


1. Introduction
More than 80 years ago, Sir Henry Dale showed that acetylcholine (ACh) could be purified from mammalian organs (Dale & Dudley, Reference Dale and Dudley1929), and went on to confirm, with others, that it is a neurotransmitter. However, for many years the fast-acting membrane receptor for this neurotransmitter remained an elusive entity. Only comparatively recently – in the early 1970s – could the detergent-solubilized receptor be purified with efficient affinity reagents (Karlsson et al. Reference Karlsson, Heilbronn and Widlund1972; Miledi et al. Reference Miledi, Molinoff and Potter1971; Olsen et al. Reference Olsen, Meunier and Changeux1972), thus opening the way for its characterization as a single protein. Following this crucial initial step, the nicotinic ACh receptor has been at the forefront of developments leading to our current understanding of ion channels. It was the first protein to yield single-channel recordings by the patch clamp technique (Neher & Sakmann, Reference Neher and Sakmann1976), and the first channel whose function could be reconstituted by reinsertion into lipid membranes (Nelson et al. Reference Nelson, Anholt, Lindstrom and Montal1980). Its four polypeptide chains were sequenced completely, using recombinant methods (Noda et al. Reference Noda, Takahashi, Tanabe, Toyosato, Kikyotani, Furutani, Hirose, Takashima, Inayama, Miyata and Numa1983), before these methods became widely applicable. The creation of hybrid channels and chimeric subunits to elucidate functional properties (Imoto et al. Reference Imoto, Methfessel, Sakmann, Mishina, Mori, Konno, Fukuda, Kurasaki, Bujo, Fujita and Numa1986; Sakmann et al. Reference Sakmann, Methfessel, Mishina, Takahashi, Takai, Kurasaki, Fukuda and Numa1985) is a technique that was pioneered with this protein.
Now, with increasing effect over the last two decades, has begun an enthralling phase of structural exploration, probing the molecular design, architecture and activation mechanism of the receptor. Atomic details of the ACh-binding site have become available through the X-ray crystal structure of a water-soluble homologue (Brejc et al. Reference Brejc, Van Dijk, Klaassen, Schurmanns, Van Der Oost, Smit and Sixma2001), confirming decisively many earlier biochemical results. The receptor's complex and beautiful membrane-spanning architecture has been viewed by electron microscopy. Structures of related homomeric channels have been solved. Most recently, as elaborated in the following pages, the transient open-channel form of the receptor has been trapped and analysed in three-dimensions, illuminating directly how the whole assembly works.
Underpinning these advances in the majority of cases is a uniquely rich source of the protein: the electric organ of the Torpedo ray. Electrocytes – the flat, stacked cells from which the organ is composed – are derived from embryonic muscle cells, but lack the contractile apparatus. Their postsynaptic membranes resemble those of the neuromuscular junction, organizing into a series of invaginations or folds, analogous to the ‘junctional folds’ of skeletal muscle. ACh receptors pack at a very high density (up to ∼16 000 μm−2) at the crests of the folds, opposite the active zones in the terminals of the invading electromotor neurons where ACh is released. For biochemical experiments, ACh receptor-rich vesicles or detergent-solubilized protein can be extracted simply and quickly from these cells, and purified in high yield. Structural work has capitalized on the fact that the receptors in the isolated postsynaptic membranes re-organize readily into regular arrays, making them amenable to analysis by electron crystallography. Since the polypeptide chains composing the Torpedo ACh receptor and those of human skeletal muscle are similar (55–80% amino-acid identity), the wealth of information that has been gained from the fish-based studies also applies to the human counterpart.
The nicotinic ACh receptor is a molecular machine that has been fine-tuned through evolution to transduce a chemical signal into an electrical signal with remarkable efficiency and speed. In principle, X-ray diffraction has the power to analyse the structural mechanism in great detail. However, this approach requires the presence of perturbing influences such as detergent and other artificial reagents and usually, recombinant modification to stabilize the protein. It would therefore carry the risk of misrepresenting the true physiological closed- and open-channel states, which are inherently unstable. Electron microscopy, on the other hand, has developed by a series of steps into an alternative direct method of structure determination allowing the ACh receptor to be evaluated in its native membrane setting, and under ionic and activation conditions mimicking closely those at the synapse. Electron crystallographic analysis of tubular crystals, grown from Torpedo membranes, has played a role in and benefited from these developments, helping to provide an increasingly profound understanding of the functioning protein.
