BPRS scores (mean and s.d.) to PANSS scores using recently published conversion charts (PANSS total score = 1.538 × BPRS total score), Reference Leucht, Rothe, Davis and Engel20 thus allowing us to present also the unstandardised weighted mean difference (WMD) in PANSS total scores for all the studies combined. When a trial had two or more relevant arms we combined the data following procedures in the Cochrane Handbook. Reference Higgins and Green16 For binary data we calculated the relative risk (RR) of the unfavourable outcome, together with 95% confidence intervals, as well as the absolute risk difference and numbers needed to treat (NNT) or harm (NNH). If a trial had eligible binary data from two or more active treatment arms, we combined these into one. We used a random effects analysis for all outcomes. For the primary outcomes we also performed a sensitivity analysis using fixed effects, but not if heterogeneity was moderate or more, defined as an I 2 statistic of ≥40%. Reference Higgins and Green16
Impact of missing data
We tested the hypothesis that trials with severe rates of missing data (≥50% at end-point) had smaller drug-placebo differences on our primary outcomes than trials with less severe rates (<50%). The 50% cut-off was chosen because it marks the point at which estimated data carry more weight than actual observations, and because the National Institute for Health and Care Excellence (NICE), the Cochrane Schizophrenia Group and others often exclude trials with this degree of missing data from their reviews. Reference Hutton, Morrison, Yung, Taylor, French and Dunn6,21,Reference Bagnall, Jones, Ginnelly, Lewis, Glanville and Gilbody22 We also wished to compare studies with <25% and ≥25% attrition at end-point, Reference Xia, Adams, Bhagat, Bhagat, Bhoopathi and El-Sayeh7 but were unable to do so because no study of 6–12 weeks’ duration had less than 25% attrition. When observed case data were available we were also able to examine the impact of missing data on the primary outcome by imputing values for those who left the trial early using new guidelines provided by Ebrahim et al. Reference Ebrahim, Akl, Mustafa, Sun, Walter and Heels-Ansdell13 Their method involves testing whether the overall treatment effect is robust under four increasingly more conservative strategies – two of which we applied here. Strategy 1 is non-extreme and involves replacing missing data in both arms of each trial with the observed case mean of the control arm. Strategy 2 is more conservative yet plausible, and uses the highest observed control arm mean to replace missing control arm data, and the lowest observed intervention arm mean to replace missing intervention arm data. For both approaches we imputed the missing data treatment and placebo standard deviations with the medians of the control arms of all the included trials, as recommended. Reference Ebrahim, Akl, Mustafa, Sun, Walter and Heels-Ansdell13
Analysis of clinical significance
The minimum clinically important difference (MCID) has been defined by Jaeschke et al as ‘the smallest difference in a score in the domain of interest which patients [or providers] perceive as beneficial and which would mandate in the absence of troublesome side-effects and excessive cost, a meaningful change in the patient’s management’. Reference Thwin, Hermes, Lew, Barnett, Liang and Valley11,Reference Jaeschke, Singer and Guyatt23 An analysis of data from 14 antipsychotic trials (n = 5970) found a rater-determined MCID on the PANSS of roughly 15 points, Reference Leucht, Kane, Etschel, Kissling, Hamann and Engel10 a criterion that has since been replicated by separate analyses of two large non-industry effectiveness trials (n = 1650). Reference Thwin, Hermes, Lew, Barnett, Liang and Valley11,Reference Hermes, Sokoloff, Stroup and Rosenheck12 Data from a large naturalistic study (n = 398) suggested a lower criterion of 10 points, Reference Schennach-Wolff, Obermeier, Seemuller, Jager, Schmauss and Laux24 which is similar to the patient-rated MCID of 11 points derived from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). Reference Hermes, Sokoloff, Stroup and Rosenheck12 We tested the validity of these definitions by comparing them with the median of mean changes that the included trials were designed to detect. We assumed that trial sponsors had provided enough resources to detect with adequate power what they regarded a priori as the smallest difference between the groups that was important to detect. Reference Man-Son-Hing, Laupacis, O'Rourke, Molnar, Mahon and Chan25
Risk of bias and study quality
Two raters independently assessed both study-level risk of bias with the Cochrane Collaboration risk of bias tool, Reference Higgins and Green16 and outcome quality using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. Reference Guyatt, Oxman, Vist, Kunz, Falck-Ytter and Alonso-Coello26 Further details on method and ratings are provided in the online supplement. We tested for publication bias using funnel plots for the PANSS/BPRS total score effect sizes (Hedges’ g) of all studies. Ratings of bias and quality were used to inform interpretation of reliability and magnitude of effects.
