


Epidemiology is essentially a collective science, in which progress has always depended largely upon that made in other fields of medical science (Reference Frost and MaxeyFrost, 1941), and the struggle of today's epidemiologists to keep pace with advances in molecular genetics forms part of a long-standing tradition. Yet there is still uncertainty as to whether ‘molecular epidemiology’ should be regarded as a radical new departure, or in more modest terms simply as a helpful travelling companion (Reference Martin, Eaves and HeathMcMichael, 1994). These doubts are amplified in psychiatric epidemiology, where the customary enthusiasm for new causal models is tempered by awareness of the chequered history of psychiatric genetics, as well as of the complex nature of most forms of emotional disorder.
EARLY PSYCHIATRIC THEORIES AND THE EUGENIC MOVEMENT
The notion that mental illness arises from a combination or interaction of causes, both predisposing and provoking, can be traced far back in the psychiatric literature (Reference Cooper and KatschnigCooper, 1986). It was given expression by Esquirol in his Des maladies mentales and elaborated by Wilhelm Griesinger, who distinguished between traumatic events, prevailing external conditions and certain internal states (including hereditary predisposition), which could combine to give rise to insanity. This simple causal model, anticipating the classic epidemiological triad of specific agent, environment and host susceptibility, was adopted by many authorities, including the founding father of psychoanalysis, whose own conception of disease was (he declared) “a purely practical one and a question of summation, that predisposition and the eventualities of life must combine before the threshold of this summation is overstepped” (Reference Freud and StracheyFreud, 1909).
In European orthopsychiatry, however, interest became centred more and more on the ‘endogenous’ aspects of mental illness, and in particular — following the introduction of twin research as a way to study the relative powers of nature and nurture (Reference GaltonGalton, 1876) — on the strength of hereditary influences. Both early twin studies in psychiatry (Reference LuxenburgerLuxenburger, 1928; Reference RoseRosanoff et al, 1934) and the prototypic German and Scandinavian field studies of the inter-war years (Reference Susser, Susser and SusserStrömgren, 1950) were fuelled by a wish to demonstrate empirically the importance of heredity in mental disorders, and in Nazi Germany the research findings were misused by Ernst Rüdin and his followers to justify a debased doctrine of ‘racial hygiene’ (Reference WhalleyWeber, 1997). The eugenic movement, of which Galton was an early protagonist, had long promulgated the notion that mental illness, feeble-mindedness, criminality, alcoholism and sexual promiscuity were all expressions of national or racial degeneracy, whose remedy lay in a policy of selective birth control extending to sterilisation of those unfit to bear children. Ideas of this nature commanded widespread political support in the early 20th century, inspired new legislation in a number of countries, and under the Hitler regime were used to justify mass extermination of the mentally ill and handicapped (Reference Miki, Swensen and Shattuck-EidensMeyer-Lindenberg, 1991).
In our own day workers in human genetics have been at pains to dissociate themselves on scientific as well as moral grounds from eugenic theory, while those in the field of psychiatric genetics have striven to free their discipline from the stigma imposed upon it. Nevertheless, in global terms eugenic notions remain a major socio-political issue. In Sweden, compulsory sterilisation persisted until 1976 (Reference ArmstrongArmstrong, 1997). In the People's Republic of China, a new eugenics law was proclaimed in 1994, with the aim of preventing ‘new births of inferior quality’, particularly in underdeveloped and economically poor areas ( Lancet, 1995). In the USA and elsewhere, the growing prospects of genetic screening for job selection (Reference GostinGostin, 1991) and medical insurance (Wilkie, 1998) have led some observers to conclude that the eugenic movement has not disappeared but merely been privatised. Against this background, the importance of maintaining high ethical standards in genetic research will continue to be essential for scientific advance.
GENETIC EPIDEMIOLOGY TODAY
Although the advent of molecular biology has greatly increased the potential scope of genetic research, its impact on public health epidemiology is still hard to assess. The main achievements of medical genetics to date have been concerned with relatively rare Mendelian disorders, whereas genetic epidemiology now deals increasingly with common chronic conditions in which an inherited predisposition may interact with environmental exposures in complex ways (Reference KendlerKendler, 1995). Hence any tendency to neglect environmental research in favour of a reductionist ‘molecular epidemiology’ is bound to be retrogressive (Reference Martin, Eaves and HeathMcMichael, 1994). Moreover, the new genetic-marker strategies, like the older family, twin and adoption study designs, are not central to epidemiological method, but rather overlap with it to greatly varying extents. Linkage studies in particular are usually restricted to — or at least over-sample from — families multiply affected by the target disorder, with no defined population base (Reference Ritsher, Warner and JohnsonRisch, 1997). This approach can be regarded as clinical epidemiology in the broad sense, but cannot make use of population denominators.
Allelic frequency counts have been conducted in large-scale population studies, most notably for rare single-gene conditions such as Tay—Sachs disease and Gaucher's disease (Reference McCabe and McCabeMcCabe & McCabe, 1997), but also in studying ‘susceptibility’ genes. Evaluation of the research findings becomes more difficult in allelic association studies of complex polygenetic disorders, causing some workers to prefer family-based studies to population surveys because of their greater feasibility and accuracy of case—control comparison (Reference Strömgren and SjögrenSmeraldi & Macciardi, 1995). The epidemiological paradigm, in short, is an ideal to which genetic-marker research aspires but often cannot attain.
Despite important applications to infectious disease (e.g. detecting human variants that modify susceptibility to human immunodeficiency virus infection, or subtyping drug-resistant strains of tuberculosis), molecular genetic research has been largely concentrated on predisposing factors of noninfectious disease. Here the hereditary component of the nature—nurture model has come to be equated for practical purposes with the human genome: “a linear arrangement of approximately three billion deoxyribonucleotides partitioned into 22 autosomes and two sex chromosomes” (Reference Ellsworth, Hallman and BoerwinkleEllsworth et al, 1997). The aim of the Human Genome Project, as its name implies, is to identify the 60 000 to 80 000 genes which encode for polypeptides and ultimately to establish all the estimated 3.2 billion base-pair sequences. Currently (late 1999) some 428 million of these sequences had been completed, while the programme Online Mendelian Inheritance in Man (OMIM) has identified over 10 000 genes related to known human characteristics, including many disease phenotypes. Screening tests are already available for single-gene disorders such as cystic fibrosis (Reference BrockBrock, 1996) and Duchenne and other muscular dystrophies (Reference DaviesDavies, 1997), while predisposing genetic factors have been identified in diseases less evidently familial, such as breast and ovarian cancers (Reference Munk-J⊘rgensen and EwaldMiki et al, 1994).
