


Anxiety disorders are the most common mental disorders, with a combined 12-month prevalence of 18.1% and a lifetime prevalence of 28.8% in the USA. Reference Kessler, Chiu, Demler and Walters1,Reference Kessler, Berglund, Demler, Jin, Merikangas and Walters2 Owing to their high prevalence, combined with an often early onset and chronic course, Reference Kessler, Berglund, Demler, Jin, Merikangas and Walters2–Reference Bruce, Yonkers, Otto, Eisen, Weisberg and Pagano4 anxiety disorders are the second most important cause of disability worldwide within the group of mental and behavioural disorders. Reference Whiteford, Degenhardt, Rehm, Baxter, Ferrari and Erskine5 Antidepressants, including the second-generation selective serotonin reuptake inhibitors (SSRIs) and serotonin–noradrenaline reuptake inhibitors (SNRIs), have been found to be efficacious in the treatment of most anxiety disorders, including generalised anxiety disorder (GAD), social anxiety disorder (SAD), obsessive–compulsive disorder (OCD), post-traumatic stress disorder (PTSD) and panic disorder. Reference Hidalgo, Tupler and Davidson6–Reference Andrisano, Chiesa and Serretti10 However, research in major depressive disorder has suggested that the efficacy of antidepressants depends on the baseline severity of depression. One meta-analysis of the Food and Drug Administration (FDA) database of randomised controlled trials (RCTs) of second-generation antidepressants showed that trials with higher mean baseline severity scores were more likely to be positive, Reference Khan, Leventhal, Khan and Brown11 and another found a significant interaction between baseline severity and treatment group in predicting improvement, such that the drug–placebo difference is relatively small at low levels of initial severity. Reference Kirsch, Deacon, Huedo-Medina, Scoboria, Moore and Johnson12 Similarly, an additional meta-analysis found no statistically significant difference between antidepressants and placebo in people with subthreshold (minor) depression. Reference Barbui, Cipriani, Patel, Ayuso-Mateos and van Ommeren13 Of four analyses using individual patient data, three found that baseline severity of depression was associated with antidepressant efficacy, Reference Khan, Bhat, Faucett, Kolts and Brown14–Reference Fournier, Derubeis, Hollon, Dimidjian, Amsterdam and Shelton16 whereas another large study did not find a significant association. Reference Gibbons, Hur, Brown, Davis and Mann17
Much less is known about the relationship between baseline severity and antidepressant efficacy in the context of anxiety disorders. For OCD, a meta-analysis of 24 antidepressant RCTs found that baseline severity predicted greater improvement in both placebo and drug groups, but there was no evidence of an interaction between baseline severity and treatment group. Reference Ackerman and Greenland18 Recently a meta-analysis of 12 RCTs of paroxetine for GAD and panic disorder found no evidence of an interaction effect. Reference Sugarman, Loree, Baltes, Grekin and Kirsch19 However, pooling trials for different disorders may obscure differences between disorders; it also necessitated the use of a secondary outcome for panic disorder, rather than the primary, panic-specific outcome in these trials. Other single trials and small pooled analyses that have examined this question have reached contradictory conclusions. Reference Stein, Stein and Pitts20–Reference Pollack, Meoni, Otto and Hackett22 The evidence to date, therefore, is (perhaps with the exception of OCD) conflicting, minimal or absent altogether. To our knowledge, no study has comprehensively investigated whether baseline severity predicts antidepressant efficacy in all anxiety disorders. If antidepressant efficacy does depend on baseline anxiety severity, this has important consequences for the continued development of guidelines for the treatment of these disorders. We therefore conducted a meta-analysis and meta-regression of RCTs of SSRIs and SNRIs for the short-term treatment of anxiety disorders using trials submitted to the FDA.