Registration of review protocol and subsequent changes
The review protocol was registered in advance with the International Prospective Register of Systematic Reviews (PROSPERO), protocol CRD4201100165. Subsequent changes, in addition to those outlined above, are detailed in the online supplement. We abandoned the use of the response rate hierarchy used by Leucht et al, Reference Leucht, Arbter, Engel, Kissling and Davis4 given their recently expressed concerns that response rate estimates are particularly vulnerable to selective reporting bias, Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter19 and used only the top of the hierarchy instead (50% or more reduction in PANSS/BPRS score). This criterion is now recommended for use in studies of acutely ill patients with nonrefractory illness, Reference Leucht, Davis, Engel, Kissling and Kane27 and we used the method of Furukawa et al to impute this data when not reported or provided. Reference Furukawa, Cipriani, Barbui, Brambilla and Watanabe15 This method has been recently validated using individual patient-level data from 16 antipsychotic trials, Reference Samara, Spineli, Furukawa, Engel, Davis and Salanti28 and has the additional advantage of allowing for the use of adjusted PANSS and BPRS total scores when calculating percentage change, thus avoiding underestimation of response. Reference Leucht, Davis, Engel, Kissling and Kane27 Additional changes included using meta-regression to assess the association between study duration (measured in weeks) and year of publication on total symptoms and clinically significant improvement. These were conducted in Stata version 9 using the Metareg command and Knapp-Hartung variance estimator. Reference Harbord and Higgins29
Results
The process of selecting studies is detailed in Fig. 1. We identified 15 relevant trials, 11 of which assessed short-term efficacy (n = 2259). Lundbeck provided us with summary reports for two unpublished 12-week placebo-controlled studies, 30,31 both of which were terminated early owing to the inefficacy of the investigational drug (bifeprunox; quetiapine IR was an active comparator in these trials). AstraZeneca, the makers of quetiapine, provided us with a considerable amount of additional unpublished data in relation to many of their trials. They decided not to provide us with the report for one unpublished long-term trial comparing therapeutic and subtherapeutic doses of quetiapine, Reference Arvanitis and Scott32 arguing that the lack of a placebo control meant it did not meet our original inclusion criteria. However, we managed to acquire an extract detailing the main results, and other published summaries allowed us partly to assess risk of bias. We therefore included data from a total of 15 studies. An overview of included studies is provided in Table 1, and excluded studies are listed in the online supplement, together with a table of trial characteristics and baseline demographic data.

Fig. 1 Study search procedure. IR, immediate release; RCT, randomised controlled trial.
Table 1 Included studies and Cochrane risk of bias ratings

| Year published/ completed |
Primary publication available? |
Clinical study report synopsis or extract available? |
Random sequence generation (selection bias) |
Allocation concealment (selection bias) |
Performance bias (masking of participants and personnel) |
Detection bias (masking of assessments) |
Incomplete outcome data (attrition bias) |
Selective reporting (reporting bias) |
Other biasFootnote a |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Arvanitis & Miller | 1997 | Yes Reference Arvanitis and Miller38 | YesFootnote b | Unclear | Unclear | Unclear | Unclear | High | Unclear | High |
| Small et al | 1997 | Yes Reference Small, Hirsch, Arvanitis, Miller and Link41 | YesFootnote b | Low | Unclear | Unclear | Unclear | High | Unclear | High |
| Borison et al | 1996 | Yes Reference Borison, Arvanitis and Miller42 | YesFootnote b | Unclear | Unclear | Unclear | Unclear | High | High | High |
| Kahn et al | 2007 | Yes Reference Kahn, Schulz, Palazov, Reyes, Brecher and Svensson43 | Yes | Unclear | Unclear | Unclear | Unclear | High | High | Unclear |
| Canuso et al | 2009 | Yes Reference Canuso, Dirks, Carothers, Kosik-Gonzalez, Bossie and Zhu37 | YesFootnote c | Low | Low | Unclear | Unclear | Low | Unclear | Unclear |
| Potkin et al | 2006 | Yes Reference Potkin, Gharabawi, Greenspan, Mahmoud, Kosik-Gonzalez and Rupnow36 | No | Low | Low | Unclear | Unclear | Low | High | Unclear |
| Chen et al | 2010 | Yes Reference Chen, Hui, Lam, Chiu, Law and Chung45 | No | Low | Low | Unclear | Unclear | High | High | High |
| Lindenmayer et al | 2008 | Yes Reference Lindenmayer, Brown, Liu, Brecher and Meulien39 | Yes | Unclear | Unclear | Unclear | Unclear | High | High | Unclear |
| Cutler et al | 2010 | Yes Reference Cutler, Tran-Johnson, Kalali, Astrom, Brecher and Meulien44 | Yes | Unclear | Unclear | Unclear | Unclear | High | High | Unclear |
| Hough et al | 2011 | Yes Reference Hough, Natarajan, Vandebosch, Rossenu, Kramer and Eerdekens51 | Yes | Low | Low | Unclear | High | Low | High | Unclear |
| Chapel et al | 2009 | Yes Reference Chapel, Hutmacher, Haig, Bockbrader, de Greef and Preskorn52 | No | Unclear | Unclear | Unclear | High | Unclear | High | Unclear |
| Findling et al | 2012 | Yes Reference Findling, McKenna, Earley, Stankowski and Pathak40 | Yes | Low | Low | Unclear | Unclear | High | Low | Low |
| Study 11915A | 2009 | No | Yes 30 | Unclear | Unclear | Unclear | Unclear | High | High | High |
| Study 11916A | 2009 | No | Yes 31 | Unclear | Unclear | Unclear | Unclear | High | High | High |
| Arvanitis & Scott (Study 15) |
1995 | No | Partially Reference Arvanitis and Scott32 | Unclear | Unclear | Low | Low | High | High | High |
a. Not including financial conflict of interest of sponsor or researcher.
b. AstraZeneca supplied extract.
c. Pfizer supplied extract.