Impressive as these achievements are, it is already clear that mapping the human genome is only the first step towards understanding its functions and must be followed by what Anthony (Reference Anthony2001, this supplement) refers to as “an emerging ‘Human Proteome Initiative’ ”, namely systematic studies of gene functions in order to establish a database of protein expression and effects. This is an objective now becoming technologically feasible, although fraught with difficulties (Reference Cantor and LittleCantor & Little, 1998). Meanwhile, the assumption that individual risk exposure due to any specific gene product is simply the product of zygosity and years of life has to be regarded with great caution.
The impact of genetic technology on public health practice has been assessed by Khoury (Reference Khoury1997) in terms of improved knowledge of host susceptibility and its implications for personal prevention. Epidemiology's contribution, in his opinion, will consist in replacing straightforward case—control comparison of groups with and without specified environmental exposures by more complex designs in which genotypic variants will be ascertained and evidence of interaction with environmental risk factors routinely tested for. A fundamental implication for disease control is that, where such quests succeed, public health measures can be focused on ‘phenotypic prevention’; that is to say, on avoiding the health consequences of a genetic anomaly, rather than on ‘genotypic prevention’ aimed at stopping its transmission to the next generation.
The concept of gene—environment interaction has thus come to occupy a central position in the epidemiological search for causes. While it is a truism that any disease is in the broadest sense 100% genetic and at the same time 100% environmental, this general model acquires clinical and public health utility only once there is empirical evidence that susceptibility to certain environmental noxae is mediated by specific gene products or their lack. Concrete examples include the effect of dietary phenylalanine on mental development in children with phenylalanine hydroxylase deficiency; dietary iron overload causing multiple organ failure in persons with the hereditary haemochromatosis gene; and the combined effect of maternal cigarette-smoking and transforming growth factor alpha polymorphisms in producing congenital oral defects (Reference KhouryKhoury, 1997; Reference Yang and KhouryYang & Khoury, 1997). In each of these examples the conclusion for public health policy is that preventive action should be focused on reducing the specific environmental exposure for those at high risk.
Technological progress, including use of the polymerase chain reaction and automated methods of analysis, have resulted in the mapping of a dense web of polymorphic genetic markers. As the number of identified susceptibility genes, and the databases on their associations with specific diseases, grow more rapidly, new problems are being created by the sheer volume of scientific data. Even for single-gene diseases, many allelic variants have been found. For complex, polygenetic conditions the quantity of information generated as the human genome is sequenced seems likely to be astronomical (Reference KhouryKhoury, 1997). To proceed from linkage to identifying and cloning the genes involved will be an enormous task.
In addition to these methodological problems, there is growing concern over the socio-political implications of the Human Genome Project and related research. Advocates of gene mapping tend to present genes as the principal determinants of health and sickness, and genomics as the key to medicine's future. In the words of one leading authority (John Sulston, then director of the Sanger Centre, reported in the Observer, London, 3 October 1999):
“Think of the human genome as the Book of Life. We are about to read the first chapter, as important an accomplishment as discovering the Earth goes round the sun or that we are descended from apes.”
While such claims can be dismissed as public relations ‘hype’, many critics believe that they are symptomatic of a dangerously hubristic attitude of mind. If DNA is seen as the ‘book of life’, human diseases may come to be regarded simply as typographical errors. In the words of Lippman (Reference Lippman1992):
“This new cartography, by adopting the blueprint as a metaphor for genes, leads to restricted conceptions of health and illness, reinforces inequities in the distribution of health and, by privatizing and individualizing responsibility for health, creates and legitimizes a new area for social control.”
The issue is one calling for wider debate, and it seems vital that public concerns about the ethical, legal and social implications of genetic technology, including the commercial pressures in favour of predictive testing, should be met by greater openness in government as well as an improved quality of scientific reporting in the media.
DEVELOPMENTS IN PSYCHIATRIC GENETICS
The same doubts and anxieties, both scientific and ethical, are shared today by many psychiatrists. The heady optimism of the early 1990s, when advances in genomics were welcomed as heralding a revolution in psychiatric research and practice, has given way in more recent reviews to a distinctly cautious tone. Rutter & Plomin (Reference Shaffer, Philips and Enzer1997) qualify their own generally up-beat assessment as “more of a promissory note than an account of accomplishments with proven clinical applications”. Despite some dramatic successes of molecular genetics, declare Merikangas & Swendsen (Reference Meyer-Lindenberg1997), application to psychiatric disorders has proved disappointing as a result of critical differences which exist between these conditions and the diseases to which genetic marker techniques have been successfully applied. According to Vieland et al (Reference Wahlberg, Wynne and Oja1995), the molecular basis of genetic effects on complex, non-Mendelian disorders has proved elusive and gene mapping in that context is an uphill struggle. Peltonen & Lönnqvist (Reference Plomin1995) note that candidate gene strategies have so far been unsuccessful, probably because of incorrect selection of the genes in question, and that we still lack the basic biological knowledge about most mental disorders which is a prerequisite for success in such endeavours.
Concerns about the ethical and social implications of the new biology have also been strongly voiced by some psychiatrists. In the words of Thomas et al (Reference Toomey and Wagenaar1996):
“Biological psychiatry plays a dual role, seeking out the causes of mental illness with the aspiration of cure and at the same time absolving society of any responsibility for mental illness… we have unwittingly devalued the importance of societal factors…. Unemployment, poor housing, homelessness, poverty, racial discrimination, are assumed to be irrelevant. We are [thus] excused from asking awkward questions of ourselves and our political leaders.”
Thus, despite all scientific and technological advances of the intervening years, the wry comment by Penrose (Reference Peltonen and Lönnqvist1971), that genetics and psychiatry make strange bedfellows, has not yet entirely lost its pertinence. It should be noted, however, that all the cited comments refer to ‘psychiatric disorders’ as a collective entity, whereas in fact the prospects for genetic research may vary greatly from one diagnostic category to another. While it is not possible here to examine this question for the whole spectrum of psychiatric disorders, the four broad categories reviewed in this supplement by Whalley (Alzheimer's disease), Tsuang et al (schizophrenia), Heath et al (alcohol dependence) and Goldberg (the common, prevalent forms of depression and anxiety) taken together give some indication of the extent of this diversity.