Method
We obtained drug approval packages (reviews) for all SSRIs and SNRIs approved by the FDA for the short-term treatment of five anxiety disorders: GAD, SAD, OCD, PTSD and panic disorder. Reference Roest, de Jonge, Williams, de Vries, Schoevers and Turner23 Reviews were downloaded from the FDA website when available or requested from the FDA's Division of Freedom of Information. Reference Turner24 From the FDA reviews we extracted data on the duration of the trial, drug dose, number of participants, mean score on the primary outcome measure at baseline and end-point (with standard error (s.e.) or standard deviation (s.d.) if available), and the mean change in the primary outcome measure (with s.e. or s.d. if available), for drug and placebo groups separately. For GAD the primary outcome measure in all included trials was the change from baseline on the Hamilton Rating Scale for Anxiety (HRSA); for SAD it was the change on the Liebowitz Social Anxiety Scale (LSAS); for OCD, the change on the Yale–Brown Obsessive Compulsive Scale (Y-BOCS); and for PTSD, the change on the Clinician-Administered PTSD Scale, part 2 (CAPS-2). For panic disorder the primary outcome for most trials was the number of panic attacks over a period of 1–3 weeks, dichotomised into 0 (remitted) or ⩾1 (not remitted); four trials, however, used the change in number of panic attacks as a primary outcome measure. Therefore, for this disorder we also extracted data on the proportion of participants who were free from panic attacks at end-point (remission rate). Furthermore, panic disorder trials reported a baseline score as the number of panic attacks in the 1, 2 or 3 weeks before baseline; we converted all scores to a 2-week time frame.
Data were extracted from the (modified) intention-to-treat analyses only, which used the last observation carried forward (LOCF) method to handle missing data from those leaving the study. Data were extracted preferably from the statistical review; however, we gave preference to other documents within the drug approval package (e.g. the medical review or administrative correspondence) if they provided more complete data (e.g. the s.e. or s.d.) as well as the mean change. If complete data were not available in the FDA review we attempted to obtain the missing data from secondary sources (trial registries and published journal articles).
Outliers and missing data
Exploration of mean baseline scores, stratified by disorder, revealed no outlier in the data-sets for GAD, SAD, OCD and PTSD. For panic disorder, however, one placebo group and one drug group (from two separate trials) had mean baseline scores more than 2 s.d. greater than the overall mean baseline score (after log-transformation to normalise the distribution); we therefore excluded these groups (but not other groups within the same trial) from our analysis. We were able to obtain data on the mean change score and its s.e. or s.d. (or the remission rate for panic disorder) for 46 out of 56 trials. For 40 trials the FDA review provided complete data; for 2 trials we obtained data on the s.e./s.d. from the GlaxoSmithKline trial registry and for 4 trials we obtained data on the s.e./s.d. (3 trials) or the remission rate (1 trial) from the matching published journal articles, Reference Roest, de Jonge, Williams, de Vries, Schoevers and Turner23 after verifying that mean baseline and change scores matched those in the corresponding FDA review. For 10 out of 56 trials not all required data could be obtained from any source. Information on the s.e./s.d. of the change score was missing for 1 PTSD and 6 OCD trials, whereas information on the remission rate was missing for 3 panic disorder trials. For all 10 trials, however, information on the mean change score was available. For PTSD and OCD the change score itself was strongly correlated with its s.d. in groups without missing data; we therefore imputed these missing s.d.s based on the change score, group membership and their interaction. For panic disorder, end-point score was strongly correlated with the remission rate, and we therefore imputed missing remission rates based on end-point score, group membership and their interaction. Imputation was performed separately per disorder in SPSS 20, using multiple imputation with fully conditional specification based on linear regression in order to create ten imputed data-sets.