Risk of bias and quality ratings
Table 1 provides the main risk of bias ratings, and the right-hand columns of Tables 2 and 3 provide the outcome quality ratings for the main primary and safety outcomes. Ratings for secondary outcomes and additional safety outcomes are provided in the online supplement. Our rationale for the ratings is also provided online, alongside ratings produced by other research groups (where available). In our judgement the main problem with these trials is a somewhat high risk of selective reporting bias in relation to secondary outcomes and adverse effects, coupled with a very high risk of attrition bias for most outcomes. We also judge unblinding due to sedative effects to be likely, Reference Perlis, Ostacher, Fava, Nierenberg, Sachs and Rosenbaum33 and the double-blind design might not protect against the risk of researchers adopting a high threshold for recording effects (e.g. adverse effects) where the desired outcome is ‘no difference’. Reference Treadwell, Uhl, Tipton, Shamliyan, Viswanathan and Berkman34 There is also evidence from documents released through legal proceedings in the USA that AstraZeneca have historically not published all
Table 2 Primary efficacy outcomes
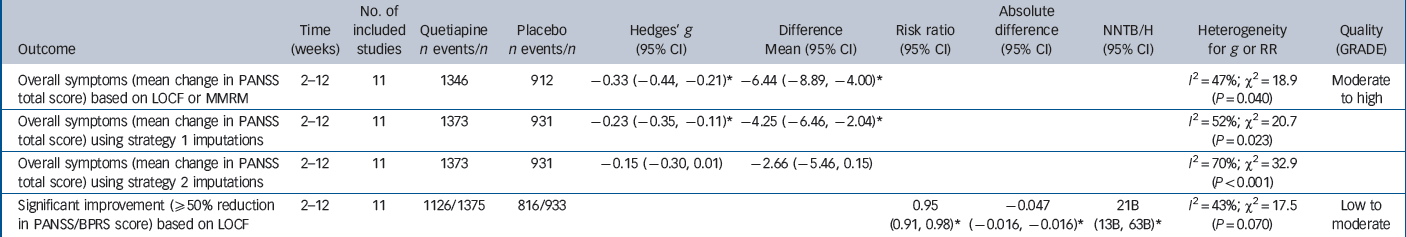
| Outcome | Time (weeks) |
No. of included studies |
Quetiapine n events/n |
Placebo n events/n |
Hedges’ g
(95% CI) |
Difference Mean (95% CI) |
Risk ratio (95% CI) |
Absolute difference (95% CI) |
NNTB/H (95% CI) |
Heterogeneity for g or RR |
Quality (GRADE) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall symptoms (mean change in PANSS total score) based on LOCF or MMRM |
2–12 | 11 | 1346 | 912 | –0.33 (–0.44, –0.21)Footnote * | –6.44 (–8.89, –4.00)Footnote * |
I
2 = 47%; χ2 = 18.9 (P = 0.040) |
Moderate to high |
|||
| Overall symptoms (mean change In PANSS total score) using strategy 1 imputations |
2–12 | 11 | 1373 | 931 | –0.23 (–0.35, –0.11)Footnote * | –4.25 (–6.46, –2.04)Footnote * |
I
2 = 52%; χ2 = 20.7 (P = 0.023) |
||||
| Overall symptoms (mean change In PANSS total score) using strategy 2 Imputations |
2–12 | 11 | 1373 | 931 | –0.15 (–0.30, 0.01) | –2.66 (–5.46, 0.15) |
I
2 = 70%; χ2 = 32.9 (P < 0.001) |
||||
| Significant Improvement (≥50% reduction in PANSS/BPRS score) based on LOCF |
2–12 | 11 | 1126/1375 | 816/933 | 0.95 (0.91, 0.98)Footnote * |
–0.047 (–0.016, –0.016)Footnote * |
21B (13B, 63B)Footnote * |
I
2 = 43%; χ2 = 17.5 (P = 0.070) |
Low to moderate |
||
BPRS, Brief Psychiatric Rating Scale; LOCF, last observation carried forward; MMRM, mixed-models repeated measures; NNTB/H, number needed to treat (benefit/harm); PANSS, Positive and Negative Syndrome Scale; RR, relative risk.
* P < 0.05.
active-comparator quetiapine trials, or have not reported all outcomes. Reference Spielmans and Parry35
Validation of MCID criterion
Researchers and trial sponsors designed their trials to detect with adequate power a mean change in PANSS total score or equivalent of approximately 12 points (range 9.0–15.5), which corresponds to an SMD of 0.55, and is similar in magnitude to the empirically derived estimate of MCID of 11–15 points. Reference Leucht, Kane, Etschel, Kissling, Hamann and Engel10–Reference Hermes, Sokoloff, Stroup and Rosenheck12
Primary efficacy outcomes
Moderate- to high-quality evidence suggested that quetiapine IR was statistically superior to placebo from 2 weeks to 12 weeks in terms of reducing overall symptoms, but the effect was small (WMD = –6.5 points, 95% CI –8.89 to –4.00; SMD = –0.33, 95% CI –0.46 to –0.21) and the 95% confidence intervals excluded the MCID of 11–15 points (Table 2, Figs 2 and 3). Low- to moderate-quality evidence suggested the NNT for much improvement was 21 (95% CI 13 to 63).

Fig. 2 Mean change in Positive and Negative Syndrome Scale (PANSS) total scores or equivalent, using mostly original last observation carried forward estimates to impute missing data. PLA, placebo; QUE, quetiapine. *Severe attrition (≥50%).

Fig. 3 Relative risk of not achieving at least 50% reduction in Positive and Negative Syndrome Scale (PANSS) total scores or equivalent, based mostly on last observation carried forward data; F estimated using Furukawa method. PLA, placebo; QUE, quetiapine. *Severe attrition (≥50%).
