


Since their introduction into clinical practice 40 years ago, the use of benzodiazepines has become widespread, because of their efficacy, safety and tolerability. However, their mechanism of action was unknown until 1977, when was it discovered that they interacted with specific receptors in the central nervous system (CNS). In 1987 this receptor, the GABAA—benzodiazepine receptor, was cloned. Now sophisticated techniques in molecular biology and in neuroimaging are giving us precise information not only into how — and where — benzodiazepines and other anxiolytics/hypnotics act, but also further insights into the pathophysiology of anxiety and related disorders including benzodiazepine dependence and addiction.
THE GABAA—BENZODIAZEPINE RECEPTOR
Following the clinical impact of the first benzodiazepines in the mid-1950s considerable effort went into investigating their mechanism of action. It was not until 1974 that researchers at Roche and in the USA independently showed that there was a highly specific potentiation of gamma-aminobutyric acid (GABA) by benzodiazepines. In 1977 it was discovered that the benzodiazepines interacted with a specific binding site in the CNS, which turned out to be an integral part of the GABAA receptor complex (reviewed in Reference HaefelyHaefely, 1978). The receptor complex was isolated and sequenced in 1987 (Reference Schofield, Darlison and FujitaSchofield et al, 1987) and was visualised by electron microscopy in 1994 (Reference Nayeem, Green and MartinNayeem et al, 1994) (see Fig. 1a). The GABAA—benzodiazepine receptor comprises five protein sub-units (Fig. 1b), arranged like a rosette around a central pore, crossing the cell membrane, which is permeable to chloride and other anions. In addition to GABA and the benzodiazepines, other psychoactive compounds, such as barbiturates and anaesthetic steroids, can also bind to the receptor and open the chloride channel (see Fig. 1c). Benzodiazepine site ligands, however, do not act directly to open the channel, but rather modulate the capacity of GABA to do so, resulting in augmentation or diminuition of its inhibitory effects (Reference Barnard, Skolnick and OlsenBarnard et al, 1998).

Fig. 1a The GABAA—benzodiazepine receptor complex, visualised by electron microscopy, showing five protein sub-units arranged around a central core (from Nayeem et al (Reference Nayeem, Green and Martin1994), with permission).
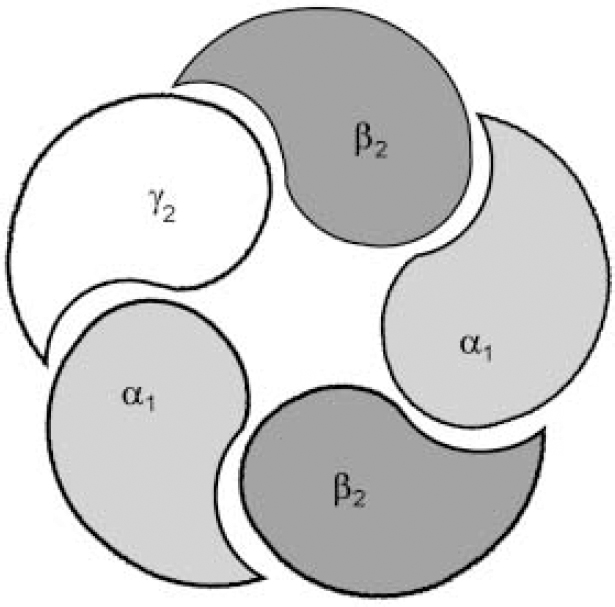
Fig. 1b The most important and most prevalent GABAA—benzodiazepine receptor in the brain is made up from α1, β2 and γ2 sub-units, encoded by the same cluster of genes on chromosome 5. The composition of the receptor sub-units, particularly α and γ sub-units, seems to determine the benzodiazepine pharmacology of the receptor, with different subtypes having different sensitivities to benzodiazepine receptor ligands.
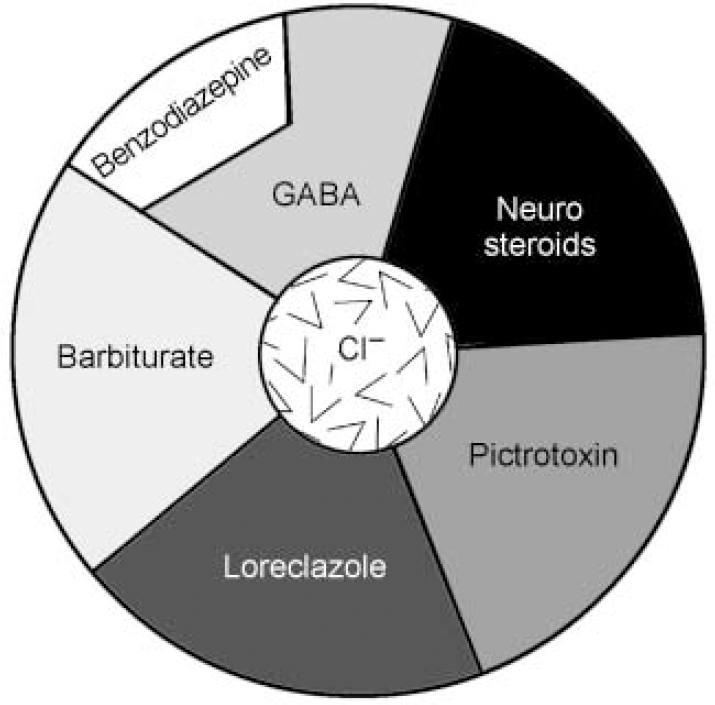
Fig. 1c Schematic representation of the binding sites on the GABAA—benzodiazepine receptor complex. Note that agonists binding to the benzodiazepine receptor site do not open the chloride channel directly, but rather augment the capacity of GABA to do so. (This is a schematic diagram and does not correspond directly to the protein sub-units seen in Fig. 1b.)