Statistical analysis
For our main analysis we calculated effect sizes separately for each treatment group, as a single-group pre–post effect size. This approach allowed us to investigate not only the relationship between baseline severity and antidepressant efficacy (drug–placebo difference) but also its underlying cause (i.e. change in the placebo response v. change in the drug response). For GAD, SAD, OCD and PTSD, the standardised mean difference (SMD) was first calculated based on the (within-group) change score and its standard deviation according to the formula SMD = D/SD D, where D signifies the change score and SD D the standard deviation of the change score. Reference Morris and DeShon25 By using this formula we assume that the correlation between baseline and end-point scores is 0.5, as the true correlation is unknown. We then applied Hedges' correction for small sample size, where n indicates the number of participants in a group: Reference Morris and DeShon25
The standard error of Hedges' g was computed as follows: Reference Morris and DeShon25
For panic disorder we selected the remission rate itself as our effect measure. To obtain a single effect size for the antidepressant arms of fixed-dose studies, we used a fixed effects, inverse variance-weighted model to pool these drug groups into one estimate of effect size with its standard error for GAD, SAD, OCD and PTSD. A pooled remission rate was derived for panic disorder by calculating the sample size-weighted average of the remission rates in the different dose groups. For all disorders a pooled baseline score was derived by calculating the sample size-weighted mean of the baseline scores in the different dose groups. Pooling different dose groups may not be appropriate in the presence of a dose–response relationship, but with the exception of venlafaxine such relationships usually cannot be demonstrated with second-generation antidepressants. Reference Adli, Baethge, Heinz, Langlitz and Bauer26
All analyses were performed in Stata 13. We performed meta-analyses using the metan command, applying a random effects (DerSimonian–Laird) model to obtain summary statistics by disorder and group. To measure heterogeneity, the heterogi module within Stata was used to calculate I 2 and its 95% confidence interval. Reference Ioannidis, Patsopoulos and Evangelou27 Meta-regression was then performed separately per disorder using the metareg command. All meta-regressions were based on a mixed effects model, used restricted maximum likelihood (REML) estimation of the residual between-study variance, and included group, baseline severity and their interaction as predictors. The dependent variables were Hedges' g for GAD, SAD, OCD and PTSD and remission rate for panic disorder. Studies were weighted according to the inverse of their variance. For OCD, PTSD and panic disorder, meta-regression estimates from the ten multiply imputed data-sets were combined using the mi suite of commands in Stata.
Sensitivity analyses
As a secondary analysis we calculated Hedges' g for the drug–placebo difference directly from the exact P-value for the statistical test performed at end-point (or alternative methods as required; see Roest et al). Reference Roest, de Jonge, Williams, de Vries, Schoevers and Turner23 The trial baseline severity score was calculated as the sample size-weighted average of all groups (drug as well as placebo) included in the trial. We performed meta-regressions separately for each disorder using the metareg command in Stata. The dependent variable was the drug–placebo difference (Hedges' g) for all disorders, and baseline severity was the only predictor in this analysis. Additionally, to increase statistical power and improve generalisability of results, we expanded our data-set by including extra trials. We included active comparator arms and trials that were not conducted for the purpose of marketing approval, such as trials of other medications (e.g. antipsychotics) in which the antidepressant was used as an active comparator. Trials were obtained from the most recent meta-analyses examining (pharmacological) treatment of anxiety disorders. Reference Soomro, Altman, Rajagopal and Oakley Browne8,Reference Bandelow, Reitt, Rover, Michaelis, Gorlich and Wedekind28,Reference Hoskins, Pearce, Bethell, Dankova, Barbui and Tol29 As this introduced trials with small sample sizes, additional heterogeneity and likely reporting bias, this expanded set of trials was examined as a secondary analysis only. We included parallel-group, placebo-controlled trials with a similar duration (8–16 weeks) to our primary set. Trials with a sample that partially or fully overlapped with a trial in the primary set were excluded, as were trials that did not use a compatible outcome (e.g. HRSA, LSAS, Y-BOCS, CAPS or number of panic attacks as a primary or secondary outcome). We repeated our primary analysis for this expanded set of trials.