Sensitivity analyses and meta-regression
We identified a significant effect of study duration (weeks) on the effect size for total PANSS score (B = –0.04, 95% CI –0.08 to –0.01; P = 0.02), with a more treatment-favourable outcome associated with longer duration. Treatment duration did not significantly moderate the effect for treatment response (B< –0.01, RR = 1.00, 95% CI 0.98 to 1.01; P = 0.70). Excluding the two 2-week studies (Potkin et al, Canuso et al) was associated with a marginal increase in average PANSS change (WMD = –7.7 points, 95% CI –10.0 to –5.3; SMD = –0.38, 95% CI –0.50 to –0.26) and response rates (NNT = 19, 95% CI 11 to 59). Reference Potkin, Gharabawi, Greenspan, Mahmoud, Kosik-Gonzalez and Rupnow36,Reference Canuso, Dirks, Carothers, Kosik-Gonzalez, Bossie and Zhu37 Year of publication did not significantly predict outcomes for total PANSS (B = 0.01, 95% CI –0.01 to 0.04; P = 0.28), but there was a small association for treatment response (B = 0.01, RR = 1.01, 95% CI 1.00 to 1.02; P = 0.01), with a less treatment-favourable outcome associated with a more recent publication date. Meta-regression bubble plots are provided in the online supplement.
Removing data from arms employing doses smaller than 400 mg reduced the contribution made by two trials, Reference Arvanitis and Miller38,Reference Lindenmayer, Brown, Liu, Brecher and Meulien39 but this had little effect on the overall estimate of average change (WMD = –6.3, 95% CI –8.7 to –3.8; SMD = –0.32, 95% CI –0.44 to –0.20) or response rates (NNT = 20, 95% CI 13 to 53). Excluding the study with an adolescent sample (Findling et al) also had little effect on estimates (WMD = –6.3, 95% CI –8.9 to –3.6; SMD = –0.32, 95% CI –0.45 to –0.19; NNT = 22, 95% CI 13 to 83). Reference Findling, McKenna, Earley, Stankowski and Pathak40
Impact of missing data
Overall, the seven 2–12 week trials with less than 50% attrition had a mean PANSS advantage of –5.4 points (95% CI –8.0 to –2.9; SMD = –0.29, 95% CI –0.44 to –0.15) and an NNT of 42 (19, 250H; H = harm), whereas the four 6-week studies with 50% or more attrition had a mean PANSS advantage of 9.2 points (95% CI –15 to –3.4; SMD = –0.39, 95% CI –0.62 to –0.17) and an NNT of 13 (95% CI 7 to 250). The three 6-week studies with less than 50% attrition had a mean advantage of 6.1 points (95% CI –9.9 to –2.3; SMD = –0.27, 95% CI –0.43 to –0.12) and a non-significant NNT of 35 (95% CI 12 to 42H).
Strategy 1 of the Ebrahim approach involved testing whether the overall results would be different if we assumed that participants who withdrew early from both groups had the same degree of change as participants in the control group who stayed until the end. Reference Ebrahim, Akl, Mustafa, Sun, Walter and Heels-Ansdell13 To illustrate this, consider the study by Small et al. Reference Small, Hirsch, Arvanitis, Miller and Link41 Here the mean change for the 49 quetiapine and 39 placebo group participants who completed the trial was, after conversion of BPRS to PANSS scores, –23.3 points (s.d. = 17.7) and –14.9 points (s.d. = 17.7) respectively – a between-group difference of around 8.4 points. Carrying forward the last available scores of the 104 people who did not complete this trial reduced the quetiapine estimate to –13.5 points (s.d. = 24.5; n = 94) and the placebo estimate to –1.5 points (s.d. = 24.0; n = 94), and increased the
between-group difference to around 12 points. These are the figures we used in the main analysis. Introducing the strategy 1 assumption that those who did not complete the trial had a similar outcome to those in the placebo group who did complete it (–14.9 points) reduced the overall estimate for the quetiapine group to –19.1 points (s.d. = 18.2) and reduced the advantage over placebo to 4.2 points. We repeated this procedure for the other five trials for which we had completer data and where no usable MMRM estimate was provided, Reference Arvanitis and Miller38,Reference Lindenmayer, Brown, Liu, Brecher and Meulien39,Reference Borison, Arvanitis and Miller42–Reference Cutler, Tran-Johnson, Kalali, Astrom, Brecher and Meulien44 and entered the revised estimates into the overall meta-analysis. Table 2 shows that the overall advantage for quetiapine over placebo fell to 4.3 points (95% CI –6.5 to –2.0; SMD = –0.23, 95% CI –0.35 to –0.11).
Strategy 2 of the Ebrahim approach involved testing whether the overall results were robust to assuming, first, that participants in the quetiapine non-completers group had the smallest treatment response observed, and second, that those in the placebo non-completers group had the largest placebo response observed. In the study by Small et al this involved assuming the 47 people in the quetiapine non-completers group had the same degree of response as the quetiapine completers group in the study by Lindenmayer et al (–17.4 points), Reference Lindenmayer, Brown, Liu, Brecher and Meulien39 and that the 57 people in the placebo non-completers group had the same degree of response as the placebo completers group in the 2007 study by Kahn et al (–23.1 points). Reference Kahn, Schulz, Palazov, Reyes, Brecher and Svensson43 The revised quetiapine and placebo estimates were –20.4 (s.d. = 18.2) and –19.8 points (s.d. = 18.3) respectively, leading to a between-group difference of 0.6 points. As shown in Table 2, applying strategy 2 to the six trials for which we had completer data reduced the overall advantage for quetiapine to 2.7 points (95% CI –5.5 to 0.2; SMD = –0.15, 95% CI –0.30 to 0.01). Revised forest plots for strategies 1 and 2 are provided in the online supplement.
