



Introduction
A universally acknowledged truth is that all biological organisms age with time. Maintaining seed quality despite the passage of time is the basis for the preservation of plant genetic resource as well as the dynamics within soil seed banks. Seeds can survive for years, and there are examples of extreme longevity (Steiner and Ruckenbauer, Reference Steiner and Ruckenbauer1995; Daws et al., Reference Daws, Davies, Vaes, van Gelder and Pritchard2007; Sallon et al., Reference Sallon, Solowey, Cohen, Korchinsky, Egli, Woodhatch, Simchoni and Kislev2008). However, all seeds eventually age, lose quality, and die. The decrease of seed quality during ageing, from seed vigour to DNA stability, is a significant impediment to efficient seed storage. However, changes in seed quality can be difficult to detect using standard germination tests during the initial phase of the ageing process or in seeds that are dormant. For example, some species show highly irregular germination during storage due to endogenous dormancy cycles (Froud-Williams et al., Reference Froud-Williams, Hilton and Dixon1986; Rawat and Thapliyal, Reference Rawat and Thapliyal2003). In those cases, germination assays under-represent the number of viable seeds and might also impose selection on the germplasm when it is regenerated (Pérez-García et al., Reference Pérez-García, González-Benito and Gómez-Campo2007; Mira et al., Reference Mira, González-Benito, Ibars and Estrelles2011b; van Hintum and van Treuren, Reference van Hintum and van Treuren2012). Thus, the evaluation of seed quality during storage would benefit from additional tests.
Though critical to ecological processes and conservation protocols, research to understand ageing mechanisms in seeds is limited. Seed deterioration may occur through different mechanisms depending on species and storage conditions, for example, low and high humidity, due to differences in molecular mobility within the cytoplasm (Walters, Reference Walters1998, Reference Walters2015; Ballesteros and Walters, Reference Ballesteros and Walters2019). Accumulation of reactive oxygen species and lipid peroxidation isgenerally considered as major contributors to seed deterioration (Merritt et al., Reference Merritt, Senaratna, Touchell, Dixon and Sivasithamparam2003; Bailly, Reference Bailly2004; Mira et al., Reference Mira, Estrelles, González-Benito and Corbineau2011a). Under humid conditions, such as in natural soil seed banks or seeds stored short term for agriculture purposes, ageing is also associated with controlled biochemical activities, such as programmed cell death (Kranner et al., Reference Kranner, Chen, Pritchard, Pearce and Birtić2011). Ageing processes result in changes in the structural and functional properties of membrane lipids, key proteins, and nucleic acids (McDonald, Reference McDonald1999; Kurek et al., Reference Kurek, Plitta-Michalak and Ratajczak2019).
One significant factor determining seed longevity is the level of DNA stability in the embryo, marked by DNA damage during ageing and repair response upon imbibition (Waterworth et al., Reference Waterworth, Bray and West2015). Structural damage, such as single and double strand breaks of DNA, or point mutations accumulate in ageing seeds (Navashin, Reference Navashin1933; Roberts, Reference Roberts1978; McDonald, Reference McDonald1999; Tuteja et al., Reference Tuteja, Singh, Misra, Bhalla and Tuteja2001). DNA fragmentation has been correlated with lower seed germination (Osborne, Reference Osborne2000; Kranner et al., Reference Kranner, Birtić, Anderson and Pritchard2006; Donà et al., Reference Donà, Balestrazzi, Mondoni, Rossi, Ventura, Buttafava, Macovei, Sabatini, Valassi and Carbonera2013). While ageing processes occur, seeds might still be able to germinate. To do so, DNA repair mechanisms are activated in the embryo during early germination steps (Balestrazzi et al., Reference Balestrazzi, Confalonieri, Macovei, Donà and Carbonera2011). It is unclear whether DNA alterations are retained within the seedling or subsequent plant. If there are lasting effects, our ability to provide germplasm with quality-assured genetic composition will be impacted.
During seed ageing, DNA alterations could compromise epigenetic stability. Cytosine DNA methylation (mC) is an epigenetic modification that is important for regulating gene expression in higher plants and other organisms (Gehring and Henikoff, Reference Gehring and Henikoff2007) and the molecular mechanism controlling gene expression by methylation–demethylation is shared among animal and plants systems (Law and Jacobsen, Reference Law and Jacobsen2010). Methylation of cytosine in vertebrates occurs only in CG dinucleotides. In plants, methylation sites are predominantly at CG dinucleotides but can also occur in any sequence context (Vanyushin and Ashapkin, Reference Vanyushin and Ashapkin2011). Cytosine methylation in DNA plays a key role in the regulation of gene expression, allowing cells to adapt to new conditions without affecting the DNA sequence, controlling growth, development, and responses to environmental stresses in plants (Qiao and Fan, Reference Qiao and Fan2011) and animals (Bird, Reference Bird1993). Epigenetic mechanisms involved in longevity and ageing in animals have been studied in multiple systems (Sen et al., Reference Sen, Shah, Nativio and Berger2016). A comprehensive genome-wide study in human beings has shown that changes in the methylation level of cells highly correlate with chronological age, detecting a decrease in transcription activity with age due to hypermethylation in promotor regions, and hypomethylation in gene bodies (Åsa et al., Reference Åsa, Enroth and Gyllensten2013). In plants, DNA methylation changes between juvenile and mature plant stages, suggesting that the methylation status of genomic DNA can vary as plants mature (Yuan et al., Reference Yuan, Sun, Guo, Yue and Gu2014; Ogneva et al., Reference Ogneva, Dubrovina and Kiselev2016). The relationship between DNA integrity and epigenetic changes during seed ageing and, perhaps more importantly, upon subsequent plant generation is poorly understood.