Results
A total of 21 reviews were obtained, concerning nine formulations of SSRIs and SNRIs: escitalopram, duloxetine, fluoxetine, fluvoxamine, fluvoxamine controlled release (CR), paroxetine, paroxetine CR, sertraline and venlafaxine extended release (XR). These reviews comprised 59 RCTs: 11 for GAD, 11 for SAD, 13 for OCD, 7 for PTSD and 17 for panic disorder (see online Table DS1). However, we excluded 2 trials for SAD and 1 trial for panic disorder (Table DS1) because baseline severity scores were not comparable with the other trials. Specifically, the excluded SAD trials did not use the LSAS and the excluded panic disorder trial that failed to distinguish between full and limited-symptom panic attacks. Consequently, 56 trials were included in this study. We used only data concerning doses recommended by the FDA; therefore, for paroxetine we excluded the 20 mg dose in one OCD trial and the 10 mg and 20 mg doses in one panic disorder trial, as only doses of 40 mg or higher were judged to be effective for these disorders.
All included trials were short-term, randomised, double-blind and placebo controlled. Some trials also used active comparators, but data from these were not included in our primary analyses. Trial duration was 12 weeks for all SAD and PTSD trials, 8–10 weeks for GAD trials, 8–16 weeks for OCD trials and 10–12 weeks for panic disorder trials. Trials included adults of 18 years or older (with the exception of 5 OCD trials that included adolescents together with adults) who met DSM-III-R or DSM-IV criteria for the anxiety disorder under investigation. The majority of trials (71%) used a flexible dose design, allowing investigators to titrate the dose according to the patient's response; the remainder (29%) used a fixed-dose design with patients randomised to one of two or three different dosage groups.
Participants and baseline severity
The 56 trials included 55 placebo groups and 78 drug groups, which were pooled into 56 drug groups. The total number of participants was 14 710, of whom 6386 were randomised to receive placebo and 8324 were randomised to the drug groups. Baseline severity was generally in the moderate to severe range. Participant numbers and baseline severity scores are shown in online Table DS2. A complete list of included trials, with baseline and change scores, is provided in online Table DS3.
Meta-analysis
We performed a meta-analysis of effect sizes, stratified by disorder and group (Table 1). The placebo group effect size was smallest for OCD (0.49), followed by SAD (0.65). These placebo effects were 59% and 60% respectively of the effects found for the corresponding drug groups. In contrast, for GAD and PTSD the placebo effect sizes were 1.03 and 0.97 respectively; these effects were 76% and 83% respectively of the effects found for the corresponding drug groups. For panic disorder the placebo remission rate was 45%, which was 76% of the rate found in the drug group. Substantial heterogeneity was present for nearly all groups, with I 2 ranging from 46% for the placebo groups in SAD trials to 89% for the drug groups in PTSD trials, although confidence intervals were generally wide.
Table 1 Meta-analysis of effect sizes
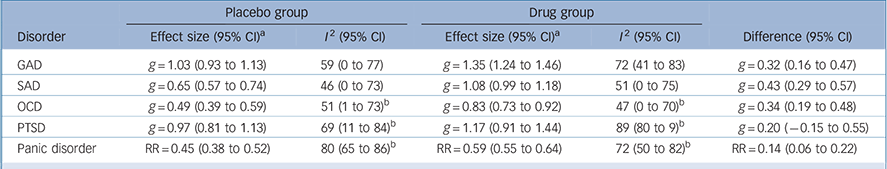
| Placebo group | Drug group | ||||
|---|---|---|---|---|---|
| Disorder | Effect size (95% CI) a | I 2 (95% CI) | Effect size (95% CI) a | I 2 (95% CI) | Difference (95% CI) |
| GAD | g = 1.03 (0.93 to 1.13) | 59 (0 to 77) | g = 1.35 (1.24 to 1.46) | 72 (41 to 83) | g = 0.32 (0.16 to 0.47) |
| SAD | g = 0.65 (0.57 to 0.74) | 46 (0 to 73) | g = 1.08 (0.99 to 1.18) | 51 (0 to 75) | g = 0.43 (0.29 to 0.57) |
| OCD | g = 0.49 (0.39 to 0.59) | 51 (1 to 73) b | g = 0.83 (0.73 to 0.92) | 47 (0 to 70) b | g = 0.34 (0.19 to 0.48) |
| PTSD | g = 0.97 (0.81 to 1.13) | 69 (11 to 84) b | g = 1.17 (0.91 to 1.44) | 89 (80 to 9) b | g = 0.20 (−0.15 to 0.55) |
| Panic disorder | RR = 0.45 (0.38 to 0.52) | 80 (65 to 86) b | RR = 0.59 (0.55 to 0.64) | 72 (50 to 82) b | RR = 0.14 (0.06 to 0.22) |
GAD, generalised anxiety disorder; OCD, obsessive-compulsive disorder; PTSD, post-traumatic stress disorder; RR, remission rate; SAD, social anxiety disorder.