Alzheimer's disease
Leaving aside Huntington's disease, whose Mendelian pattern of transmission has been known for decades, a particularly favourable candidate for molecular genetic research is Alzheimer's disease (AD), since its neuropathological and neurochemical characteristics are now quite well defined. A number of studies published in the 1980s indicated a cumulative risk for dementia in first-degree relatives approaching 50% in the highest age group, suggesting that the genetic factor could be a single autosomal dominant gene with age-dependent penetrance (Reference Folstein and PowellFolstein & Powell, 1984). Subsequent research has failed to support this hypothesis, pointing instead to causal heterogeneity.
Although twin studies in old age are hedged about with difficulties, several reports from twin cohort projects in the USA and Scandinavian countries have confirmed that there is an important genetic component in AD (Reference Breitner, Welsh and GauBreitner et al, 1995; Reference Reiss, Hetherington and PlominRaiha et al, 1996; Reference Bergem, Engedal and KringlenBergem et al, 1997; Reference Gatz, Pedersen and BergGatz et al, 1997). In these studies concordance rates were consistently found to be higher in monozygotic (MZ) than in dizygotic (DZ) twins, but rates among MZ twins varied widely (for probandwise concordance, in particular, from 21% to 83%). Hence, although the disparities are largely understandable in terms of differences in research designs and methods, no firm conclusions can be drawn about the strength of genetic effects. For vascular and mixed dementias, the evidence in favour of any genetic influence is less clear.
Studies of multiply affected families with early-onset AD have so far identified three gene mutations of causal significance: the amyloid precursor protein (APP) gene on chromosome 21 and the so-called presenilin (PS) genes 1 and 2 on chromosomes 14 and 1 respectively (Reference Lendon, Ashall and GoateLendon et al, 1997). The total number of documented cases is small, and these mutations together are thought to account for only a small fraction of prevalence in the general population. Thus Cruts et al (Reference Cruts, van Duijn and Backhovens1998), investigating a population-based series of 101 unrelated cases of familial and sporadic early-onset AD, detected none with APP mutations and only seven (6.9%) with PS1 or PS2 missense mutations.
In addition, susceptibility to AD is known to be influenced by the apolipoprotein E (APOE) locus on chromosome 19, persons who are homozygotic for the ϵ4 allele carrying a greatly increased risk. In a re-analysis of pooled autopsy studies (Reference NeiderhiserNalbantoglu et al, 1994), the odds ratio associating the ϵ4 allele with presence of AD changes was estimated at 6.2 and the population-attributable risk owing to this factor at 53%. Autopsy series are, however, unrepresentative, and the strength of association found in area surveys of unselected elderly populations in Finland (Reference Kuusisto, Koivisto and KervinenKuusisto et al, 1994) and Australia (Reference Henderson, Easteal and JormHenderson et al, 1996) is less impressive. In the latter study, estimated prevalence of dementia (all causes) by age 90 years was only around 50% for APOE ϵ4 homozygotes, indicating that this allele is not a sufficient cause of AD.
Risk of AD is thus probably determined in part by environmental exposures, but their nature remains unknown, with the exception of earlier brain trauma. In a re-analysis of seven case—control studies (Reference Van Os, Park and JonesVan Duijn et al, 1994), both a history of head injury and late maternal age at birth were associated with an increase in risk, whether a family history of dementia was present or not. No evidence was found for an interaction of other risk factors with the genotype as indicated by family history, the findings being consistent with a model in which these determinants act independently of one another. Indeed, the only evidence of gene—environment interaction to emerge from this meta-analytic study was a negative association between cigarette-smoking and incidence of AD among those with a family history of dementia. A community-based case—control study in New York (Reference Merikangas and SwendsenMayeux et al, 1993) yielded more positive evidence of interaction, but here the relative excess of previous brain trauma among dementia sufferers, compared with matched controls, was entirely due to head injuries incurred after the age of 70 years and less than 5 years before the diagnosis of clinical dementia, so that this may have been a prodromal rather than a causal phenomenon.
More recently, interest in the long-term sequelae of brain trauma has been stimulated by a report of amyloid beta-protein deposition, similar to that characterising AD, in the brains of nearly one-third of younger patients who had survived for varying periods after severe head injury (Reference Rosanoff, Handy and PlessetRoberts et al, 1994). A significantly raised frequency of the APOE ϵ 4 allele was found in cases with beta-protein deposition when compared with other cases, suggesting an interaction between a predisposing genetic risk factor and the effects of brain trauma (Reference PenroseNicoll et al, 1996). Whether these findings model the pathogenesis of Alzheimer's disease remains to be seen.
Schizophrenia
Although the overwhelming majority of all relatives of patients with schizophrenia are not themselves suffering from schizophrenia, evidence from family, twin and adoption studies has consistently pointed to an important hereditary influence. Kendler (Reference Kendler1988) concluded, from family studies using blind diagnoses, control groups, personal interviews and operational diagnostic criteria, that the risk in close relatives is 5-15 times that in the general population. Similarly, Gottesman (Reference Gottesman1991), on the basis of pooled data from some 40 family studies, found an average risk ratio of 9.3 among first-degree relatives. Adoption studies have provided further supporting evidence, although small case numbers and the use of ‘schizophrenia spectrum’ diagnoses have made for difficulty in interpreting the findings (Reference Tambs and MournSusser & Susser, 1987).
Estimates of the strength of genetic effect vary greatly between studies. On one hand it has been argued that apparently non-genetic variation may be due simply to “stochastic events affecting gene expression or structure” (Reference McMichaelMcGuffin et al, 1994). On the other hand, register-based twin studies from the Nordic countries have indicated generally moderate MZ/DZ likelihood ratios that leave a large part of the attributable risk unexplained. Kringlen (Reference Kringlen1995), for example, computed mean pairwise concordance rates of 25.6% and 7.5% and mean probandwise concordance rates of 39.0% and 14.0% for MZ and DZ twins, respectively. The extent to which the high proportion of ‘normal’ MZ cotwins can be explained in terms of reduced penetrance hinges on unresolved methodological problems, but on balance the evidence in favour of some environmental influence seems compelling.
Given the evidence for a genetic role in schizophrenia, it is unsurprising that much research has focused on identifying specific markers. The successes of linkage studies of neuropsychiatric disorders have not yet been matched in schizophrenia or affective psychosis, for which no disease-related gene mutation has been firmly identified. Nevertheless, linkage studies have identified several chromosomal regions with a possible linkage to schizophrenia. Hopes were high in the late 1980s, when two groups (Reference Bassett, McGillivray and JonesBassett et al, 1988; Reference Smeraldi and MacciardiSherrington et al, 1988) reported familial cases with linkage to a gene on chromosome 5. Unfortunately, attempts at replication failed and the findings came to be regarded as false positives. This has become a familiar pattern, linkage studies suggesting a number of possible gene loci which cannot be subsequently replicated, while the newer approach of scanning the human genome for DNA markers has so far produced only equivocal findings. Tsuang et al (Reference Vaillant and Milofsky2001, this supplement) observe, however, that within the past few years evidence for linkage has been strengthened by replication of positive findings at loci on chromosomes 6p and 8p, while others have been reported on 10p, 13q, 15q and 22q.