GABA: REINING IN THE NERVOUS SYSTEM
Gamma-aminobutyric acid is quantitatively the most important inhibitory transmitter in the CNS. GABAergic neurons are distributed widely in the CNS, but are virtually absent outside the brain and the spinal cord. GABA controls the state of excitability in all brain areas and the ongoing level of neuronal activity is regulated by the balance between excitatory inputs (mostly glutamatergic) and inhibitory GABAergic activity. If the balance swings in favour of GABA, then sedation, amnesia and ataxia appear. On the other hand, the mildest attenuation of the GABAergic system results in arousal, anxiety, restlessness, insomnia and exaggerated reactivity. It has been estimated that, depending on the brain region, 20-50% of all central synapses use fast-signalling GABAA—benzodiazepine receptors.
AGONISTS, ANTAGONISTS, INVERSE AGONISTS
When GABA binds with the GABAA—benzodiazepine receptor complex, it acts as an agonist: inducing conformational changes, which increase the permeability of the central pore to chloride ions. The resulting chloride flux hyperpolarises the neuron, reducing its excitability and producing a general inhibitory effect on neuronal activity. Classical benzodiazepines in clinical use act to enhance the effectiveness of GABA in a unique way, by lowering the concentration of GABA required for opening the channel. Benzodiazepine binding allosterically changes the receptor complex to increase the efficiency of GABA, so enabling the GABAergic circuits to produce a larger inhibitory effect. The action of benzodiazepines is thus markedly different from drugs such as barbiturates, chloral hydrate, chlormethiazole and ethanol, which, as well as enhancing GABA, can also directly open the chloride channel. The propensity of these latter drugs to be fatal in overdose probably reflects this direct action on the chloride channel. Benzodiazepines are safer, perhaps because the vital brain circuits cannot be inhibited over and above the level that would be achieved by natural GABAergic effects.
Further insights into the sophistication of the benzodiazepine site came 15 years ago with the discovery of drugs which bind to it, yet have the opposite effects to the classical benzodiazepine receptor agonists. These drugs decrease the probability of the chloride channel opening in response to GABA and have stimulant, anxiogenic and proconvulsant properties. They are now termed ‘inverse agonists’. Subsequently, benzodiazepine receptor antagonists, notably flumazenil, have been discovered which block the activities of both agonists and inverse agonists. Moreover, the recent discovery of both partial agonists and partial inverse agonists shows that the benzodiazepine receptor mediates a spectrum of different actions (see Fig. 2).