a. Effect sizes (inverse variance-weighted) are expressed as Hedges' g or remission rate (for panic disorder only).
b. Derived as mean estimates over ten imputed data-sets.
Meta-regression
For both GAD and SAD neither baseline severity nor the interaction between group and baseline severity was a statistically significant predictor of the effect size, although group membership was (P = 0.001 and P<0.001 respectively) (Table 2 and Fig. 1). For OCD the interaction between baseline severity and group membership was not significant, but the main effects of both group membership and baseline severity were (group P<0.001; baseline P = 0.001), indicating that the (positive) slope of the association between baseline severity and effect size was similar in the placebo and drug groups. For PTSD none of the predictors achieved statistical significance. For panic disorder we modelled the relationship between baseline severity and the remission rate; in this model the interaction between group membership and baseline severity was not significant (Table 2 and Fig. 2). Group membership was a significant predictor of the remission rate (P<0.01) but baseline severity was not. Paralleling these results, inclusion of treatment group in the model reduced between-group heterogeneity for GAD (I 2 decreasing from 83% to 67%), SAD (84% to 49%), OCD (75% to 52%) and panic disorder (86% to 77%), although it did not reduce heterogeneity for PTSD (85% to 84%). Including the main effect of baseline reduced heterogeneity for OCD only (52% to 14%), whereas including the interaction did not reduce heterogeneity further for any disorder.

Fig. 1 Meta-regression analysis for (a) generalised anxiety disorder, (b) social anxiety disorder, (c) obsessive–compulsive disorder and (d) post-traumatic stress disorder. Data points are sized in proportion to the inverse of their standard error.
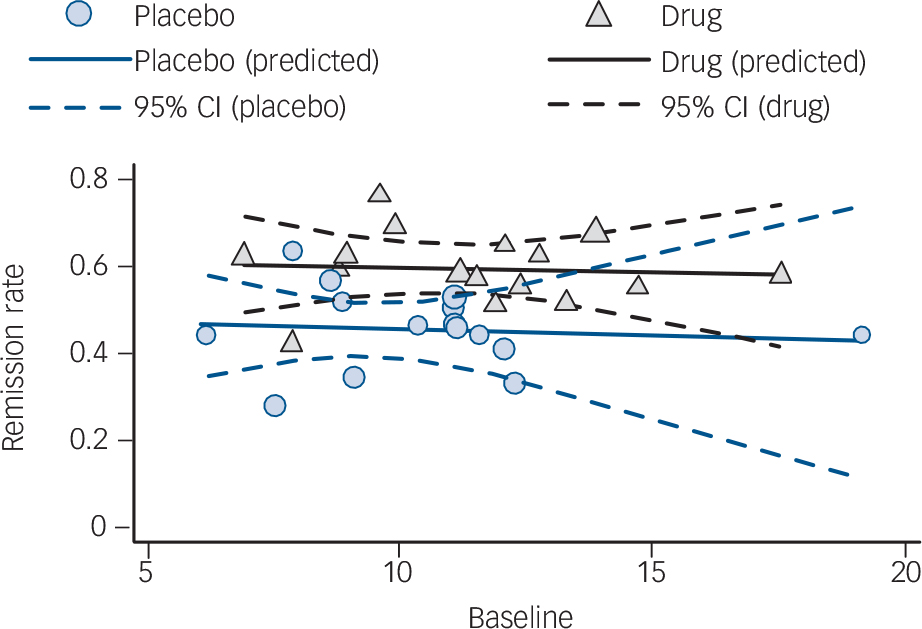
Fig. 2 Meta-regression analysis for panic disorder. Data points are sized in proportion to the inverse of their standard error.