Because of the lack of established genetic markers, attempts to detect and quantify gene—environment interaction have to rely on family, twin or adoption study designs into which some measures of specific environmental exposures can be incorporated. The hypotheses relating to obstetric complications and to maternal influenza during the second trimester of pregnancy (see Tsuang et al, Kunugi et al and Reference Nalbantoglu, Gilfix and BertrandMunk-Jørgensen & Ewald, 2001, this supplement) are obvious candidates, raising a question as to whether the effects of genetic predisposition to schizophrenia might be mediated through an increased vulnerability to prenatal or perinatal exposures.
Tsuang et al (Reference Vaillant and Milofsky2001, this supplement) propose a neurodevelopmental model, according to which prenatal maldevelopment affecting key neural networks forms a substrate for schizophrenia. As yet clear evidence is lacking, and the work of Murray's group suggests that obstetric complications are more often implicated in cases with no positive family history (Reference Dassa, Sham and van OsDassa et al, 1996). To make matters more complicated, women diagnosed with biological mothers suffering from schizophrenia have been shown in record-linkage studies to be at increased risk for complications of childbirth and reproductive pathology more generally (Reference Jablensky, Zubrick and MorganJablensky et al, 1999), so that genetic and environmental influences may indeed be closely interlinked.
That early-life exposures also play an important role is suggested by findings of the Finnish Adoptive Family Study of Schizophrenia (Reference WeberWahlberg et al, 1997), in which adoptees with biological mothers suffering from schizophrenia have been compared with other adoptees, and the adoptive parents of both groups investigated for evidence of psychopathology and ‘communication deviance’. Although the index-group children as a whole do not appear to be at increased risk, those among them whose adoptive parents have high communication-deviance scores are particularly vulnerable as shown by evidence of thought disorder: a conclusion consistent with a genetically determined vulnerability to psychosocial influences in childhood. These results are intriguing, but the study's heavy reliance on Rorschach Test scores in assessing both the children and their adoptive parents gives rise to difficulties of interpretation.
Chronic alcoholism and alcohol dependence
It is a commonplace that alcoholism differs fundamentally from both AD and schizophrenia in having a known environmental factor — i.e. alcohol consumption — built into the diagnostic concept. Both alcohol consumption and the prevalence of alcoholism show strong cohort effects, socially determined changes in national drinking patterns since the Second World War being associated with an increased probability of dependence among alcohol users (Reference GrantGrant, 1997). Moreover, the frequencies of such alcohol-related problems as road traffic accidents, violent behaviour and liver cirrhosis are known to be reducible by changes in alcohol taxation and licensing laws (Reference Tsuang, Stone and FaraoneToomey & Wagenaar, 1999). These facts underline the importance of societal forces, rather than biological variation, in determining the extent of the public health problem and pointing the way to preventive action.
Nevertheless, evidence has steadily accumulated of a familial aggregation in alcohol abuse and dependence, which seems to be in part genetically determined. Merikangas & Swendsen (Reference Meyer-Lindenberg1997), reviewing control studies published up to that time, computed an average prevalence ratio of 7.0 for first-degree relatives, and noted an excess of cases among male MZ co-twins compared with DZ co-twins (ratio 1.6 : 1). In adoption studies of both genders, the biological relatives of affected persons manifested an average prevalence ratio of 2.4 compared with their adoptive relatives.
Research based on the Virginia Twin Register is of particular interest because of the large size of the samples and their representative nature (Reference Kendler, Neale and HeathKendler et al, 1994; Reference Raiha, Kaprio and KoskenvuoPrescott & Kendler, 1999). Here also, pairwise concordance was found to be higher among MZ than among DZ twins, both for women (47% v. 32%) and for men (55% v. 31%). The computations were, however, based on extremely high ‘life-time’ prevalence estimates for alcohol misuse (in the case of men, 34% for MZ and 38% for DZ twins), which must place a question mark against the method of case identification.
The search for specific genetic markers by means of linkage analysis and allelic association studies has so far yielded mainly inconclusive results, with pointers to susceptibility loci on chromosomes 1, 2, 7 and 16 but no firm evidence. Most association studies have concentrated either on dopamine and serotonin receptors or, more specifically, on the aldehyde dehydrogenase (ALDH) and alcohol dehydrogenase (ADH) genes (Reference GoldmanGoldman, 1995). Heath et al (Reference Heath, Whitfield and Madden2001, this supplement) point out that alcoholism research benefits from the advantage of having already identified polymorphisms of these two genes which in populations of Asian ancestry are associated with differences in alcohol dependence risk. Possession of a single ALDH2*2 allele causes raised blood acetaldehyde concentrations and a characteristic flushing response after ingestion of alcohol, and is associated with a decreased risk of alcohol dependence. Individuals who are ALDH2*2/*2 homozygotes have such an adverse reaction to even moderate doses of alcohol that this genotype has not been found in large series of people with alcoholism in Japan. Since, for better or worse, the allele in question is rarely found among persons of European ancestry, this does not help to explain variations in risk within such populations.
What stands out clearly from the above is that any genetic influence on alcoholism can only be partial and must in some way combine or interact with environmental exposures. Evidently there is potential scope for studies in which both genetic and environmental indicators are measured and entered into multivariate analyses. Current prospects for such research might seem most favourable with respect to early predisposing factors, since a set of childhood environmental variables is known to be highly predictive of alcohol misuse in adult life (Reference McCord and McCordMcCord & McCord, 1960; Reference Van Duijn, Clayton and ChandraVaillant & Milofsky, 1982). In practice, demonstration by means of family studies is problematic, both because the risk factors in question (e.g. parental marital conflict, low family cohesion and poor attachment to parents) are difficult to measure reliably and because they themselves may be attributed to inherited characteristics. Hence adoption study designs are required which can separate the effects of family behaviour patterns from those of biological inheritance.