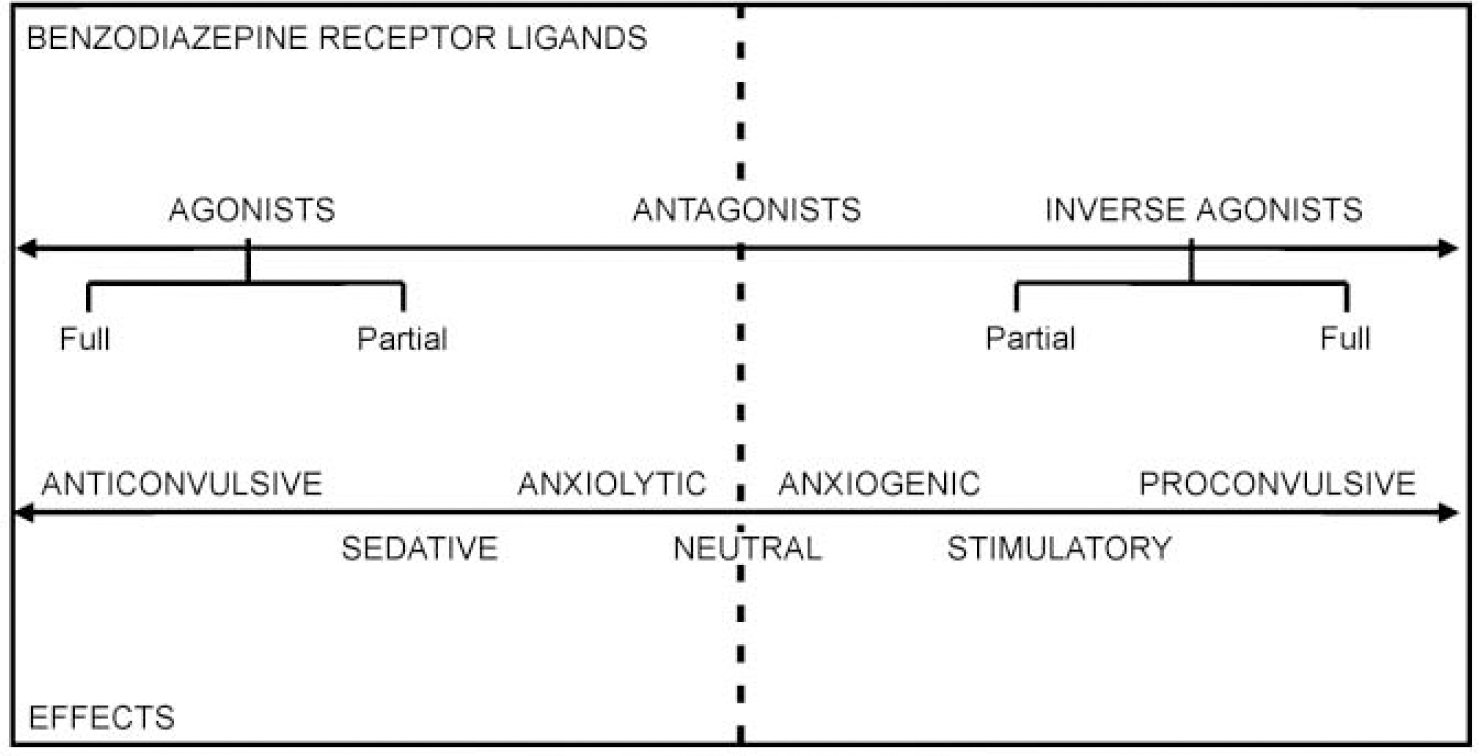
Fig. 2 The benzodiazepine receptor mediates a spectrum of actions, via a range of ligands, including full and partial agonists and inverse agonists.
WHY DOES THE BENZODIAZEPINE SITE EXIST?
The benzodiazepine site is the most evolutionary recent part of the GABAA complex discovered (Reference Nielsen, Braestrup and SquiresNielsen et al, 1978). So what evolutionary pressures have led to the emergence of benzodiazepine receptors and their widespread presence in neurons? One theory is that they are necessary to regulate anxiety, and that the brain itself produces an anxiety-reducing compound (an ‘endogenous agonist’). Anxiety states and insomnia could be the result of a deficiency in the production of this compound and, by analogy with other deficiency diseases (such as insulin in Type 1 diabetes), might require continual, long-term replacement therapy. Attempts to isolate an endogenous agonist have led to some interesting findings. Benzodiazepines are found in the (paraffin-preserved) brains of individuals who died long before the first laboratory synthesis of benzodiazepines (Reference Sangameswaran, Fales and FriedrichSangameswaran et al, 1986). Endogenous benzodiazepine agonists (endozapines) are found in the rare familial condition, idiopathic recurrent stupor (Reference Tinuper, Montagna and PlazziTinuper et al, 1994) and possibly in hepatic encephalopathy (Reference Cossar, Hayes and O'CarrollCossar et al, 1997). Plants, notably Aspergillus fungi, can make a range of benzodiazepines, and these naturally occurring benzodiazepines can also be stored in human brains after being eaten — so it is possible that the receptors evolved to take advantage of these naturally occurring anxiolytics.
It is also possible that benzodiazepine receptors exist for exactly the opposite purpose — to mediate the activities of endogenous inverse agonists. Such compounds could keep brain arousal optimal and if levels fell, sleep could result. In the search for an endogenous benzodiazepine receptor ligand, several compounds with inverse agonist activity have emerged. One early candidate, ethyl-β-carboline-3-carboxylate (β-CCE), was the first compound shown to promote anxiety by a direct action at a receptor in the brain (Reference Braestrup, Nielsen and OlsenBraestrup et al, 1980). But it was later shown that β-CCE was not endogenous, being formed in the extraction process. Another, called tribulin (Reference Hucklebridge, Doyle and PangHucklebridge et al, 1998), is found in urine and its levels are elevated in many conditions where there is increased anxiety (post-traumatic stress disorder and alcohol withdrawal, for example; reviewed by Reference GloverGlover, 1998). However, its structure has not yet been determined and its status remains uncertain.