The major aim of this study was to investigate the overall nucleic acid profile during seed ageing and the ultimate goal of the work is to identify reliable markers of seed deterioration. DNA profiles visualized by RAPDs and MSAPs analysis were compared between non-aged and aged seeds of Mentha aquatica and plants produced by germinating seeds from these seed lots. M. aquatica seeds are an interesting study system because it is difficult to evaluate longevity during storage because seeds display endogenous dormancy cycles that can mask viability results obtained by a standard germination test. Dormancy can be broken using gibberellic acid to prevent incorrect classifying of dormant and dead seeds, allowing a clear correlation between viability loss and nucleic acid stability. We hypothesized that ageing affects genetic stability and global methylation of genomic DNA. The work contributes to understanding the complex network of molecular events associated with seed ageing, which will benefit seed conservation efforts through improved detection of seed quality changes.
Materials and methods
Seed storage experiment
M. aquatica L. produces four-sectioned schizocarp fruits with one-seeded nutlets that are its dispersion and germination units (Morales, Reference Morales, Castroviejo, Aedo, Cirujano, Laínz, Montserrat, Morales, Muñoz, Navarro, Paiva and Soriano2007). Lamiaceae nutlets possess mainly a schlerenchimatic pericarp, little endosperm, and a fully developed embryo, so embryo tissue proportion is bigger than both pericarp and endosperm (Martín Mosquero et al., Reference Martín Mosquero, Pastor Díaz and Juan Rodríguez2003; Bonzani et al., Reference Bonzani, Bravi and Barboza2011). Experiments were carried out with nutlets (hereafter called ‘seeds’) received from Semillas Cantueso S.L. (Córdoba, Spain). Seeds were stored at laboratory conditions (20°C, 35% RH in darkness) until use.
For the ageing treatment, seeds were placed over a saturated solution of NaCl (75% RH) at 25°C for 3 days (Vertucci and Roos, Reference Vertucci and Roos1993) and then hermetically sealed in aluminium foil bags (S-156, Valsem Industries SAS, Lachelle, France) with 125 μm thickness of polyester, aluminium, and polyethylene layers. Seeds were then stored for 28 days at 35°C. The storage experiment was replicated in two aluminium bags. Water content (wc) was calculated by the low-constant-temperature-oven method (ISTA, 2017) using two replicates. Water contents were expressed as percentage of g H2O/g dry weight.
Germination assays of non-aged and aged seeds were performed by placing eight replicates of 25 seeds in 9 cm Petri dishes on top of two discs of filter paper moistened with distilled water. Distilled water was added to the filter paper regularly when required. Incubation conditions were 25°C with a 16-h photoperiod provided by cool white fluorescent tubes with an irradiance of 35 μmol m2/ s. The emergence of the radicle was the criterion for germination. Radicle length following 21 days of incubation and the number of days required to reach 50% of final germination (MGT) were calculated as vigour parameters. A solution of gibberellic acid (1000 mg/l-) was added to the non-germinated seeds to stimulate germination of dormant seeds. Differences in total germination or vigour before and after storage were confirmed by a Chi-square test or t-test, respectively.
DNA extraction and analysis
Two replicates for DNA extractions were done for each treatment (non-aged and aged) of seeds and seedlings. Each seed DNA replicate was isolated from 30 mg (550 seeds per sample). The two replicates from aged seeds were taken from the two aluminium bags stored (one replicate per bag). Each seedling DNA replicate was isolated from 20 mg (50 seedlings per sample). Seedlings were bulked among the eight replicates of germination Petri dishes per treatment (non-aged and aged seeds) after 21 days of incubation. Plants with a similar developmental stage were used for DNA extraction.
Samples were pounded in liquid nitrogen and DNA was extracted using the DNeasy Plant Mini Kit (Qiagen®, Netherlands), according to the manufacturer's instructions.
Fungal contamination can be a problem when working with seeds and seedlings, especially after high humidity storage. To discard samples that show fungal contamination on DNA extracts, we tested for PCR amplification products of fungus-specific DNA regions using the internal transcribed spacer (ITS) region of the ribosomal DNA repeat cluster, as well as markers for the specific detection of three fungi genera commonly found in seeds: Fusarium, Penicillium, and Aspergillus (Gálvez et al., Reference Gálvez, Gil-Serna, García, Iglesias and Palmero2016; Gil-Serna et al., Reference Gil-Serna, Gálvez, París and Palmero2016). The universal primer pair ITS1-ITS4 was tested (5′-TCCGTAGGTGAACCTGCGG-3′ for ITS1 and 5′TCCTCCGCTTATTGATATGC-3′) for ITS4 (White et al., Reference White, Burns, Lee, Taylor, Innis, Gelgard, Sninsky and White1990) and other markers specific included an elongation factor specific for Fusarium (5′-ATGGGTAAGGAGGACAAGAC-3′/5′-GGAAGTACCAGTGATCATGTT-3′) (O'Donnell et al., Reference O'Donnell, Cigelnik and Casper1998) and betatubuline-specific for Penicillium and Aspergillus (5′-GGTAACCAAATCGGTGCTGCTTTC-3′/5′-ACCCTCAGTGTAGTGACCCTTGGC-3′ respectively) (Glass and Donaldson, Reference Glass and Donaldson1995).
DNA integrity analysis
Random amplified polymorphic DNA (RAPD) analysis was carried out to assess DNA stability (Callow et al., Reference Callow, Frod-Lloyd and Newbury1997). A total of 16 random 10-base primers (sequences from Operon Technology, Spain) were used to screen variation in the amplicon product between non-aged and aged seeds and seedlings produced after 21 days of incubation (sequences are listed in Table 1).