In a US cross-sectional study of 300 adoptees aged 18-40 years (Reference Cutrona, Cadoret and SuhrCutrona et al, 1994), no direct effects for either genetic or environmental factors were found to predict the development of alcohol abuse or dependence. Among female adoptees, though, evidence of adoptive parental conflict and psychopathology based on the Family Environment Scale did appear to interact with a biological family history of alcohol problems to increase the risk of alcoholism significantly. Although this finding suggests a relatively specific form of gene—environment interaction, at least among women, it must be viewed with caution. The interaction factor was significant only at the 5% level and, as in the Virginia Twin Register studies cited above, the frequency of reported alcohol misuse cases was so high as to suggest either strong selectivity in recruitment or else the use of a very low threshold in identifying cases.
Common forms of depression and anxiety
Epidemiological and genetic research into the affective disorders is still handicapped by problems of classification. Whereas bipolar (manic—depressive) psychosis shows evidence of hereditary influence at least as strong as that in schizophrenia, evidence bearing on both non-psychotic, unipolar forms of depression and the anxiety disorders (generalised anxiety state, phobic and panic disorders) is on the whole less compelling. Merikangas & Swendsen (Reference Meyer-Lindenberg1997), while confirming strong familial aggregation of both unipolar depressive and anxiety disorders, computed relatively modest differences in probandwise concordance between MZ and DZ twins: 1.5 : 1 for depression, 2.4 : 1 for panic disorder and 1.6 : 1 for social phobia. Andrews et al (Reference Andrews, Neilson and Hunt1990) in an Australian study found no evidence of genetic effects on mild depression and anxiety, and Tambs & Moum (Reference Thomas, Romme and Hamelijnck1993) in a large-scale questionnaire survey in Norway also reported only a weak degree of heritability.
From a genetic standpoint, any gain in studying these conditions as separate entities is questionable. Kendler et al (Reference Kendler, Heath and Martin1987) in a study of nearly 4000 Australian twin pairs found no evidence for genes pre-disposing to depression which did not also predispose to anxiety. More recently the same group, collaborating with Swedish researchers, arrived at the same broad result from a massive control study based on the Psychiatric Twin and National Twin Registries in Sweden (Roy et al, 1995). Their conclusion that generalised anxiety disorder and major depressive disorder share common genetic determinants but have partly differing environmental causes is consistent with the model for ‘common mental disorders’ proposed by Goldberg & Huxley (Reference Goldberg and Huxley1992). One should note that these extensive surveys share a weakness of method in their reliance on mailed questionnaires or telephone interviews to establish the diagnosis.
Because the common affective disorders seem to fit dimensional models as readily as categorical ones, some investigators prefer to regard both depression and anxiety as continuously distributed population variables, with individual differences governed by the concerted action of a number of genes, each having only a small contributory effect. Both linkage and association methods have been extended to the study of such variables, whose microbiological correlates are sought for at ‘quantitative triat loci’. Technological advances have made it possible to scan the whole human genome for loci associated with defined traits, by means of high-capacity automated techniques allowing thousands of DNA samples to be analysed simultaneously.
The quantitative trait locus (QTL) approach has definite advantages in studying more or less permanent characteristics, the analysis of continuous variables yielding greater statistical power than that conferred by diagnostic categories and allowing interaction terms to be quantified. The search for genes associated with the general intelligence factor ‘g’ exemplifies this approach in cognitive psychology (see Reference Prescott and KendlerPlomin & Craig, 2001, this supplement). Its applications to remittent morbid conditions presents greater difficulty, and in the case of depressive and anxiety states this tends to be side-stepped by substituting a latent trait such as ‘neuroticism’ (Reference Henderson and BlackwoodHenderson & Blackwood, 1999). The strategy has its drawbacks, in that trait—state correlations are only partial, and the psychological trait measures in question are generally crude. Neuroticism scores, for examples, are usually based on yes/no responses to questions such as ‘Does your mind often wander while you are trying to concentrate?’, ‘Are you inclined to be moody?’ and ‘Are you sometimes bubbling over with energy and sometimes very sluggish?’ (Reference EysenckEysenck, 1958). Drawing conclusions about the genetic basis of affective disorders from material of this nature must be a perilous undertaking.
The importance of distinguishing affective states from underlying traits is neatly illustrated by a study based on the Virginia Twin Registry (Reference Kendler, Kessler and WaltersKendler et al, 1995), in which information on occurrence of stressful life events and the onset of major depressive episodes over 1 year was collected for a population-based sample of over 1000 female twin-pairs. Occurrence of any of four types of severe event (death of a close relative, assault, divorce or marriage break-up, and serious marital conflict) was associated with a more than 10-fold increase in depression onsets within the same month. Genetic predisposition, as gauged by twin concordance, also played a part, suggesting a strong gene—environment interaction. Thus the 1-month probability of a depression onset was calculated for those with unaffected co-twins as 0.5% and 6.2%, respectively, and for those with affected co-twins as 1.1% and 14.6%, respectively, depending on the absence or presence within that month of a severe life event. The best-fitting model of interaction suggested a genetic control of sensitivity to the psychological impact of life events. Making allowances for the limitations of data gathered retrospectively by telephone interview, these findings represent an advance in our understanding of the causal mechanisms involved. As a next step, a careful differentiation of ‘independent’ and ‘dependent’ types of life events, along the lines pioneered years ago by the Bedford College group (Reference Brown and HarrisBrown & Harris, 1978) could be applied to meet the frequently raised objection that exposure to stressful life events is itself largely under genetic control (Reference Peltonen and CraigPlomin, 1994).
In summary, this brief review of four diagnostic groups, drawn from across the broad spectrum of psychiatric morbidity, indicates how sketchy in most areas is our knowledge of both genetic and environmental causal factors, and how divergent are the lines of research that must be pursued in gathering evidence on the different categories.
DEFINING THE ‘ENVIROME’: ENVIRONMENTAL RISK FACTORS OF MENTAL DISORDER
Behavioural scientists who are reluctant to accord a major independent role to environmental factors have adopted a number of different standpoints. Of these, perhaps the most extreme is to claim that the deviation of a complex psychiatric disorder such as schizophrenia from clear-cut Mendelian transmission can be explained purely in terms of ‘incomplete penetrance’ and ‘variable expressivity’ of the genes (Reference McMichaelMcGuffin et al, 1994). Even without positive evidence for environmental influences, the rates of concordance reported from modern twin and adoption studies make these arguments implausible.
An alternative response has been to recognise environmental effects, but to treat them in effect as background noise rather than as signals. No attempt need then be made to explore their nature, beyond partitioning variance in twin and family studies between ‘shared’ and ‘non-shared’ or unique environment: a procedure that can be useful only if it serves as a starting point for direct microenvironmental research.