A third theory suggests that there is no endogenous benzodiazepine receptor ligand and that the site may simply be a particular protein conformation that ‘fine tunes’ GABA function, possibly altering maximal efficacy, or the rate of desensitisation. Recent evidence suggests that the benzodiazepine receptor spectrum is not fixed and that the ‘set-point’ — where drugs bind, but have no effect — can be moved, perhaps as a result of differential sub-unit expression (see below). Among the effects of a set-point shift could be tolerance of, and/or dependence on, benzodiazepine receptor agonists, differential sensitivity to alcohol, anxiety predisposition, panic disorder and stress responsiveness (see Fig. 3). Genetically determined factors may also lead to different positions of the set-point (see also benzodiazepine dependence).
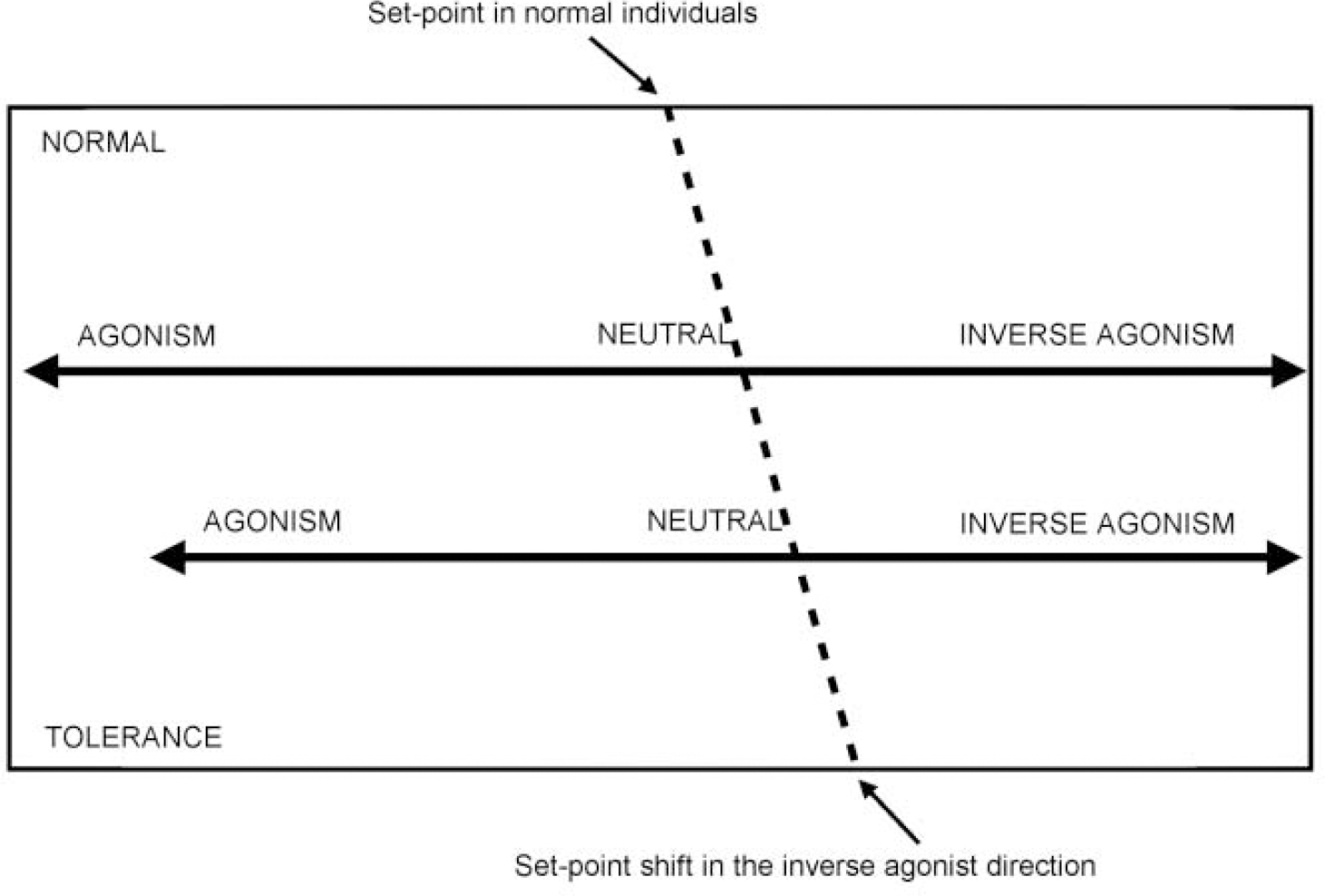
Fig. 3 Recent evidence suggests that the benzodiazepine receptor spectrum is not fixed and that the ‘set-point’ — where drugs bind, but have no effect — can be moved. Anxiety states, particularly panic disorder, may be due to receptor abnormalities which lead to a movement of the set-point in the inverse agonist direction.
ANXIETY: A BENZODIAZEPINE RECEPTOR ABNORMALITY?