Table 1. Primers and number of monomorphic and polymorphic PCR amplification products obtained from RAPD analysis of non-aged and aged M. aquatica seeds and plants produced by them
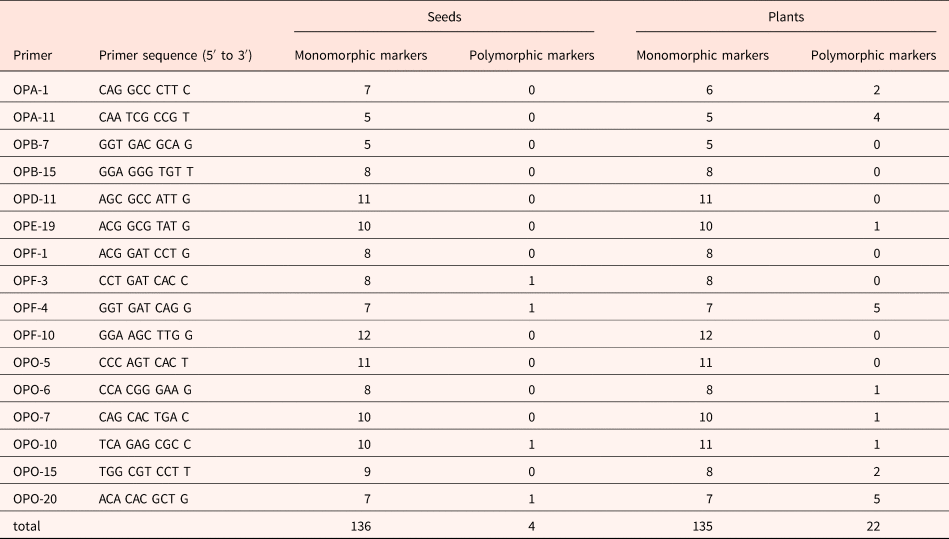
DNA amplification reactions were performed in 25 μl tubes containing approximately 10 ng total DNA, 0.8 μM of a single decanucleotide, 5 μl of PCR buffer, and 1.25 U MyTaq DNA polymerase (Bioline, UK). The PCR amplifications were performed in a SimpliAmpl Thermal Cycler (Thermo Fischer Scientific, Waltham, Massachusetts, USA) using one cycle of 1 min at 95°C, followed by 35 cycles of 45 s at 92°C, 1 min at 37°C and 2 min at 72°C, and a final cycle of 10 min at 72°C. RAPD products were electrophoresed in 1.5% (w/v) agarose gels in 1× TBE buffer at 100 V for 2 h, stained with ethidium bromide (0.5 μg/ml) for 7–10 min and photographed under UV light. The size of the amplified bands was related by reference to the molecular size marker (100 Base- Pair Ladder, GE Healthcare, Chicago, Illinois, USA). All the reactions were repeated at least twice to monitor the reproducibility of banding patterns, and the polymorphism presented were repeatable.
The reproducible RAPD bands were scored in binary characters and a similarity matrix was constructed using the Jaccard coefficient, which was further subjected to clustering unweighted pair group method analysis (UPGMA) to eventually generate a dendrogram. The analysis was performed using the NTSYS-PC software package version 2.
Epigenetic stability
To study epigenetic stability during storage, we used a methylation-sensitive amplified polymorphism (MSAP) technique, which is a modification of the amplified fragment length polymorphism (AFLP) method that uses methylation-sensitive restriction endonucleases followed by an amplification of digested fragments. We selected the MSAP protocol involving methylation-sensitive isoschizomers HpaII and MspI (Reyna-Lopez et al., Reference Reyna-Lopez, Simpson and Ruiz-Herrera1997). The adapter and primer for the enzyme EcoRI were the same as that used in standard AFLP analysis, while the HpaII/MspI adapter was designed according to Xiong et al. (Reference Xiong, Xu, Maroof and Zhang1999) who modified it for the use in plant species.
To identify epigenetic markers, two sets of restriction/ligation reactions were simultaneously carried out. To perform the digestion, approximately 500 ng of the extracted DNA was digested with 10 U of EcoRI and 10 U of MspI or 6 U of HpaII, in a final volume of 35 μl containing buffer R-L (10×). The mixture was incubated at 37°C for 3 h and the reaction stopped by incubation at 65°C for 5 min and then the samples were cooled for 30–45 min at room temperature. The ligation mixture was carried out by adding to the digested DNA 5 μl of ligation mix (5 pmol EcoRI adaptor, 50 pmol MspI/HpaII adaptor, 10 mM ATP, 1.2 U T4 DNA ligase (Boehringer, Germany), and 10× buffer R-L). The ligation was incubated for 3 h at 37°C and overnight at 4°C. After the incubation, the product of this double reaction was diluted 1:4 to be used as a template for the first amplification reaction.
The pre-selective PCR reaction was performed in 20 μl containing 30 ng of each primer EcoRI + A and HpaII/Msp I + A, 0.2 mM of each dNTP, 1.5 mM MgCl2, 0.4 U of Taq DNA polymerase (Biotaq, Bioline), and 3 μl of diluted fragments in the 1× PCR buffer provided by the manufacturer (Bioline).
The pre-selective amplification was performed with the following profile: 28 cycles of 30 s at 94°C, 1 min at 60°C, and 1 min at 72°C. Preamplified fragments were diluted tenfold to be used as the starting material for selective amplification. In this amplification, only the EcoRI primer was labelled with FAM fluorochrome; both the EcoRI and the HpaII/MspI primers contain the same sequences as those used in the preamplification but with three selective nucleotides at the 3′ end for HpaII/MspI primers and two nucleotides for EcoRI primers.
Selective amplification was carried out using a total of five primer combinations obtained with two EcoRI primers in combination with three HpaII/MspI primers (EcoRI + AC–HpaII/MspI + ATC; AC/ACT; AA/AAT; AA/ATC; AA/ACT). The PCR reaction was performed in a volume of 20 μl containing 6 ng of labelled EcoRI primer, 30 ng of HpaII/MspI primer, 0.1 mM of each dNTP, 1.5 mM MgCl2, 0.4 U of Taq DNA polymerase, and 5 μl of diluted preamplified DNA in the 1× PCR buffer provided by the manufacturer (Ecogen, UK). Reaction was performed with the following profile: 1 cycle of 30 s at 94°C, 30 s at 65°C, 1 min at 72°C, followed by 12 cycles in which the annealing temperature decreases 0.7°C per cycle, followed by 23 cycles of 1 min at 94°C, 30 s at 56°C and 1 min at 72°C.