Third, probands and family members may be questioned about their social behaviour and attitudes on general topics and the resulting scores entered into multivariate analyses as ‘sociocultural correlates’ of the biological variables. Since, however, on the basis of twin studies it has been claimed that the behaviours and attitudes in question are themselves inherited (e.g. Reference Mayeux, Ottman and TangMartin et al, 1986), such analyses are in danger of yielding tautological results.
A fourth position, commonly adopted by behavioural psychologists, is to accept the importance of environmental exposures but to recognise only those that are cognitively mediated, as in the case of life events perceived to be threatening or to signify loss (e.g. Reference Felsner, Jason and MoritsuguFelsner et al, 1983), and hence to neglect chemical, biological — and indeed also social — risk factors of which individuals may be unaware.
The fifth standpoint emphasises that while environmental hazards can be important, individual probabilities of exposure to them are largely genetically determined, first because both genes and early surroundings are normally provided by the family of origin (passive correlation), and second because a person's genetic endowment governs both the selection of a personal environment and the person's relationship with it (active and evocative correlation) (Reference Shaffer, Philips and EnzerRutter & Plomin, 1997; see also Reference Nicoll, Roberts and GrahamNeiderhiser, 2001, this supplement).
Finally, it is argued that we know as yet too little about the mode of action of environmental risk factors for gene—environment studies to make real progress until much basic research has been undertaken. Thus Plomin (Reference Peltonen and Craig1994) in the course of an informative review commented that, just as genetic effects must be explained ultimately at a molecular level, so environmental effects need to be explained in terms of molecular regulatory processes, and added:
“For the environment, there is nothing comparable to the laws of heredity worked out by Mendel, to DNA, or to the triplet code…. Although much remains to be learned about genetics, understanding of genetic processes seems to be light years ahead of our understanding of environmental processes.”
This assessment pays no regard to the achievements of public health medicine over the past two centuries, which have been based primarily on environmental epidemiology rather than detailed analysis of pathogenetic mechanisms. None the less, one must agree that research in this field is hampered by lack of a sound theoretical foundation. “The environment”, declared Einstein, “is everything which isn't me” (cited by Reference McGuffin, Asherson and OwenMcDowall, 1987), thus laying claim to it as a personal construct. A biologist might, however, argue that to each of Einstein's own genes the environment consisted of the rest of his DNA complement, and a behavioural geneticist that the great man's selection of Princeton as a habitat, in preference to Berlin, provides a fine example of active gene—environment correlation. Evidently we stand in need of a clear pragmatic concept for defining the human environment and classifying the hazards it presents to health in general and mental health in particular.
Anthony, who together with his colleagues coined the term ‘psychiatric enviromics’ (Reference Anthony, Eaton and HendersonAnthony et al, 1995) defines it in this supplement as “a deliberately complementary search for specific environments or environmental processes and conditions that promote mental health and reduce the occurrence of psychiatric disturbances” — i.e. a positive concept — but concedes that for practical purposes the best starting point is likely to be the negative concept of preventing mental illness, and proposes a catalogue of environmental exposures and conditions damaging to mental health, based on the manual by Ernest Gruenberg (Reference Gruenberg and Last1980). In addition, however, a number of more recent guidelines could also contribute to the development of an up-to-date inventory (World Health Organization, 1988a , b , Reference Yang and Khoury1991; Reference Sharpley, Hutchinson and McKenzieShaffer et al, 1989; Reference Rutter and PlominRoyal College of Psychiatrists, 1993).
Table 1, which draws on these various publications, is restricted to forms of risk for which relatively specific measures, largely targeted on individual prophylaxis, are at least potentially available, and is certainly not intended to be comprehensive. One may surmise that in purely quantitative terms, macrosocial influences such as mass unemployment, poverty, inner-city decay, ethnic conflicts and enforced migration are more powerful determinants of psychiatric morbidity as a whole. Against these forces, however, preventive action needs to be targeted on whole populations or population subgroups, rather than on individuals, so that specific gene—environment interactions are likely to prove less significant.
Table 1 Preventive actions aimed at reducing mental health damage caused by environmental exposures or their consequences

| Prenatal and neonatal interventions |
| Immunisation against maternal rubella and influenza |
| Voluntary screening for HIV and syphilis infection |
| Dietary supplementation during pregnancy, including folate supplements |
| Antenatal monitoring and care to prevent teratogenic toxicity, foetal alcohol syndrome and neonatal narcotic dependency |
| Measures in infancy and childhood |
| Immunisation against measles, pertussis and herpes simplex encephalitis |
| Eradication campaigns against meningococcal meningitis and congenital syphilis |
| Iodine supplementation in deficiency-endemic areas (orally and in salt) |
| Detection and dietary treatment of phenylketonuria and other metabolic errors |
| Detection of non-accidental injury, physical and sexual abuse |
| Monitoring of prolonged medication (e.g. anti-epileptic drugs) |
| Reduction of neurotoxic exposures (e.g. lead pollution from petrol additives) |
| Measures in adolescence and early adult life |
| Prevention of brain trauma through road traffic accidents (highway speed limits, drink-driving controls, seat belts, crash helmets) |
| Programmes targeting risk-taking behaviour (use of alcohol and ‘designer’ drugs, AIDS infection, etc.) |
| Crisis helplines; youth and student advisory/counselling services |
| School-leaver and welfare-to-work rehabilitation programmes |
| Occupational health and safety programmes (e.g. reducing exposure to organic solvents, organophosphates, heavy metal compounds) |
| Measures in middle and later adult life |
| Reduction of risk for cerebrovascular disease, hypertension and stroke by control of diet, alcohol consumption, etc. |
| Detection of conditions with neuropsychiatric complications (e.g. vitamin B12 deficiency and megaloblastic anaemia, hypothyroidism, Borrelia infection) |
| Avoidance of iatrogenic neuropsychiatric syndromes (e.g. confusional states, oral dyskinesia) by tailored dosage and monitoring of medication |
| Medico-social support in high-risk situations (e.g. recent bereavement, discharge from hospital, admission to long-term care) |
Clearly, the traditional dichotomy into predisposing and provoking causes of mental disorder — although not invalid — must now be regarded as an oversimplification. Systematic attempts to order and classify such a complex, disparate set of influences will call for some kind of multiaxial framework. How might this be constructed? Adapting the simple typology put forward by McDowall (Reference McGuffin, Asherson and Owen1987), environmental health hazards can be classified as:
-
(a) natural physico-chemical;
-
(b) man-made physico-chemical;
-
(c) biological/organic (hitherto mainly natural, but in future presumably also man-made);
-
(d) macrosocial;
-
(e) micro- or psychosocial.