Publication bias
We detected some asymmetry in the funnel plot of clinically significant change, but not in relation to mean change in overall symptoms or most other outcomes. Funnel plots for the primary outcomes are provided in the online supplement.
Secondary efficacy outcomes
Full details concerning secondary efficacy outcomes are given in sections H and K of the online supplement.
Relapse, exacerbation and need for hospital care
Evidence from one study indicated that quetiapine IR was effective for prevention of symptom exacerbation in people with early psychosis who had responded to quetiapine, Reference Chen, Hui, Lam, Chiu, Law and Chung45 but an unpublished study suggested there was no effect of therapeutic dose (300–600 mg) over a subtherapeutic dose (75 mg) in relapse prevention in chronic schizophrenia. Reference Arvanitis and Scott32 The combined estimate was therefore heterogeneous (I 2 = 87%) and not significant (NNT = 5, 95% CI 2 to 13H). Quetiapine IR was associated with a marginally reduced need for hospital care after 2–6 weeks in three RCTs (NNT = 19, 95% CI 10 to 143). Reference Potkin, Gharabawi, Greenspan, Mahmoud, Kosik-Gonzalez and Rupnow36,Reference Canuso, Dirks, Carothers, Kosik-Gonzalez, Bossie and Zhu37,Reference Cutler, Tran-Johnson, Kalali, Astrom, Brecher and Meulien44 One trial suggested quetiapine IR had a small effect over 52 weeks in relation to reducing readmission to hospital due to relapse (NNT = 11, 95% CI 6 to 143), Reference Chen, Hui, Lam, Chiu, Law and Chung45 but the results were not robust to changing assumptions about the outcome of those leaving early. Overall the relapse and readmission data were very low to low in quality.
Other outcomes
There was a small effect on positive symptoms (SMD = –0.32, 95% CI –0.44 to –0.20; moderate-quality evidence) and a marginal to small effect on negative symptoms (SMD = –0.21, 95% CI –0.32 to –0.10; moderate-quality evidence) over 2–12 weeks, and a marginal effect on depression over 2–6 weeks (SMD = –0.13, 95% CI –0.23 to –0.02; low-quality evidence). Forest plots are provided in the online supplement. We did not investigate whether these estimates were robust to missing data, but the sensitivity analyses for the primary outcome of total symptoms suggest that this is unlikely. Those taking quetiapine IR had a marginally reduced need for additional sedative medication after 2–6 weeks (NNT = 34, 95% CI 13 to 53H; six RCTs), but we judged the evidence as low quality because of selective reporting and missing data, and no reduced need for antipsychotic medication was observed in the two 6-week RCTs where additional medication was not restricted (NNT = 24, 95% CI 7 to 19H; moderate-quality evidence). Reference Potkin, Gharabawi, Greenspan, Mahmoud, Kosik-Gonzalez and Rupnow36,Reference Canuso, Dirks, Carothers, Kosik-Gonzalez, Bossie and Zhu37
Pooled self-report end-point data from the two 12-week trials did not indicate any benefit of quetiapine IR on quality of life, 30,31 as measured by the Schizophrenia Quality of Life (S-QoL) scale (SMD = 0.11, 95% CI –0.15 to 0.36), but we judged the evidence to be very low in quality owing to early termination of the trials, missing data and possible selective reporting from the other trials. No significant effect was observed on any of the subscales, including psychological well-being (SMD = –0.02, 95% CI –0.28 to 0.24) or family relationships (SMD = 0.01, 95% CI –0.25 to 0.28). Since only observed case S-QoL data were reported, we imputed missing data using strategy 1 from Ebrahim et al. Reference Ebrahim, Akl, Mustafa, Sun, Walter and Heels-Ansdell13 This reduced the overall effect from 0.11 to 0.06 (95% CI –0.14 to 0.27). Long-term quality of life data from two RCTs remain unpublished. Reference Arvanitis and Scott32,Reference Chen, Hui, Lam, Chiu, Law and Chung45
An analysis of data from three RCTs (one studying adolescents, two with adult samples) covering a period of 6–12 weeks found quetiapine IR had a small to moderate benefit on functioning, 30,31,Reference Findling, McKenna, Earley, Stankowski and Pathak40 as assessed by a combination of Children’s Global Assessment Scale data and Personal and Social Performance (PSP) data (SMD = 0.39, 95% CI 0.18 to 0.60). Global Assessment of Functioning data were also reported, but unlike the PSP this measure assesses symptom severity as well as functioning. After imputing missing PSP data using strategy 1, the effect size was small (SMD = 0.28, 95% CI 0.09 to 0.46). One study found no benefit of 12 months of quetiapine IR maintenance treatment over placebo in relation to employment status. Reference Chen, Hui, Lam, Chiu, Law and Chung45 Overall, the functioning and employment data were very low in quality owing to selective reporting, early termination of studies, imprecision and missing data. High-quality evidence from 11 trials suggested quetiapine IR had a marginal effect on rates of early discontinuation over a period of 2–6 weeks (NNT = 21, 95% CI 10 to 333H).