Amplification products were analysed in an automated ABI3730 sequencer by the company SECUGEN S.L. (Madrid, Spain). The resulting electropherograms were analysed using GeneMarker® v1.90 software (SoftGenetics, LLC, PA, USA) and Peak Scanner V1.0 software.
MSAP markers were classified as type 1/1, type 1/0 and type 0/1 accordingly to the presence (1) or absence (0) of the corresponding amplified fragment with HpaII and MspI, respectively. Methylation sensitivity criteria are as follows (Bardini et al., Reference Bardini, Labra, Winfield and Sala2003): HpaII only recognizes sites that are hemi-methylated at the external cytosine (mCCGG) and MspI only recognizes sites being hemi- or fully methylated at the internal cytosine (CmCGG). Sites that are fully methylated at the external cytosine (mCCGG) or hemi- or fully methylated at both internal and external cytosines (mCmCGG) are not cut by either enzyme. However, CCGG-sequences being free of any methylation are digested by both. HpaII/MspI markers were scored as the presence (1) or the absence (0), and a matrix was arranged for further evaluation. Fisher's exact test was performed to verify the statistical significance of the results.
Results
Seed viability and vigour
A 54% decrease in M. aquatica seeds viability after 28 days of storage at 35°C and 12% wc was statistically significant (P < 0.001) (Fig. 1). About 48% and 70% of the non-aged seeds completed germination without and with, respectively, the application of gibberellic acid. The remaining 30% of non-germinated seeds were empty (i.e. no embryo), which is consistent with a previous report for the species (Martín Mosquero et al., Reference Martín Mosquero, Pastor Díaz and Juan Rodríguez2003) and may be related to the high incidence of vegetative multiplication of the species. About 21 and 32% of the aged seeds completed germination without and with, respectively, the application of gibberellic acid.
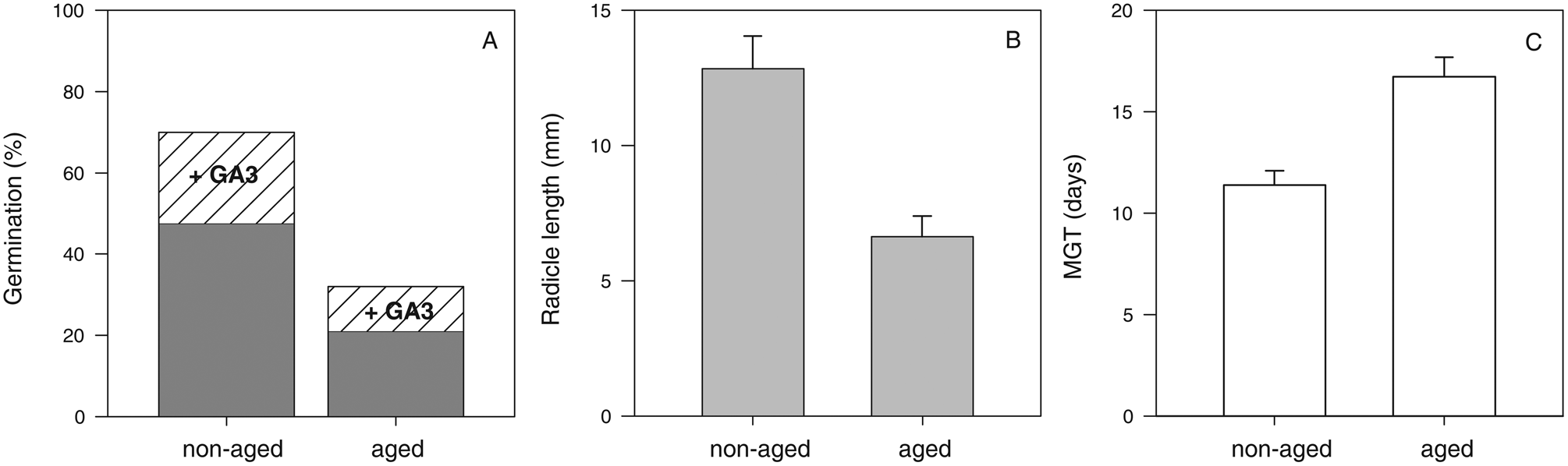
Fig. 1. Seed viability of M. aquatica before (non-aged) and after storage for 28 days at 35 °C and 12% wc (aged). Germination before and after gibberellic acid was added (A) and vigour parameters: radicle length growth (B) and MGT (number of days required to reach 50% of final germination) (C). Data are expressed as mean values ± standard error and were statistically significant (P < 0.001).
Moreover, vigour was also affected by 28 days of storage (P < 0.001) (Fig. 1B, C). A 46% decrease in radicle length was noted after storage. The average radicle length in seedlings obtained from non-aged seeds was 13 ± 1 mm of the radicle, while those obtained from aged seeds was 7 ± 1 mm. Also, germination was faster in non-aged seeds compared to aged seeds, with MGT (time to achieve half of final germination, MGT) occurring in 11 ± 1 and 17 ± 1 days, respectively.
Considering these data, we used 28 days of storage as the time for germination or radicle length to decrease to 50% of maximum, P50.
DNA integrity analysis
DNA extracted from non-aged and aged seeds and resulting seedlings were tested for fungal contamination (Fig. 2). Control samples (material donated by Dr. Palmero) produced a clear amplification product for the ITS1-ITS4 fragment (Fig. 2A), the transcription elongation factor specific for detection of Fusarium (Fig. 2B), and PCR amplification products specific to Penicillium and Aspergillus (Fig. 2C). In contrast, no amplification products corresponding to these primer sequences were observed for M. aquatica seed and seedling DNA extracts, suggesting that fungal contamination does not contribute to observed differences in DNA integrity.