Table 2 Meta-regression analysis of Hedges' g or remission rate (panic disorder only)
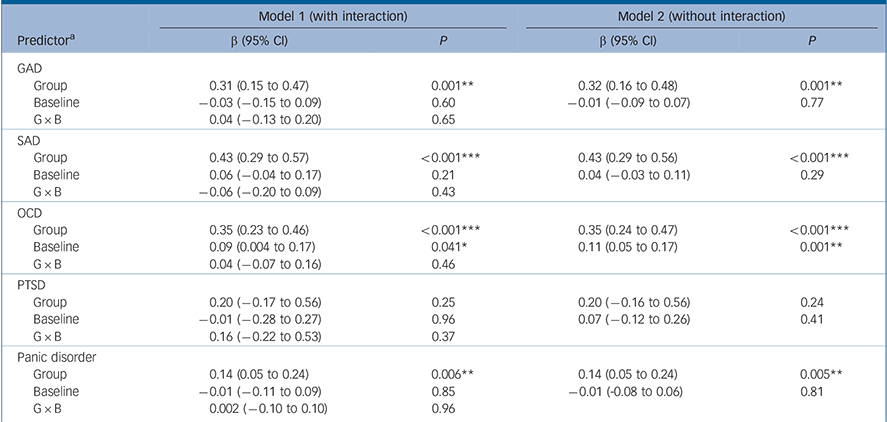
| Model 1 (with interaction) | Model 2 (without interaction) | |||
|---|---|---|---|---|
| Predictor a | β (95% CI) | P | β (95% CI) | P |
| GAD | ||||
| Group | 0.31 (0.15 to 0.47) | 0.001** | 0.32 (0.16 to 0.48) | 0.001** |
| Baseline | −0.03 (−0.15 to 0.09) | 0.60 | −0.01 (−0.09 to 0.07) | 0.77 |
| G × B | 0.04 (−0.13 to 0.20) | 0.65 | ||
| SAD | ||||
| Group | 0.43 (0.29 to 0.57) | <0.001*** | 0.43 (0.29 to 0.56) | <0.001*** |
| Baseline | 0.06 (−0.04 to 0.17) | 0.21 | 0.04 (−0.03 to 0.11) | 0.29 |
| G × B | −0.06 (−0.20 to 0.09) | 0.43 | ||
| OCD | ||||
| Group | 0.35 (0.23 to 0.46) | <0.001*** | 0.35 (0.24 to 0.47) | <0.001*** |
| Baseline | 0.09 (0.004 to 0.17) | 0.041* | 0.11 (0.05 to 0.17) | 0.001** |
| G × B | 0.04 (−0.07 to 0.16) | 0.46 | ||
| PTSD | ||||
| Group | 0.20 (−0.17 to 0.56) | 0.25 | 0.20 (−0.16 to 0.56) | 0.24 |
| Baseline | −0.01 (−0.28 to 0.27) | 0.96 | 0.07 (−0.12 to 0.26) | 0.41 |
| G × B | 0.16 (−0.22 to 0.53) | 0.37 | ||
| Panic disorder | ||||
| Group | 0.14 (0.05 to 0.24) | 0.006** | 0.14 (0.05 to 0.24) | 0.005** |
| Baseline | −0.01 (−0.11 to 0.09) | 0.85 | −0.01 (−0.08 to 0.06) | 0.81 |
| G × B | 0.002 (−0.10 to 0.10) | 0.96 | ||
GAD, generalised anxiety disorder; OCD, obsessive-compulsive disorder; PTSD, post-traumatic stress disorder; SAD, social anxiety disorder.
a. Predictors are treatment group, baseline severity and their interaction (G × B).