Evidence that anxiety disorders may be caused by abnormalities in benzodiazepine receptors has come from a series of studies using the benzodiazepine receptor antagonist, flumazenil, both as a challenge test and as an imaging ligand.
When patients with panic disorder were given an intravenous 2 mg dose of flumazenil, enough to occupy more than half of the receptors in the brain, it provoked panic in most of the patients but was quite innocuous in the control subjects (Reference Nutt, Glue and LawsonNutt et al, 1990). This finding, which has been replicated in a number of studies (though not in all) (Reference Woods, Charney and SilverWoods et al, 1991; Reference MaddockMaddock, 1998; Reference Strohle, Kellner and HolsboerStrohle et al, 1999) clearly demonstrates that panic disorder is not due to the actions of an inverse agonist, as in this case the antagonisit would reduce anxiety. The anxiogenic effect of flumazenil could reflect displacement of an endogenous agonist, but this would only be present in patients (since controls did not experience an increase in anxiety). Another, and more likely, hypothesis is that the ‘set-point’ of the benzodiazepine receptor has moved in the inverse agonist direction, making flumazenil a weak inverse agonist, thus generating anxiety. Further, we could expect the effects of full agonists to be reduced in patients with panic disorder and this is indeed the case. It has been shown that patients with panic disorder are subsensitive to the central effects of diazepam (Reference Roy-Byrne, Cowley and GreenblattRoy-Byrneet al, 1990) and we also know, from clinical experience, that treatment of these patients requires either a high-dose or a high-potency benzodiazepine.
Abnormality at the GABA—benzodiazepine receptor may be specific to, or more pronounced in, severe episodic anxiety — as patients with generalised anxiety disorder, post-traumatic stress disorder and depression do not panic when given flumazenil. Since there is a significant hereditary factor in panic disorder, the receptor abnormality could be due to the transmission of a defective receptor gene (see ‘Identifying the role of receptor sub-units’, below).
IMAGING STUDIES IN ANXIETY
Modern neuroimaging techniques, notably positron emission tomography (PET) and single photon emission tomography (SPET), allow us to measure the GABAA—benzodiazepine receptor complex in the living human brain. In a recent study, PET scans were used to measure brain GABAA—benzodiazepine receptor binding using flumazenil, radio-labelled with 11C. The major finding of the study was that, compared with controls, there is a significant global reduction in flumazenil binding to benzodiazepine sites throughout the brain in patients with panic disorder (Reference Malizia, Cunningham and BellMalizia et al, 1998); PET scans showing flumazenil binding in controls and patients with panic disorder are shown in Fig. 4. SPET studies using the related benzodiazepine receptor ligand 123I-iomazenil, have shown similar decreases in binding (reviewed in Reference MaliziaMalizia, 1999). A localised reduction in benzodiazepine binding in the temporal lobe has also been reported in generalised anxiety disorder (Reference Tiihonen, Kuikka and RasanenTiihonen et al, 1997).

Fig. 4 Position emission tomography (PET) scans of the brains of patients with panic disorder (right) show a significant global reduction in binding to the benzodiazepine antagonist flumazenil compared with ‘normal’ brains (left).
These findings are consistent with the idea that some anxiety disorders may be due to defective neuroinhibitory processes. The greatest decreases observed in benzodiazepine binding occurred in areas thought to be involved in the experience of anxiety in man, such as the orbitofrontal and temporal cortex and insula. Interestingly, in a recent PET study of anxiety generation, we have shown that the anxiolytic effects of the benzodiazepine most closely relate to modulation in brain metabolism in insula and orbitofrontal cortex, among others (Reference MaliziaMalizia, 2000) (see Fig. 5). Care should be taken in drawing too many conclusions from these small studies, but they do strengthen the case that abnormalities in basal or adaptive inhibitory neuromodulation are of pathological significance in anxiety disorders.

Fig. 5 Water positron emission tomography (PET) scan imaging shows that the anxiolytic effects of the benzodiazepines (in this case, midazolam) relate to significant modulation in brain metabolism in the insula/orbitofrontal cortex (OF/I), frontal cortex (F), thalamus (T) and cerebellum (C).
ROLE OF GABA IN MEMORY AND ANXIETY
As well as its apparent involvement in anxiety, the central GABAergic system is also critically involved in cognitive processes, especially memory. Many human and animal studies show that benzodiazepine receptor agonists impair memory formation. Conversely drugs that decrease GABA function can have memory-enhancing properties. Since anxiety conditioning involves both the experience of anxiety and the formation of an association (a form of memory) it can be postulated that inhibition of GABAergic transmission is a powerful mechanism in predisposing an organism to increased and sometimes inappropriate anxiety reactivity. Conversely, increased GABAergic stimulation would be powerfully anxiolytic by inhibiting both the experience of anxiety and the aversive reinforcement.