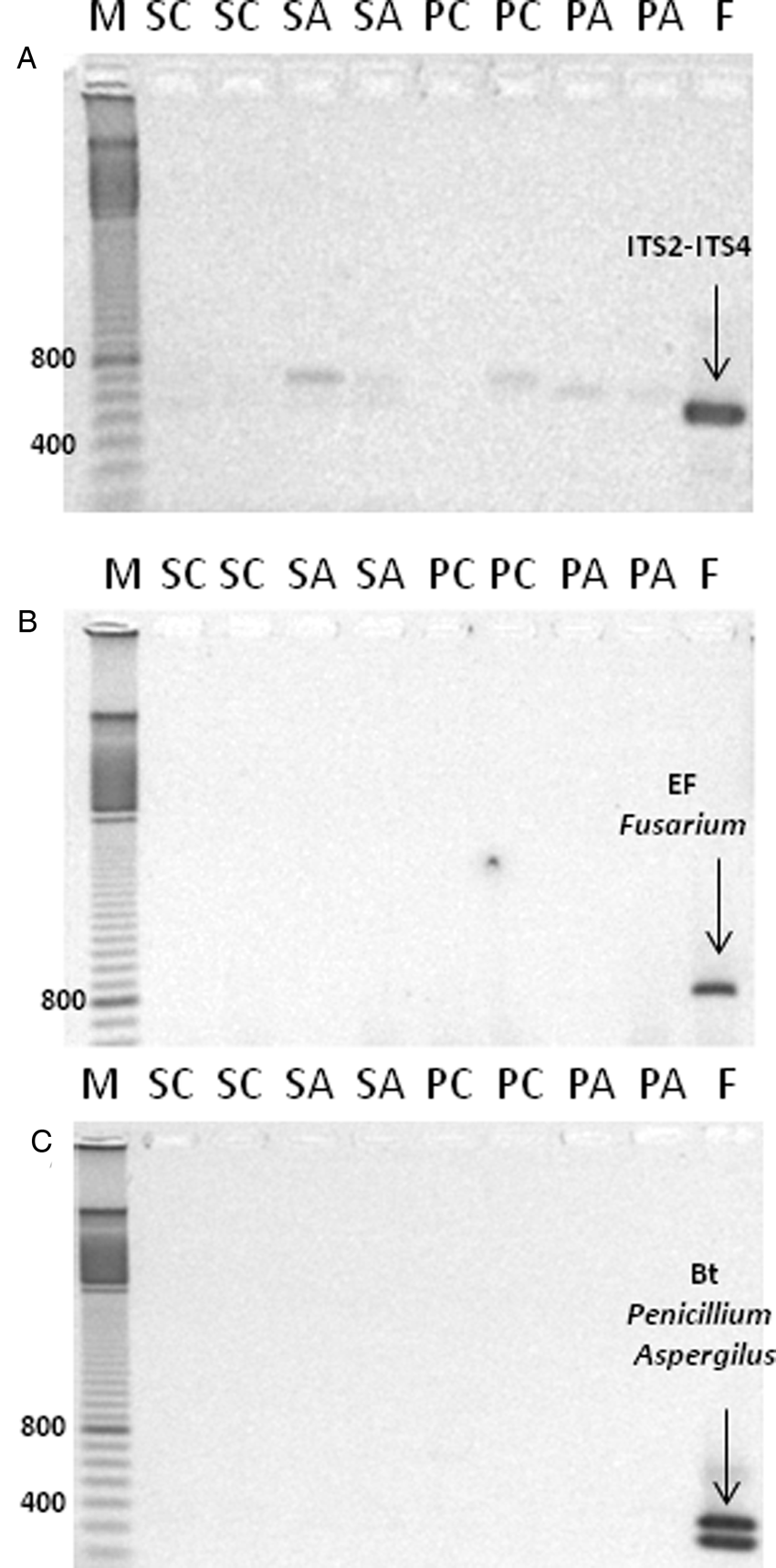
Fig. 2. Evaluation of fungal contamination by the presence of fungus-specific DNA markers in DNA extracted from seeds and plants of M. aquatica. PCR amplification products using the primer combination ITS1-ITS4 (A); elongation factor specific for detection of Fusarium (B); and betatubuline marker specific for detection of Penicillium and Aspergillus (C). Lanes ‘M’: bp marker; ‘SC’: seeds control; ‘SA’: seeds aged; ‘PC’: plants control; ‘PA’: plants aged; ‘F’ fungal DNA used as control.
Specific and reproducible amplification products were obtained using 16 RAPD primers (Table 1), allowing a comparison of DNA profiles from non-aged and aged seeds and resulting seedlings. The size of RAPD-amplified DNA fragments ranged from 200 to 2000 base pairs (bp). Amplification produced between 5 (primer OPB-7) and 12 (primers OPF-4, OPO-10 and OPO-20) bands depending on the sample. A representative RAPD profile, obtained with the primer OPF-4, illustrates differences in the DNA band pattern from non-aged and aged seeds and resulting seedlings (Fig. 3A).