Safety outcomes
Safety outcomes are detailed in Table 3 and Figs 4, 5, 6. There was low-quality evidence that quetiapine IR was associated with a small to moderately increased risk of non-serious adverse effects over the short term (NNH = 11, 95% CI 8 to 22). There was no evidence of extrapyramidal side-effects and no evidence of an increased risk of serious adverse events. Moderate-quality evidence suggested no need for additional anti-Parkinsonian medication in quetiapine-treated participants in the short term, but longer-term data were not reported. Data from 12 trials suggested quetiapine IR had a moderate to large effect on weight gain over 2–12 weeks (SMD = 0.64, 95% CI 0.43 to 0.85). Participants gained an extra 1.75 kg (95% CI 1.10 to 2.40) on average, but we rated the evidence as very low quality because of non-reporting of variance parameters in 7 out of 12 studies, high rates of withdrawal and high heterogeneity. Moderate-quality evidence suggested that around 12% of participants treated with quetiapine experienced a clinically significant increase in weight over 2–12 weeks, compared with 4% of those taking placebo (NNH = 13, 95% CI 9 to 23), and 35% reported sedation or somnolence as an adverse effect compared with 6% of those taking placebo (NNH = 9, 95% CI 7 to 13). Details on additional safety outcomes and forest plots are reported in sections I and K of the online supplement.
Table 3 Main safety outcomes
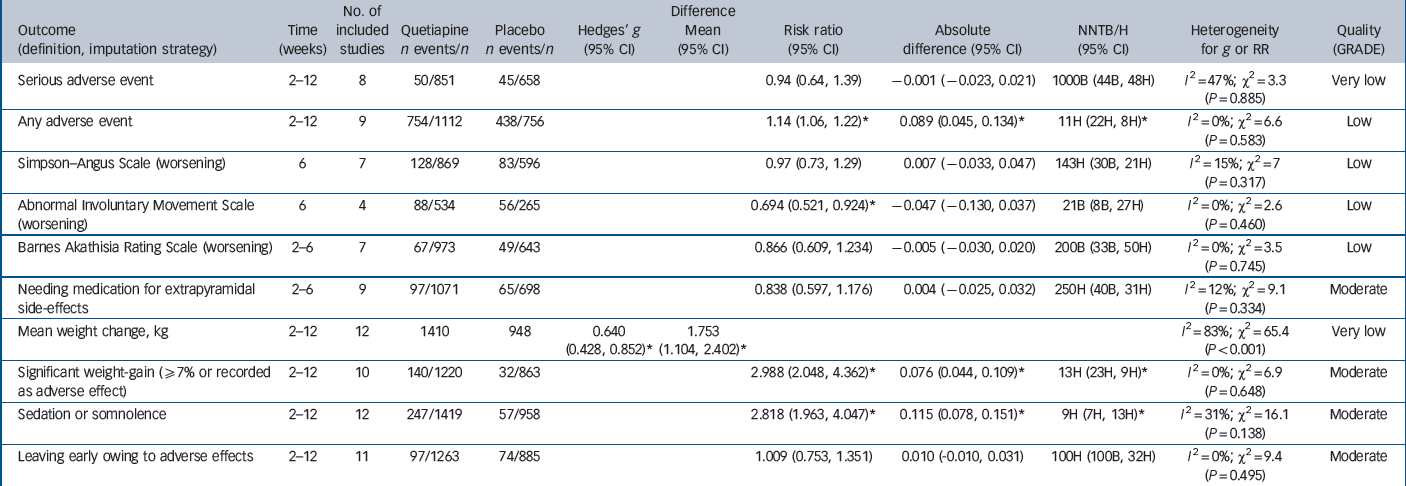
| Outcome (definition, imputation strategy) |
Time (weeks) |
No. of included studies |
Quetiapine n events/n |
Placebo n events/n |
Hedges’ g
(95% CI) |
Difference Mean (95% CI) |
Risk ratio (95% CI) |
Absolute difference (95% CI) |
NNTB/H (95% CI) |
Heterogeneity for g or RR |
Quality (GRADE) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Serious adverse event | 2–12 | 8 | 50/851 | 45/658 | 0.94 (0.64, 1.39) | –0.001 (–0.023, 0.021) | 1000B (44B, 48H) |
I
2 = 47%; χ2 = 3.3 (P = 0.885) |
Very low | ||
| Any adverse event | 2–12 | 9 | 754/1112 | 438/756 | 1.14 (1.06, 1.22)Footnote * | 0.089 (0.045, 0.134)Footnote * | 11H (22H, 8H)Footnote * |
I
2 = 0%; χ2 = 6.6 (P = 0.583) |
Low | ||
| Simpson–Angus Scale (worsening) | 6 | 7 | 128/869 | 83/596 | 0.97 (0.73, 1.29) | 0.007 (–0.033, 0.047) | 143H (30B, 21H) |
I
2 = 15%; χ2 = 7 (P = 0.317) |
Low | ||
| Abnormal Involuntary Movement Scale (worsening) |
6 | 4 | 88/534 | 56/265 | 0.694 (0.521, 0.924)Footnote * | –0.047 (–0.130, 0.037) | 21B (8B, 27H) |
I
2 = 0%; χ2 = 2.6 (P = 0.460) |
Low | ||
| Barnes Akathisia Rating Scale (worsening) | 2–6 | 7 | 67/973 | 49/643 | 0.866 (0.609, 1.234) | –0.005 (–0.030, 0.020) | 200B (33B, 50H) |
I
2 = 0%; χ2 = 3.5 (P = 0.745) |
Low | ||
| Needing medication for extrapyramidal side-effects |
2–6 | 9 | 97/1071 | 65/698 | 0.838 (0.597, 1.176) | 0.004 (–0.025, 0.032) | 250H (40B, 31H) |
I
2 = 12%; χ2 = 9.1 (P = 0.334) |
Moderate | ||
| Mean weight change, kg | 2–12 | 12 | 1410 | 948 | 0.640 (0.428, 0.852)Footnote * |
1.753 (1.104, 2.402)Footnote * |
I
2 = 83%; χ2 = 65.4 (P < 0.001) |
Very low | |||
| Significant weight-gain (≥7% or recorded as adverse effect) |
2–12 | 10 | 140/1220 | 32/863 | 2.988 (2.048, 4.362)Footnote * | 0.076 (0.044, 0.109)Footnote * | 13H (23H, 9H)Footnote * |
I
2 = 0%; χ2 = 6.9 (P = 0.648) |
Moderate | ||
| Sedation or somnolence | 2–12 | 12 | 247/1419 | 57/958 | 2.818 (1.963, 4.047)Footnote * | 0.115 (0.078, 0.151)Footnote * | 9H (7H, 13H)Footnote * |
I
2 = 31%; χ2 = 16.1 (P = 0.138) |
Moderate | ||
| Leaving early owing to adverse effects | 2–12 | 11 | 97/1263 | 74/885 | 1.009 (0.753, 1.351) | 0.010 (−0.010, 0.031) | 100H (100B, 32H) |
I
2 = 0%; χ2 = 9.4 (P = 0.495) |
Moderate | ||
GRADE, Grading of Recommendations Assessment, Development and Evaluation; NNTB/H, number needed to treat (benefit/harm); RR, risk ratio.