* P<0.05,
** P<0.01,
*** P<0.001.
Sensitivity analyses
In our secondary analysis baseline severity was not a significant predictor of the drug–placebo difference for any disorder (see online Table DS4). The expanded set of trials included 9 additional trials for GAD, 10 additional trials for SAD, 6 additional trials for OCD, 6 additional trials for PTSD and 6 additional trials for panic disorder (see online data supplement for references). The range of baseline severity was increased slightly to substantially for all disorders in this expanded set of trials. However, no statistically significant interaction effect was found for any disorder, although the main effect of baseline became statistically significant for GAD, PTSD and SAD, in addition to OCD (see online Table DS5).
Discussion
We found no evidence that baseline severity of disorder affected the efficacy of second-generation antidepressants in the short-term treatment of anxiety disorders. This finding stands in remarkable contrast to the reports of studies investigating major depressive disorder. For OCD, baseline severity did predict change in score in both placebo and drug groups but no differential effect was apparent. This suggests that patients with more severe OCD may show substantially greater improvement with antidepressant treatment than patients with milder OCD, but this is not due to improved antidepressant efficacy in severe disorder. Our sensitivity analysis including 37 additional trials suggested that a similar regression to the mean effect might also occur in other anxiety disorders, but confirmed the lack of evidence for an interaction effect in all anxiety disorders.
Effect sizes for the drug–placebo difference were unaffected by inclusion of baseline severity as a main effect or in interaction with group, but they were generally smaller than a criterion for clinical significance previously used (although without clear justification) for major depression (d = 0.5). Reference Kirsch, Deacon, Huedo-Medina, Scoboria, Moore and Johnson12 However, it has been shown that effect sizes exceeding 0.5 are not achieved by most current treatments, either in psychiatry or in general medicine. Reference Leucht, Hierl, Kissling, Dold and Davis30 Furthermore, clinical significance is both context- and disorder-specific. Reference Kraemer and Kupfer31 An empirically derived criterion of d = 0.24 has been proposed as a more meaningful threshold for clinically significant efficacy in major depression. Reference Cuijpers, Turner, Koole, van Dijke and Smit32 Although not all included anxiety disorders met that threshold in our primary analysis, the drug–placebo difference may have been underestimated slightly owing to our analytical approach. We have previously analysed the trials included in this study by conventional meta-analytic methods and found effect sizes of 0.27 and greater. Reference Roest, de Jonge, Williams, de Vries, Schoevers and Turner23 Further research is required to establish whether antidepressant efficacy in the different anxiety disorders may be considered clinically significant in all or in subsets of patients. Additionally, the expected efficacy of medication is only one of many factors that play a part in the decision to prescribe antidepressants for an individual patient. Other issues – such as the expected burden of side-effects, the burden posed by the disorder itself, the anticipated course of the disorder, and the availability, acceptability and efficacy of alternative treatment options such as psychotherapy – must also be considered. 33 These considerations might lead to different prescribing decisions for patients with mild v. severe anxiety disorders, even in the absence of differential efficacy.
Comparison with depression
For major depressive disorder it has been suggested that the threshold for ‘antidepressant-treatable’ depression should be higher, Reference Baumeister34 given that patients with mild depression do not show a strong differential response to antidepressants over placebo. It is possible that this threshold needs to be even higher in anxiety disorders, and that we would find greater efficacy of antidepressants in patients with more severe anxiety than we do in patients with less extreme anxiety. Another possible explanation lies in the difference in chronicity between anxiety disorders and major depression. Depressive episodes tend to be relatively short, with a median duration of 3–6 months, Reference Penninx, Nolen, Lamers, Zitman, Smit and Spinhoven3,Reference Spijker, de Graaf, Bijl, Beekman, Ormel and Nolen35 whereas the median duration of an anxiety episode has been estimated at 16 months. Reference Penninx, Nolen, Lamers, Zitman, Smit and Spinhoven3 However, duration of a depressive episode is positively correlated with severity. Reference Penninx, Nolen, Lamers, Zitman, Smit and Spinhoven3 This suggests that (all other things being equal) people with mild depression are more likely to achieve spontaneous remission within the short time frame of a clinical trial than people with severe depression. Since a drug effect cannot be demonstrated in spontaneous remission, efficacy would be expected to be reduced in a patient group where this is likely to occur. Consequently, the correlation between severity and episode duration may explain the increase in antidepressant efficacy with increasing depression severity. Although severity also correlates with episode duration in anxiety disorders, Reference Hendriks, Spijker, Licht, Beekman and Penninx36 this may play less of a part within the context of a short clinical trial. Given the long median duration of an episode of anxiety, spontaneous remission rates would be expected to be relatively low across the severity range.