There may be many clinical sequelae from this relationship. Traumatic memories, such as those which occur in post-traumatic stress disorder, are extremely deeply encoded, probably because of the high level of emotion and arousal at the time of the original insult as well as the negative reinforcement on re-experience of the events. Anxiety leads to rapid and profound learning of escape and avoidance behaviours, as in agoraphobia following a panic attack, or anxious avoidance after being bitten by a dog. Apart from animal studies, clinical evidence of reduced GABA in the genesis of anxiety comes from the use of pentylenetetrazol as a convulsant agent before electroconvulsive therapy was introduced in clinical practice. Pentylenetetrazol acts by blocking GABAA receptor function and has been reported to produce extreme anxiety, traumatic memories and extreme avoidance behaviour when used clinically (reviewed in Reference Kalueff and NuttKalueff & Nutt, 1997). Recently, Goddard et al (Reference Goddard, Mason and Almai2001) have reported decreased cortical GABA levels in patients with panic disorder using magnetic resonance spectroscopy.
INSIGHTS FROM MOLECULAR BIOLOGY
Each of the five sub-units of the GABAA—benzodiazepine receptor complex is a different protein. These can be classified into families according to their structural similarities. The principal families have been coded the α, β and γ sub-units. GABA binds to the β sub-unit, whereas benzodiazepines and related drugs, for example, zopiclone, zolpidem and zaleplon, bind to a site on the α sub-unit (Reference DobleDoble, 1999).
At the last count, 19 subtypes coded by different genes had been found in mammals. Six different isoforms of the α sub-unit have so far been identified and different subtypes have different sensitivities to benzodiazepine receptor ligands. Receptors with the α6 sub-unit, for example, are essentially insensitive to all hypnotic/anxiolytic benzodiazepine agonists. The type of γ sub-unit is also critical as only the γ2 sub-unit gives GABAA—benzodiazepine receptor which is responsive to benzodiazepines. The genes that encode for these various subtypes seem to occur in clusters, suggesting that there is some coherence in the expression of receptor complexes with different forms of the sub-units. The most important and most prevalent GABAA—benzodiazepine receptor in the brain is made up from α 1, β2 and γ2 sub-units, encoded by the same cluster of genes on chromosome 5.
IDENTIFYING THE ROLE OF RECEPTOR SUB-UNITS
Using ‘knock-out’ (gene deletion) and ‘knock-in’ (gene alteration) technologies in mice it is possible to produce GABAA—benzodiazepine receptors that are deficient in various types of sub-unit. The first such experiments were carried out by Mohler and colleagues who knocked out the gene for the γ2 sub-unit. Homozygous mice with both the γ2 genes knocked out are not viable, but heterozygotes survive to adult-hood and breed. However, their GABAA—benzodiazepine receptors have half the usual complement of the γ2 sub-unit and so are less sensitive to benzodiazepines. Moreover, these mice exhibit symptoms of hypervigilance and anxiety (see Fig. 6) and so represent a genetically defined model of trait anxiety that closely mimics pharmacological and behavioural features of human anxiety disorders (Reference Crestani, Lorez and BaerCrestani et al, 1999). Further, these mice show decreases in flumazenil binding throughout the brain that are similar to the decreases shown in humans with panic disorder (see ‘Imaging studies in anxiety’, above).
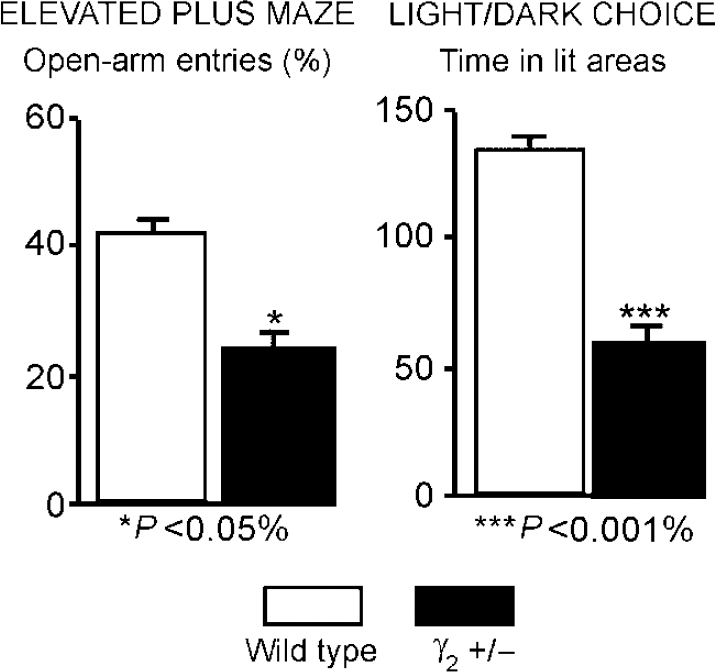
Fig. 6 Mice heterozygous for the γ2 subtype of the γ sub-unit represent a genetically defined model of trait anxiety, closely reproducing the molecular, pharmacological and behavioural features of human anxiety disorders. The figure shows significant differences from control animals in two separate measures of anxiety. Such studies suggest that GABAA—benzodiazepine receptor dysfunction could be a causative factor for a heightened harm-avoidance behaviour and a hypersensitivity to negative associations in patients. (Data from Reference Crestani, Lorez and BaerCrestani et al, 1999.)