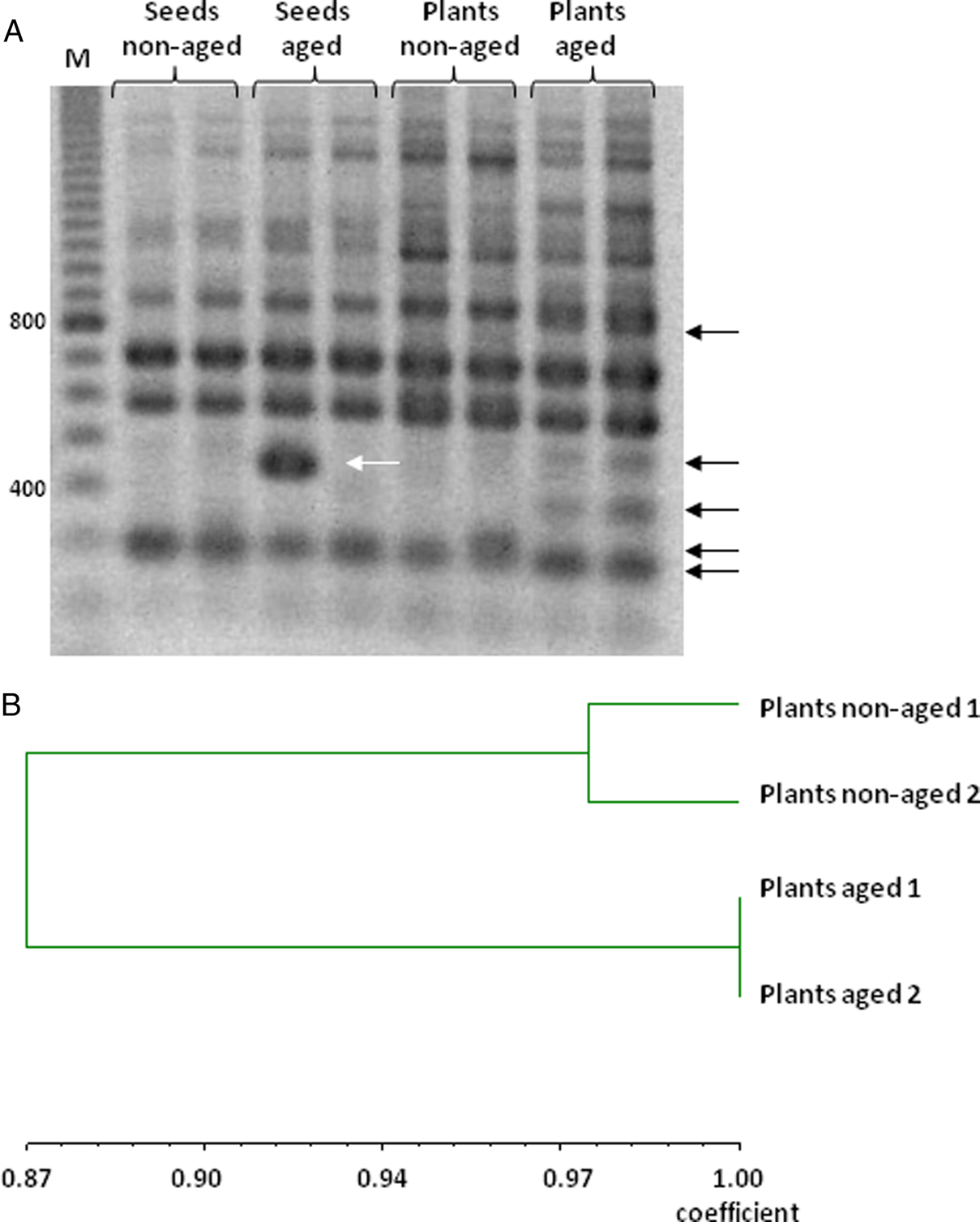
Fig. 3. (A) Representative RAPD profile of amplification products using primer OPF-4 and DNA extracted from non-aged and aged M. aquatica seeds and the plants produced by them. (B) Dendrogram for M. aquatica plants from aged and non-aged seeds generated by the UPGMA method using Jaccard's similarity coefficient using RAPD markers in (A).
In seeds, 140 amplification fragments were detected, 136 of which were monomorphic. Only four primers (OPF-3, OPF-4, OPO-10 and OPO-20) produced PCR products with polymorphisms, resulting in only four polymorphic markers (one per primer). There were few differences detected in DNA extracted from non-aged and aged seeds, and the similarity coefficient was 0.99.
In plants, 157 amplification fragments were analysed, 135 of which were monomorphic. Ageing effects on RAPD profiles were detected by 9 out of the 16 random primers used, which produced 22 polymorphic bands (14% of the total); the other 7 primers resulted in a monomorphic product for all samples. The number of polymorphic bands ranged from 1 (primers OPE-19, OPO-6, OPO-7, and OPO-10) to 5 fragments (primers OPF-4 and OPO-20).
The UPGMA cluster analysis of seedlings revealed differences between non-aged and aged treatments, with a similarity coefficient of 0.87. While the two samples of aged seedlings were identical, the two samples of non-aged seedlings showed some differences, although presented a high percentage of similarity, 97% (Fig. 3B).
Epigenetic stability after ageing
MSAP profiles were generated from non-aged and aged seeds, and seedlings obtained by them. Analysis with the 5 selective-primer combinations revealed a total number of 147 MSAP markers in seeds and 158 in seedlings, with an average of 30 markers per primer set (Table 2).
Table 2. Comparison of detected changes in MSAP in DNA extracted from non-aged and aged M. aquatica seeds and the plants produced by these seeds
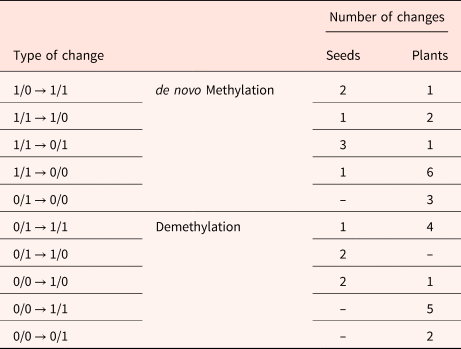
Binary code corresponds to the selective amplifications of the digested DNA with HpaII/MspI.
Aged seed samples showed 92% similarity among MSAP markers, with 135 being identical to those observed in the non-aged seeds. Twelve polymorphic markers differed between aged and non-aged seeds, from which 2 markers showed a variation from type 1/0 to type 1/1, 1 marker showed a variation from type 1/1 to type 1/0, 3 markers from type 1/1 to type 0/1, and 1 marker showed a variation from type 1/1 to type 0/0. All variations indicate a de novo methylation (Bardini et al., Reference Bardini, Labra, Winfield and Sala2003). Likewise, 2 variations from type 0/0 to type 1/0, 1 marker from type 0/1 to type 1/1, and 2 markers from type 0/1 to type 1/0 were considered as demethylation.