An additional category of occupational factors can also be distinguished, but in practice is an amalgam formed from the other five.
A second possible axis of classification relates to the tempo with which risk exposures in a population undergo change. In Table 2, physico-chemical, biological and social hazards are arranged, with illustrative examples, according to whether the change in rates is sudden and massive, as in a communal disaster; relatively rapid, as in an infectious epidemic; or slow-moving to static, as for natural neurotoxic exposures. At population level, sudden changes will tend to operate as provoking factors, and slow changes or steady states as predisposing factors, although it is not difficult to think of exceptions.
Table 2 Macroenvironmental factors of psychiatric disorder: a simple typology with examples
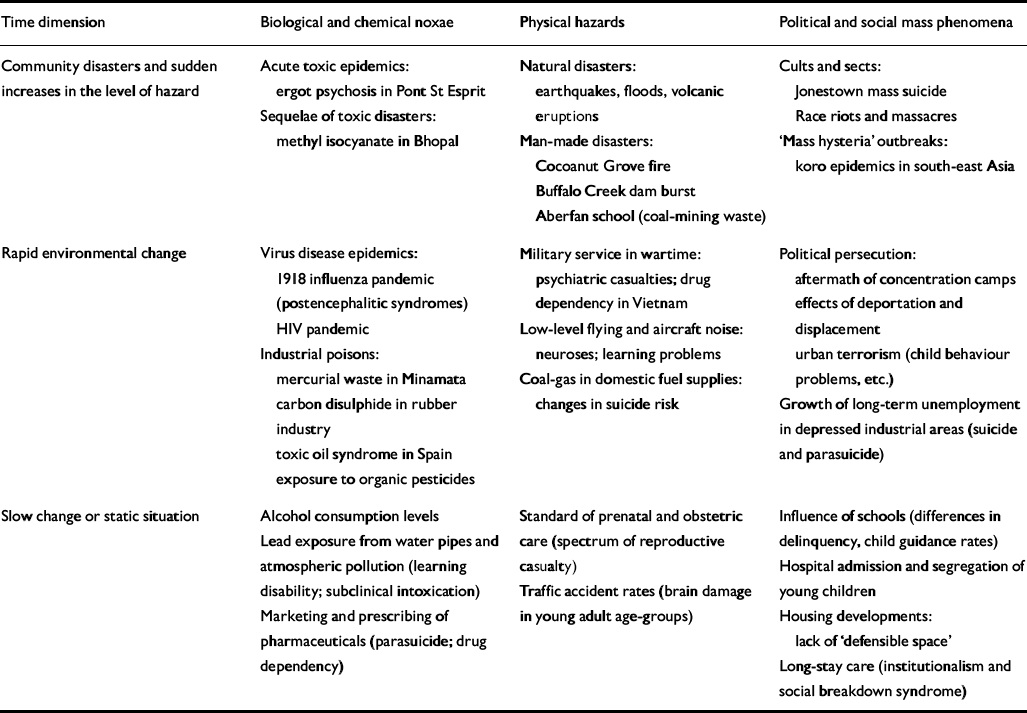
| Time dimension | Biological and chemical noxae | Physical hazards | Political and social mass phenomena |
|---|---|---|---|
| Community disasters and sudden increases in the level of hazard | Acute toxic êpidemics: ergot psychosis in Pont St Esprit | Natural disasters: earthquakes, floods, volcanic eruptions | Cults and sects: Jonestown mass suicide Race riots and massacres |
| Sequelae of toxic disasters: methyl isocyanate in Bhopal | Man-made disasters: Cocoanut Grove fire Buffalo Creek dam burst Aberfan school (coal-mining waste) | ‘Mass hysteria’ outbreaks: koro epidemics in south-east Asia | |
| Rapid environmental change | Virus disease epidemics: 1918 influenza pandemic (postencephalitic syndromes) HIV pandemic | Military service in wartime: psychiatric casualties; drug dependency in Vietnam | Political persecution: aftermath of concentration camps effects of deportation and displacement urban terrorism (child behaviour problems, etc.) |
| Industrial poisons: mercurial waste in Minamata carbon disulphide in rubber industry toxic oil syndrome in Spain exposure to organic pesticides | Low-level flying and aircraft noise: neuroses; learning problems | Growth of long-term unemployment in depressed industrial areas (suicide and parasuicide) | |
| Coal-gas in domestic fuel supplies: changes in suicide risk | |||
| Slow change or static situation | Alcohol consumption levels | Standard of prenatal and obstetric care (spectrum of reproductive casualty) | Influence of schools (differences in delinquency, child guidance rates) |
| Lead exposure from water pipes and atmospheric pollution (learning disability; subclinical intoxication) | Traffic accident rates (brain damage in young adult age-groups) | Hospital admission and segregation of young children | |
| Marketing and prescribing of pharmaceuticals (parasuicide; drug dependency) | Housing developments: lack of ‘defensible space’ | ||
| Long-stay care (institutionalism and social breakdown syndrome) |
An obvious inference from Tables 1 and 2 is that identifying concomitant risk factors in the environment, for example by means of cross-sectional case—control studies, will not suffice to delineate the ‘psychiatric envirome’: past exposures must also be identified, extending back to those that affect intrauterine development or indeed the previous generation. A number of contributions to this supplement provide supporting evidence:
-
(a) the review by Sharpley et al (Reference Sherrington, Brynjolfsson and Petursson2001, this supplement) of functional psychosis rates among first- and second-generation African—Caribbean immigrants in Britain highlights long-term consequences of migration and social disadvantage;
-
(b) Fan & Eaton (Reference Fan and Eaton2001, this supplement) demonstrate the combined influence of birth abnormalities and socio-economic status in childhood on risk for adult emotional disorder;
-
(c) the report by Ritsher et al (Reference Roberts, Gentleman and Lynch2001, this supplement) of their longitudinal study of depression in families over three generations points to inter-generational low social status rather than social selection as a causal factor;
-
(d) Lawrence Whalley's (Reference Wilkie2001, this supplement) investigation of early-onset dementia rates in Scotland reveals case-clustering in areas that were characterised by social deprivation two generations earlier; here, high mortality and morbidity, low educational achievement, unemployment, hardship and poverty seem to be intertwined in a nexus from which causal and selective influences are almost impossible to disentangle.
Each of these projects affords a tantalising glimpse of the ways in which intergenerational processes and early developmental faults can accumulate in a population to form a matrix for psychiatric morbidity.