We may also have been unable to detect a relationship between baseline severity and antidepressant efficacy owing to a restriction in range. As trials generally have a minimum severity requirement, as well as exclusion criteria that tend to exclude severely ill patients (e.g. regarding treatments received and comorbidity), baseline severity was restricted to the moderate to very severe range in our primary analyses, although the range was extended somewhat in our sensitivity analyses. However, previous studies in major depression which were able to detect a relationship between baseline severity and antidepressant efficacy had similarly restricted severity ranges. We may also have had insufficient power to find a statistically significant interaction: although the total number of included trials was large, the number of trials per disorder was limited to 7–16. However, the results do not suggest a meaningful trend towards increasing efficacy with increasing severity, except possibly in PTSD, for which we had the fewest available trials. The estimates of the interaction effect were substantially lower (⩽0.04, except for PTSD) or even in the opposite direction as the estimate previously reported for depression. Reference Kirsch, Deacon, Huedo-Medina, Scoboria, Moore and Johnson12 These estimates are small enough to be of limited clinical relevance, although some confidence intervals encompass values that might be considered clinically relevant. Furthermore, our secondary analyses, which included six to ten additional trials per disorder, also showed no evidence of increasing efficacy with increasing severity, even for PTSD. Larger samples would be required to conclusively exclude the possibility of even small interaction effects, but unfortunately the number of randomised trials that have been conducted is limited, and consequently such samples are not available.
Strengths and limitations
Among the strengths of the study is the fact that we were able to obtain a data-set that was free from the influence of publication or outcome reporting bias. An additional strength is the high quality of the included trials. As these trials were conducted for the purpose of obtaining marketing approval, they were required to meet strict standards on internal validity (masking, randomisation, etc.). This also ensured that trials conducted for the treatment of the same disorder were in general comparable. A limitation is that we were forced to use a different outcome measure for panic disorder (remission) from that used for the other disorders; consequently we cannot rule out the possibility that an interaction might have been found if we had used a continuous outcome (such as change) instead. An additional, important limitation is that we did not have access to individual patient data and hence used summary data instead. Use of summary data necessarily causes a loss of information, due to averaging out inter-individual variation within trials, and a concomitant loss of power. Although individual patient data have historically been difficult to obtain, novel initiatives such as the Yale University Open Data Access Project (http://yoda.yale.edu) and Clinical Study Data Request (https://clinicalstudydatarequest.com) are now beginning to increase the accessibility of such data to researchers. Given our current results, which suggest that the influence of baseline severity on antidepressant efficacy may be different for anxiety disorders compared with depression, future research making use of individual patient data is essential to provide a definitive answer to this important question.
Clinical implications
It has been recommended that treatment with antidepressants should be withheld for mild depression. Reference Baumeister34,37 The absence of an interaction between treatment group and baseline severity in predicting symptom change in our study shows that this advice cannot be simply extrapolated to anxiety disorders, and it would therefore be premature to recommend that antidepressants be withheld for mild anxiety. What defines a clinically relevant effect size remains a matter of debate, but if the effect of antidepressants on anxiety is considered clinically relevant, these results suggest that antidepressants may be prescribed to patients with anxiety regardless of symptom severity.
Funding
The study was supported by grant KS2011(1)-120 from the Dutch Brain Foundation to P.J. The funder had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.






eLetters
No eLetters have been published for this article.