Higher variation was detected in seedlings compared to seeds. The number of monomorphic MSAP markers in aged seedlings decreased to 84% (133 markers). Twenty-five polymorphic markers differed between aged and non-aged treatments, from which 1 marker showed a variation from type 1/0 to type 1/1, 2 markers showed a variation from type 1/1 to type 1/0, 1 marker from type 1/1 to type 0/1, 6 markers showed a variation from type 1/1 to type 0/0, and 3 markers showed a variation from type 0/1 to type 0/0. All these changes were also considered as de novo methylation. Furthermore, 1 variation from type 0/0 to type 1/0, 4 markers from type 0/1 to type 1/1, 5 markers from type 0/0 to type 1/1, and 2 from type 0/0 to type 0/1 indicated demethylation.
In summary, percentages of stability and epigenetic changes observed by comparing samples from non-aged and aged M. aquatica seeds are shown in Fig. 4. The ageing treatment resulted in an 8% epigenetic change in seeds, detected by polymorphic markers, while a 16% epigenetic change was indicated by seedlings produced by non-aged and aged seeds (P < 0.01). The number of epigenetic changes corresponding to a de novo methylation or a demethylation was almost identical in both seeds and resulting seedlings.
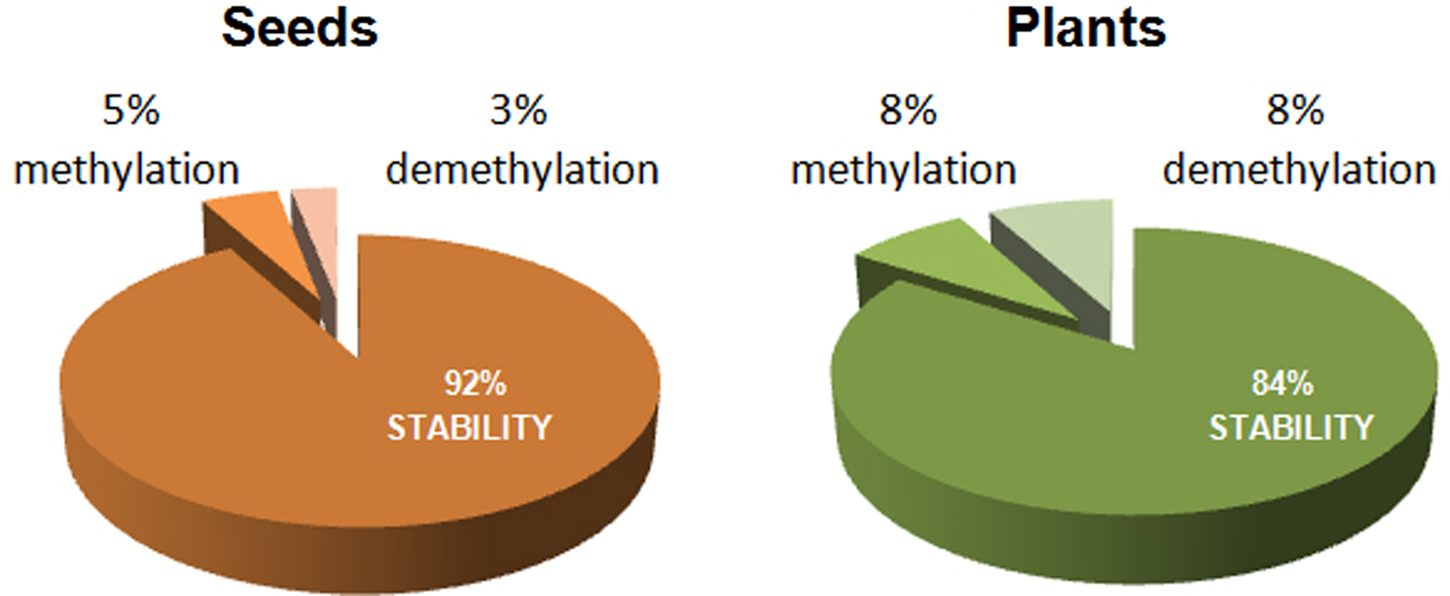
Fig. 4. Percentage of stability and epigenetic changes detected by MSAP markers between non-aged and aged M. aquatica seeds and the plants produced by them.
Discussion
The effect of storage and consequent ageing on DNA stability were studied in M. aquatica seeds and seedlings produced from these seeds. Aged seeds were studied at P50, the time when 50% of germination was lost. A 50% loss of viability is an important parameter to evaluate a seed lot storage performance (Priestley, Reference Priestley1986), and it was a key point for DNA alteration identified by RAPD analysis in sunflower (El-Maarouf-Bouteau et al., Reference El-Maarouf-Bouteau, Mazuy, Corbineau and Bailly2011). In M. aquatica, non-aged and aged seeds at P50 shared 99% similarity in RAPD profiles, but when seedlings were compared the percentage of variation in RAPD and MSAP markers was higher. We hypothesize that alterations of nucleic acids might occur in seeds during ageing, and those changes could not be detected in M. aquatica seeds but were detectable in the seedling produced by them. Alterations produced on the embryo, particularly on their meristematic cells from which the plant will develop, could have been masked in the analysis of a pool of seeds. DNA extracts from pooled seeds are a mix of genotypes, physiological stages (dead, alive and empty seeds) and also a pool of tissues, which may have different susceptibilities to ageing as well as different importance in PCR amplification. Marcos-Filho et al. (Reference Marcos-Filho, McDonald, Tekrony and Zhang1997) found that RAPD profiles were similar between non-aged and aged soybean seeds. On the other hand, working with the same species and technique, Shatters et al. (Reference Shatters, Schweder, West, Abdelghany and Smith1995) found differences between non-aged and aged treatments when analyzing individual seeds, and also detected polymorphic fragments in plants that were not observed in seeds.