* P < 0.05.
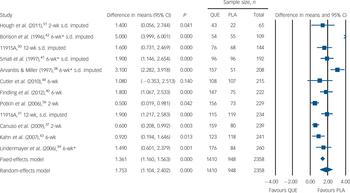
Fig. 4 Mean weight gain, kg. PLA, placebo; QUE, quetiapine. *Severe attrition (≥50%).

Fig. 5 Relative risk of clinically significant weight gain (normally ≥7% increase). PLA, placebo; QUE, quetiapine. *Severe attrition (≥50%).
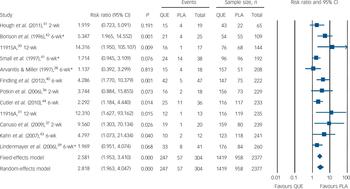
Fig. 6 Relative risk of somnolence or sedation. PLA, placebo; QUE, quetiapine. *Severe attrition (≥50%).
Discussion
Using published and unpublished data, we found the average change in PANSS total score attributable to quetiapine IR over 2–12 weeks to be small. Although the 95% confidence intervals excluded the minimum clinically important difference of 11–15 points, it should be noted that few treatments for psychosis reach this threshold. Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter19 Furthermore, as study duration increased, so did the effect size. Marginal advantages were observed at 2 weeks, whereas moderate effects that approached the threshold for change of at least minimal clinical importance were observed in two as yet unpublished 12-week trials. On the other hand, the overall effect size was smaller if we assumed that the substantial number of participants who left early would have had the same degree of change as placebo-treated participants who stayed. There was no evidence to suggest that drug-attributable benefits had been underestimated because of the severe rates of withdrawal in the older studies. Approximately 21 people needed to take quetiapine IR for 1 person to experience much improvement, defined in accordance with recent recommendations as a 50% or greater reduction in PANSS total score. Reference Leucht, Davis, Engel, Kissling and Kane27
Null to small effects were observed for depression and negative symptoms respectively. Although moderate effects on positive symptoms were observed in the two unpublished 12-week trials, the pooled effect over 2–12 weeks was small. The two 12-week trials also reported small to moderate effects on functioning but found no difference between quetiapine IR and placebo on participant-reported quality of life. Quetiapine IR caused weight gain and sedation, but did not lead to extrapyramidal side-effects. Although there was no evidence of increased serious adverse effects, the evidence was very low quality owing to imprecision and incomplete reporting.
We found some evidence that estimates of clinically significant response derived from more recent trials were lower than in older trials, which is consistent with results from other antipsychotic meta-analyses, Reference Leucht, Arbter, Engel, Kissling and Davis4,Reference Agid, Siu, Potkin, Kapur, Watsky and Vanderburg46 although no relationship between publication year and total symptoms was observed. It remains unclear whether reduced antipsychotic response in recent, large multisite trials with multiple treatment arms reflects a change in the characteristics of participants taking part, improvements in study quality or reporting, increased variability due to an increasing number of sites, Reference Leucht, Heres and Davis47 or better masking of treatment allocation due to reduced expectation of receiving placebo in such trials. Reference Mallinckrodt, Zhang, Prucka and Millen48
Older reviews of quetiapine IR have reported effect sizes of around 0.4 when compared with placebo, Reference Srisurapanont, Maneeton and Maneeton3,Reference Leucht, Arbter, Engel, Kissling and Davis4,Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter19 and NNTs of around 10 or 11. Reference Srisurapanont, Maneeton and Maneeton3,Reference Leucht, Arbter, Engel, Kissling and Davis4 However, most of the trials available at the time had high rates of participant withdrawal and examined quetiapine IR as a target drug for regulatory approval rather than as a control for other preparations. Previous reviews have not been able to account for selective reporting in relation to response rates, examine the impact of changing assumptions about missing data on estimates, or consider the clinical significance of the change attributable to quetiapine IR. A more recent review pooled 4–12 week outcome data for quetiapine IR with outcome data for the more recent extended release version of quetiapine (quetiapine XR) and reported an overall moderate effect size of 0.44. Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter19 Since quetiapine XR was judged by its manufacturer to be sufficiently novel to warrant a separate patent application and significantly greater cost, our a priori view was that pooling the data for the two formulations would give an inaccurate appraisal of both. In relation to duration, we planned to include 2-week quetiapine IR data in our review because this was the approach favoured by preceding reviews that were available at the time of protocol writing. Reference Leucht, Arbter, Engel, Kissling and Davis4 We adhered to this decision because several prescribing guidelines recommend a minimum 2-week trial, and evidence on prescribing practices suggests psychiatrists normally wait only 3–3.5 weeks before switching to another antipsychotic because of non-response. Reference Hamann, Kissling and Leucht49 Nonetheless, it is important to consider that overall efficacy was positively associated with trial duration in our review, and might have been larger still had we included quetiapine XR data. Our data may help explain why a recent meta-analysis found that quetiapine IR was significantly less effective at reducing positive symptoms than first-generation antipsychotics (nine RCTs). Reference Leucht, Corves, Arbter, Engel, Li and Davis17 In an unpublished study therapeutic dose quetiapine IR was significantly less effective than haloperidol in preventing relapse. Reference Arvanitis and Scott32