Knock-out mice with GABAA—benzodiazepine receptors deficient in other types of sub-unit have also been produced. When the gene for the expression of the β3 subunit is knocked out, for instance, it produces mice that are hyperactive, have poor motor coordination and have spontaneous seizures. They have an attenuated response to GABA and it would seem that the β3 sub-unit has been replaced by a β 2 sub-unit. Interestingly, there is also reduced sensitivity to anaesthetic agents, including halothane, enflurane, etomidate and the benzodiazepine, midazolam, the first direct evidence that GABA—benzodiazepine receptors may be important in mediating the anaesthetic effects of these agents.
More recently, knock-in technology has been used to produce mice in which the α1 subtype gene has been mutated to become insensitive to benzodiazepines but still responsive to GABA (knock-in mutations). In these mice the sedative actions of benzodiazepines are abolished but the anxiolytic, anticonvulsant and hypnotic ones remain (Reference Rudolph, Crestani and BenkeRudolph et al, 1999; Reference Tobler, Kopp and DeboerTobler et al, 2001). Subsequent mutation of two remaining subtypes has shown that the anxiolytic effect is lost if the α2 but not the α3 subtype is mutated (Reference Low, Crestani and KeistLow et al, 2000). The localisation of the α2 subtype in the limbic system supports the role of this circuit in anxiety. This growing evidence for the specificity of benzodiazepine actions being mediated via receptor subtypes is leading to the search for subtype-selective drugs such as α 2 or α3 agonists as non-sedating anxiolytics and α5 inverse agonists (which act mainly in the hippocampus) as memory enhancers (Reference Lingford-Hughes, Hume and FeeneyLingford-Hughes et al, 2001).
CURRENT CLINICAL ISSUES
Because the benzodiazepines may cause dependence and withdrawal after chronic use (see Fig. 7) and owing to their potential for misuse, there are controls on their use in most countries. This control can result in a significant number of patients being denied a therapeutic option that could be appropriate and effective. Recently, there has been an effort to reassess the place of this class of drugs in clinical practice and provide a more balanced view of their relative advantages and disadvantages (Reference Williams and McBrideWilliams & McBride, 1998). A clearer understanding of the mechanisms underlying dependence will also enable us to develop treatment regimes for using current drugs which will optimise benefits and minimise any unwanted effects. We may also be able to identify individuals who are more liable to develop dependence. As discussed above, there is evidence to suggest that the sensitivity of the GABAA—benzodiazepine receptor to various ligands is not fixed and that the set-point — where drugs bind, but have no effect — can be moved (Fig. 3). A shift in the set-point of the receptor in the inverse agonist direction has been seen in studies in animals following chronic benzodiazepine use and could explain both tolerance to benzodiazepine effects and withdrawal symptoms such as rebound insomnia, anxiety and seizures when the treatment is stopped (Reference Little, Gale and SellarsLittle et al, 1988).