Plants produced from aged seeds showed lower vigour, but phenotypes at the first stage of plantlet development were normal. Aberrant phenotypes such as chlorophyll mutants, anomalous leaf shapes, or chlorotic spots, which could indicate major DNA modifications during ageing were not detected. However, DNA variation between plants produced from non-aged or aged seeds was detected through the RAPD analysis. Our results indicate that DNA integrity was compromised during ageing and was not repaired upon imbibition, being detectable by RAPD analysis on the plants produced by aged seeds (13% dissimilarity). Both the disappearance and the appearance of new amplification products on RAPD profiles of aged plants can be due to structural changes in the DNA (extensive rearrangements of DNA) to point mutations (Karp et al., Reference Karp, Seberg and Buiatti1996). Different responses have been reported concerning DNA lesions during seed storage and ageing. At 12% wc, cytoplasm mobility would allow both enzymatic and non-enzymatic mechanisms related to ageing (Walters et al., Reference Walters, Hill and Wheeler2005), such as programmed cell death reactions, nuclease activity, and oxidative degradation of macromolecules by volatile aldehydes or free radicals (Mira et al., Reference Mira, González-Benito, Hill and Walters2010, Reference Mira, Hill, Gonzalez-Benito, Ibanez and Walters2016; Kranner et al., Reference Kranner, Chen, Pritchard, Pearce and Birtić2011; Morscher et al., Reference Morscher, Kranner, Arc, Bailly and Roach2015). Damage to chromatin nitrogen bases, sugars, and histones may lead to structural changes (strand breakage, chromosomal aberrations, rearrangements, small inserts, or deletions) and point mutations (Navashin, Reference Navashin1933; Cheah and Osborne Reference Cheah and Osborne1978; Waterworth et al., Reference Waterworth, Bray and West2015). Upon imbibition during early germination, the DNA repair mechanism will be activated, and while some DNA alterations appear to be reversible, most are considered irreversible (Waterworth et al., Reference Waterworth, Masnavi, Bhardwaj, Jiang, Bray and West2010, Reference Waterworth, Bray and West2015). A simple point mutation within the primer site can generate significant changes in RAPD patterns (Williams et al., Reference Williams, Kubelik, Livak, Rafalski and Tingey1990; Vijay et al., Reference Vijay, Dadlani, Kumar and Panguluri2009). RAPD primers scan the entire genome at random, so it is possible that most of the polymorphisms observed are located in regions that are non-expressed, not highly conserved and more likely to be altered and, therefore, allow seed germination and plant early development processes. According to the data of M. aquatica genome size (Bennett and Smith, Reference Bennett and Smith1991), our RAPD analysis has screened approximately 0.005% of the whole genome; therefore, it may be possible that other DNA sequences were not detected and could have been modified.
Epigenetic regulation, by DNA methylation, of gene expression is known to be involved in plant responses to environmental stresses (Kim et al., Reference Kim, Sasaki, Ueda, Sako and Seki2015). Methylation in seeds affected by storage and ageing has barely been studied. Our data suggest that the DNA methylation profile changed during seed storage, which is consistent with other work using orthodox and recalcitrant seeds (Michalak et al., Reference Michalak, Barciszewska, Barciszewski, Plitta and Chmielarz2013, Reference Michalak, Plitta, Tylkowski, Chmielarz and Suszka2015). Of the epigenetic changes detected in aged M. aquatica seeds and plants, half corresponded with new methylations and half with demethylations, so no clear trend was detected, but variations in the methylation state could result in changes in gene transcription, some of them responsible for age-related plant deterioration.
We found that changes on the cytosine methylation profile could be detected in seeds after storage (8%) and, in a higher proportion (13%), in plants produced from aged seeds. This indicates that stress due to high temperature and humidity of storage induced changes on the methylation state of seeds and that changes were also detectable in the regenerated plants. In pear, an increase from 3 to 5% in DNA methylation level of seeds was found in response to storage at 3°C, and seedlings derived from those seeds maintained an altered methylation profile (Michalak et al., Reference Michalak, Barciszewska, Barciszewski, Plitta and Chmielarz2013). It is known that most of the stress-induced epigenetic modification of DNA are reset upon relief from the stress, but some of the alterations might be stable and be carried forward in the form of a ‘stress memory’ that is inherited across mitotic or even meiotic cell divisions in plants (Moliner et al., Reference Moliner, Ries, Zipfel and Hohn2006; Verhoeven et al., Reference Verhoeven, Jansen, van Dijk and Biere2010). Therefore, changes in the DNA methylation level triggered by environmental stresses may be inherited by successive generations, being a potential factor in the adaptative and evolutionary mechanism in plants (Avramova, Reference Avramova2015).
We show that genetic screening methods detected differences in plants produced by aged seeds and these epigenetic changes might have important, but unknown consequences for their optimum conservation. Mutation accumulation during storage could alter the genetic profile of the conserved material during long-term storage (Schoen et al., Reference Schoen, David and Bataillon1998; Schoen and Brown, Reference Schoen and Brown2001; Chwedorzewska et al., Reference Chwedorzewska, Bednarek and Puchalski2002a, Reference Chwedorzewska, Bednarek, Puchalski and Krajewski2002b, Reference Chwedorzewska, Bednarek, Lewandowska, Krajewski and Puchalski2006). Also, epigenetic changes are known to be the first step towards phenotypic changes that could have an unknown effect on mature plants (Bird, Reference Bird2007) and perhaps emerge as a new source of phenotypic variation to improve adaptation to changing environments and drive breeding strategies (Gallusci et al., Reference Gallusci, Dai, Génard, Gauffretau, Leblanc-Fournier, Richard-Molard, Vile and Brunel-Muguet2017). Efficient methods to establish the quality of a seed lot is a critical step for ex situ conservation management of plant germplasm, and our data indicate that quality should be evaluated both phenotypically and genetically in stored seeds and the plants obtained after regeneration. Genotype screening techniques such as RAPD and MSAP, independent of genome sequence procedures for detecting DNA polymorphisms, have a strong potential as molecular indicators of nucleic acid stability during seed storage and ageing.
Acknowledgements
The authors would like to thank Dr. Daniel Palmero and his team, Departamento de Producción Vegetal (Universidad Politécnica de Madrid), for the advice on fungi DNA detection in seed extracts.
Financial support
Work supported by the project AGL2015-64899-C2-1-P (MINECO/FEDER). M. Pirredda supported by ‘Programa Propio de I+D+I, UPM’.J.E. Marchessi supported by ‘Programa BEC.AR’.