Missing data
Levels of missing data were high in the included trials. In order to reduce this, trial researchers should continue to assess participants who stop treatment early, as this will inform realistic estimates of likely outcome had they stayed, both in relation to efficacy and adverse effects. Since many early studies of other second-generation antipsychotics also suffered from severe attrition, Reference Hutton, Morrison, Yung, Taylor, French and Dunn6 the robustness of their effects to changing assumptions about missing data may also need to be examined. Although meta-regression has been used to examine the relationship between withdrawals and effect size, Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter19 such analyses are inevitably limited by the fact that few trials have low rates of missing data. Reference Hutton, Morrison, Yung, Taylor, French and Dunn6 Application of the Ebrahim approach would help prescribers and patients appreciate the extent of uncertainty in estimates of antipsychotic benefits and costs. Reference Ebrahim, Akl, Mustafa, Sun, Walter and Heels-Ansdell13 In addition to attrition bias, the proper assessment of both drug and non-drug treatments for psychosis continues to be limited by incomplete and selective reporting of outcomes, low external validity and non-publication of negative trials. Reference Leucht, Heres, Hamann and Kane8 Indeed, a recent meta-analysis found the median effect of currently available antipsychotics over placebo fell from moderate to small after adjusting for the tendency for small studies to report larger effects, Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter19 and selective reporting bias is a particular concern when, as documented by Spielmans & Parry and others, Reference Spielmans and Parry35 it biases our understanding of the severity of adverse effects.
Study limitations
We took advantage of several important developments in methodology which were published after we registered our protocol. Reference Ebrahim, Akl, Mustafa, Sun, Walter and Heels-Ansdell13,Reference Leucht, Rothe, Davis and Engel20,Reference Samara, Spineli, Furukawa, Engel, Davis and Salanti28 Changes post hoc do raise a risk of bias, but we had to balance this against taking the opportunity to increase the quality, robustness and usefulness of our estimates, and we hope we have provided enough information for readers to judge the merit of these decisions. Our claim that a score of 11–15 points is required for minimal clinical improvement might be controversial, not least because few treatments achieve such change in psychosis. Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter19 However, we note the evidence supporting this minimum threshold is now quite consistent across different populations, Reference Leucht, Kane, Etschel, Kissling, Hamann and Engel10–Reference Hermes, Sokoloff, Stroup and Rosenheck12 and we demonstrated that quetiapine trial researchers designed their trials to be able to detect with adequate power only differences of approximately 12 points. Although it has been argued that small benefits might have value at a public health level, Reference Leucht, Arbter, Engel, Kissling and Davis4 there is clearly a need for further debate on this issue. As with non-inferiority and equivalence trials, Reference Treadwell, Uhl, Tipton, Shamliyan, Viswanathan and Berkman34 researchers planning superiority trials might consider stating in advance what they believe constitutes a minimum important difference on continuous outcomes. Although this can be inferred from power calculations, it needs to be stated explicitly.
We were unable to access the full clinical study reports for each trial, which is problematic given a recent study found a much better quality of reporting in these documents when compared with registry reports or peer-reviewed publications. Reference Wieseler, Kerekes, Vervoelgyi, McGauran and Kaiser50 Although we have acquired a large amount of previously unpublished data, access to the reports would have raised the quality of many of the outcomes, in particular the assessments of mean weight gain and response rates. Acquiring unpublished data was challenging, and we doubt we would have been successful had a public debate on publication bias in clinical trials not been taking place at the time. This is an unsatisfactory and unsustainable situation, and a change in the law is clearly required to ensure that all trials past and present are registered, and their full methods and summary results reported, as advocated by the Alltrials campaign (www.alltrials.net).
Funding
This work was not funded. P.H. has been a co-investigator on a National Institute of Health Research (NIHR) funded pilot trial of cognitive therapy for people with psychosis who refuse antipsychotics, and is a co-investigator on another NIHR-funded pilot trial of cognitive therapy v. antipsychotics in early psychosis; J.M. is co-chairperson of the Critical Psychiatry Network.
Acknowledgements
We thank the following individuals and their organisations for providing us with unpublished data from various clinical trials: Dr Carla Canuso from Johnson & Johnson, Rakesh Kantaria, John Ramsey, Jasmine Lichfield and Craig Shering from AstraZeneca and Mads Kronborg and Dr Andrew Roberts from Lundbeck. We also thank Giovana Pezzi from AstraZeneca, Jan Yonge and Dr Chris Bushe from Eli Lilly, Sanofi (Medical Information), the UK Medicines and Healthcare Regulatory Authority, the Danish Medicines Agency, Dr Ben Goldacre, Edd Howard and Kerry Roberts.











eLetters
No eLetters have been published for this article.