GENE-ENVIRONMENT CORRELATION AND INTERACTION
Although gene—environment relationships are now highly topical in epidemiology, few substantive findings have so far been reported with regard to mental disorders. This is readily understandable when one considers that such research is in its infancy and is still handicapped by an absence of well-established candidate genes for most conditions, together with the lack of any firm conceptual framework for environmental exposures.
Attempts to date at characterising gene—environment correlation (G r E) and interaction (G × E) have been of limited heuristic value. Some confusion still exists between the statistical concept of interaction as departure from a multiplicative model, and the epidemiological concept of it as co-participation in the same causal mechanism of disease development, with departure from an additive model (Reference Yang and KhouryYang & Khoury, 1997). The six patterns of interaction which these authors distinguish on theoretical grounds have yet to be demonstrated for psychiatric disorders, as also in great measure has the differentiation of passive, active and evocative correlation, mentioned above.
So far most of the available evidence has come from family and twin studies in which family history and MZ/DZ concordance are employed as indicators of genetic inheritance. Such studies will continue to be of critical importance, especially when representative population samples can be drawn and DNA testing carried out. Their potential scope is already indicated by a series of findings on the risk for affective disorders, which call for further investigation:
-
(a) a common, partly genetic susceptibility may combine with different types of environmental exposure to result in either depressive or anxiety states (Reference Kendler, Heath and MartinKendler et al, 1987);
-
(b) shared genetic inheritance and early upbringing can have disparate predisposing effects within nuclear families, because of differential parenting behaviours (Reference RischReiss et al, 1995);
-
(c) genetically influenced susceptibility, as gauged by twin concordance, can interact with stressful events in adult life to provoke the onset of new major depressive episodes (Reference Kendler, Kessler and WaltersKendler et al, 1995).
Adoption research designs have inevitably focused on biological family history (G) and psychosocial risk factors in the adoptive families (E). The latter, including family conflict, poor cohesion and deviant communication, appear not to be diagnosis-specific, but largely shared in common by categories including schizophrenia (Reference WeberWahlberg et al, 1997), conduct disorder (Reference Cadoret, Yates and TroughtonCadoret et al, 1995) and alcoholism (Reference Cutrona, Cadoret and SuhrCutrona et al, 1994). Adoption studies have been influential in demonstrating interaction effects, but their findings cannot be directly extrapolated to the wider population, and their usefulness may in future become more limited as the frequency of adoption at a very early age declines.
Gene marker studies, whether based on direct analysis of DNA or on gene expression products, have so far provided only fragmentary evidence, chiefly because of the lack of candidate genes with established pathogenic relationships to the target conditions. Supportive findings include increased risk of an Alzheimer ‘cascade’ following head injury, in persons carrying the APOE ϵ4 allele (Reference PenroseNicoll et al, 1996); an influence of D2 dopamine receptor alleles on the consequences of family stress for children's cognitive functioning (Reference Berman and NobleBerman & Noble, 1997); and the putative effect of a serotonin transporter gene polymorphism on risk for affective symptoms, which, however, has not been confirmed by a systematic study (Reference Jorm, Henderson and JacombJorm et al, 1998). Clearly, such tentative findings do not amount to more than straws in the wind, and it is too early yet to judge how important advances in molecular genetics will prove to be for this field of enquiry.
CONCLUSION
Overall, firm knowledge concerning either genetic or environmental causes of mental disorder is still at a premium, although progress has varied a good deal among the main diagnostic categories. That the environmental aspects have as a rule been dealt with only perfunctorily by behavioural geneticists can be explained in part by the lack of a firm conceptual framework to order the principal types of ‘candidate’ hazard — physico-chemical, biological, macrosocial and psychosocial — and the nature of exposures to these which may serve to increase host susceptibility or provoke illness onset. Such candidate hazards should be plausible on biological or psychological grounds, and are likely to differ between diagnostic groups.
Once environmental risks have been more firmly defined and exposures to them made quantifiable, the foundation for truly scientific gene—environment interaction studies will have been laid, and we may then expect to see real progress. The kind of multi-disciplinary initiative proposed by Anthony (Reference Anthony2001, this supplement) could be of great help, though the Human Genome Project seems an unlikely analogy in terms of scale and size of research funding — not to mention prospects for patenting and commercial exploitation. How far ‘psychiatric enviromics’ will prove to be a distinct area of research within environmental epidemiology remains to be seen.
Future developments in this whole field cannot yet be predicted with any confidence. What few pointers there are suggest that genetically controlled interaction will be identified with features of the physico-chemical and biological environments, and will prove critical in some neuropsychiatric syndromes with clearly demonstrable central nervous substrates. For most other psychiatric disorders, evidence of such ‘lock and key’ effects may emerge in due course, but it seems at least equally probable that genetic factors will be shown to operate in less specific ways: by influencing the developmental consequences of early adversity, increasing sensitivity to stressful events in adult life, or modifying the clinical features of illness.
How radically mental-illness epidemiology should be adapted to meet the challenge of genomic science is open to debate. Henderson & Blackwood (Reference Henderson and Blackwood1999) in a recent editorial affirmed that psychiatric epidemiology is currently becalmed, and that failure to resolve the difficulties of incorporating molecular genetics into its agenda could leave much of the discipline stranded in the 20th century. The danger should not be dismissed, but other scenarios can also be envisaged. Application of the new technology to mental health problems may prove disappointing and disillusion follow, as so often in psychiatry. Interest may move on to gene expression and proteomics, as Anthony suggests (Reference Anthony2001, this supplement). The impending global population and ecological crises may in the decades ahead bring about a major shift in medical research priorities and a renewed awareness of the need for public health measures based on systems-level change, rather than personal prevention. On the whole, McMichael's (Reference Martin, Eaves and Heath1994) assessment of ‘molecular epidemiology’ as a helpful travelling companion is probably the right one.
Should reductionist designs in causal research now be replaced by some kind of integrative synthesis? Steven Rose (Reference Rosanoff1997, and Reference Roy, Neale and Pedersen2001, this supplement) has argued forcefully that it is time to abandon the outworn nature—nurture dichotomy, and that in psychiatry attention should be focused instead on the life trajectories of people with mental illnesses. His standpoint, which in essence is close to the psychobiological teaching of Adolf Meyer, is one with a strong appeal for many clinicians and therapists, and carries implications for population research that may pose one of the great future challenges for psychiatric epidemiology. Yet the underlying urge to isolate and quantify different aetiological components will not disappear, partly because analytical reductionism is central to modern scientific method, but also because in psychiatric thinking the concepts involved are so closely bound up with fundamental issues concerning the deterministic or dynamic nature of mental illnesses.



eLetters
No eLetters have been published for this article.