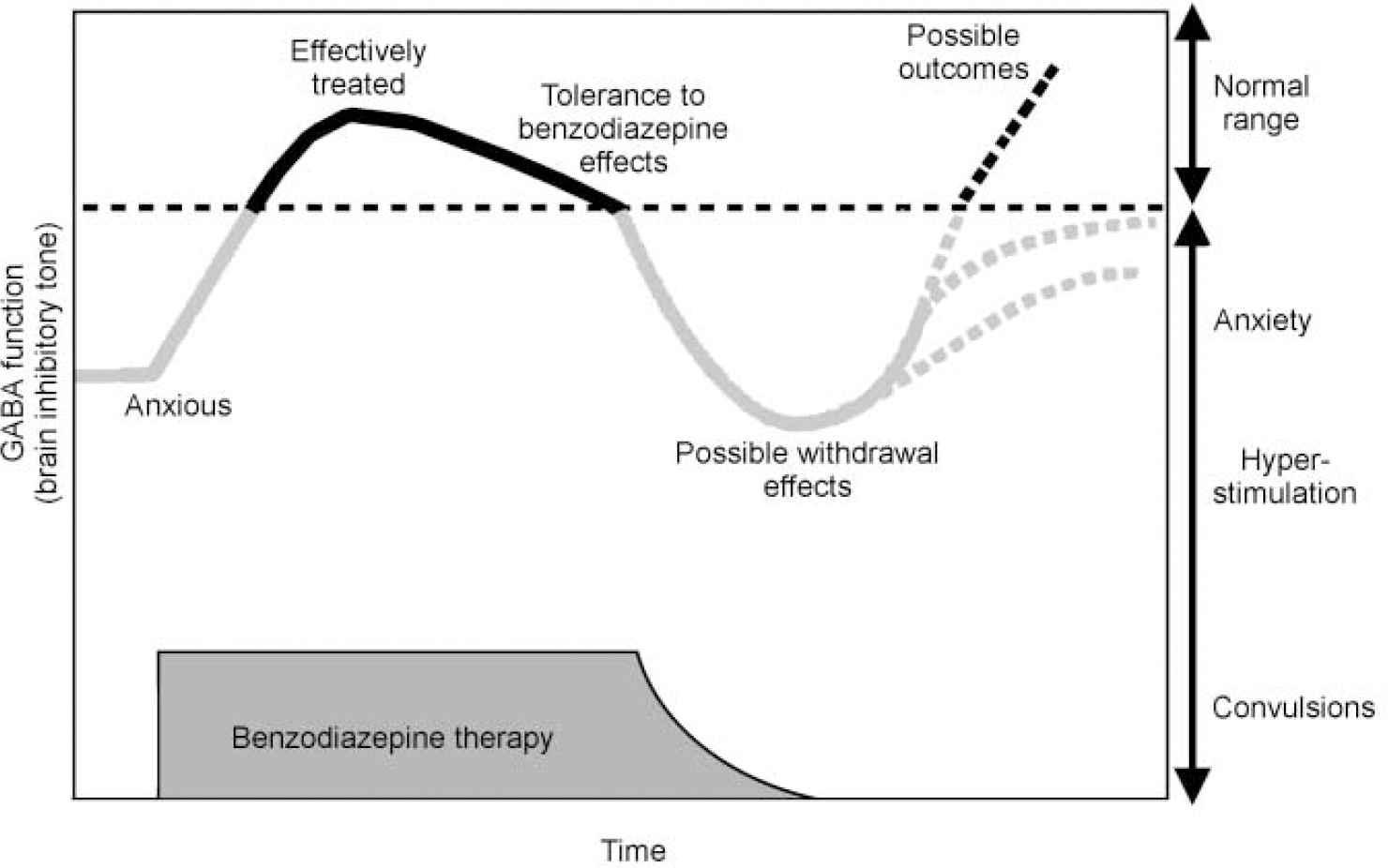
Fig. 7 Schematic diagram of changes in GABAA receptor function during benzodiazepine treatment and withdrawal.
WHY SHOULD RECEPTOR SENSITIVITY CHANGE?
One possibility is that there is substitution of one sub-unit subtype for another. In rats given benzodiazepines chronically, the common α 1 γ2 sub-units are down-regulated, while rarer sub-units are elevated proportionately (Reference Holt, Bateson and MartinHolt et al, 1999). It is suggested that transcription of the gene cluster on chromosome 5 (which encodes for α1 β2 γ2 sub-units) is inhibited on chronic benzodiazepine administration, while the transcription of the gene cluster on chromosome 15 is upregulated (Reference Holt, Bateson and MartinHolt et al, 1999). In certain brain regions, the chromosome-5-encoded receptor sub-unit proteins are replaced by those encoded in chromosome 15, which show less sensitivity. When benzodiazepines are present this manifests as tolerance, but when they are stopped GABA function is effectively reduced, leading to withdrawal.
NEW THERAPEUTIC HORIZONS
Both neuroimaging and molecular biology are giving us fresh insights into the role of the GABAA—benzodiazepine receptor in anxiety. The latest data confirm the importance of the GABAergic system in the pathogenesis of anxiety states and help explain the important role of drugs that bind to the GABAA—benzodiazepine receptor in treating such disorders. Molecular biological and gene manipulation studies have given major clues to the mechanisms involved in tolerance, dependence and withdrawal. This research should ultimately enable us to develop regimens for using current drugs which will optimise their benefits and to find new drugs with enhanced anxiolytic effects and with fewer undesirable side-effects. It would also benefit patients with other conditions responsive to GABAA—benzodiazepine ligands, such as seizures, spasticity and insomnia.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
• The benzodiazepines work at specific receptor sites on the GABAA receptor complex in the mammalian brain, and subtypes of these receptors mediate different actions of these drugs.
-
• Abnormalities of these benzodiazepine receptors may underlie some anxiety disorders.
-
• Drugs targeted at specific receptor subtypes may offer the hope of anxiolytics without unwanted side-effects.
LIMITATIONS
-
• Imaging studies need replication and extension to other anxiety disorders.
-
• Mutations in benzodiazepine receptor genes have not yet been linked to human anxiety.
-
• The effects of new anxiolytics in mouse may not be replicated in humans.










eLetters
No eLetters have been published for this article.