In this review, I discuss these results from experiments on the Torpedo membrane tubes in the context of findings from biochemical, biophysical and other structural studies, with the aim of achieving a reasonably integrated description of the ACh receptor and of its mode of action at the synapse. I pay particular attention to the imaging and analysis methods, giving a brief account of their development. Although in some aspects uniquely suited to the Torpedo system, these imaging methods may well find wider application in the future. The muscle ACh receptor is still the only transmitter-gated ion channel – and the only heteromeric ion channel – whose fully intact, membrane-associated structure has been investigated, and it remains to be seen how widely applicable are some of the insights and principles to emerge from the imaging approach. The biophysical properties of this receptor, and the roles played by specific amino-acid residues, have been examined extensively, and are covered in more depth in other recent reviews (Colquhoun et al. Reference Colquhoun, Shelley, Hatton, Unwin, Sivilotti and Abraham2003; Engel et al. Reference Engel, Ohno and Sine2003; Karlin, Reference Karlin2002; Sine, Reference Sine2012).
2. A model transmitter-gated ion channel
Nicotinic ACh receptors are present not only in the postsynaptic membranes of muscle cells and Torpedo electrocytes: a similar ‘neuronal-type’ ACh receptor is found in the central nervous system, relaying messages from one nerve cell to the next. Both are members of an important superfamily of cation- or anion-selective synaptic ion channels activated by different neurotransmitters (for recent reviews, see: Lester et al. Reference Lester, Dibas, Dahan, Leite and Dougherty2004; Sine & Engel, Reference Sine and Engel2006; Millar & Gotti, Reference Millar and Gotti2009; Thompson et al. Reference Thompson, Lester and Lummis2010), and which have their origins in related ligand-gated ion channels found in prokaryotes (Corringer et al. Reference Corringer, Poitevin, Prevost, Sauguet, Delarue and Changeux2012; Tasneem et al. Reference Tasneem, Lyer, Jakobsson and Aravind2005). They include the vertebrate 5HT3, GABAA, glycine receptors, and the invertebrate glutamate receptor, GluCl, which respond to serotonin, γ-aminobutyric acid, glycine and glutamate respectively. All members of this superfamily are pentamers of homologous (sometimes identical) subunits delineating a central ion path through the membrane. Each subunit contains the so-called Cys-loop – a thirteen amino acid loop flanked by disulphide-bonded cysteines – which gives the superfamily its name. Within each superfamily member there may be a large number of paralogous subunits, and the variation in subunit type, together with the possibility of different subunit combinations, permits a wealth of functional diversity to meet a wide range of physiological needs. Cys-loop receptors function in the central and peripheral nervous system, and are pharmaceutical targets for numerous human diseases and psychiatric disorders, including myasthenia gravis, epilepsy, depression, nicotine addiction, schizophrenia and Alzheimer's disease.
The muscle ACh receptor is a heteropentamer composed of four polypeptide chains: it has two α subunits, and one each of β, γ and δ (in adult human muscle, the embryonic γ subunit is replaced by the homologous ∊ subunit). As in electric tissue, this receptor is concentrated at the crests of invaginations or folds in the postsynaptic membrane, lying opposite active zones in the pre-synaptic nerve terminal from which ACh is released. The small (146 Da) ACh molecules, released in bursts, diffuse almost instantly across the narrow synaptic cleft and bind to the α subunits of the receptor at their interfaces with neighbouring γ and δ subunits. The channel of the receptor opens within a few microseconds (Chakrapani & Auerbach, Reference Chakrapani and Auerbach2005; Maconochie et al. Reference Maconochie, Fletcher and Steinbach1995), and cations flow through it down their electrochemical gradients (mainly influx of Na+) at a high rate (∼20 000 ions ms−1). Within a further millisecond or so, the concentration of ACh in the synaptic cleft decreases sufficiently, as a result of hydrolysis by acetylcholinesterase and through diffusion, that the weakly bound ACh molecules dissociate from their binding sites and the channel reverts to its initial closed, or resting state. The opening and closing events are therefore extremely fast, enabling rapid initiation and termination of the postsynaptic response. This is crucial for neuromuscular transmission, where closely spaced temporal patterns of nerve impulses have to be communicated with high fidelity.
The release process triggered by a single nerve impulse is mediated by 100–300 synaptic vesicles, and raises the local concentration of ACh in the synaptic cleft to ∼0.3 mM (Kuffler & Yoshikami, Reference Kuffler and Yoshikami1975), which then rapidly falls off due to hydrolysis and diffusion. The consequent transient opening of many channels depolarizes the muscle membrane from its resting value, of about −90 mV towards 0 mV. Very rapidly a threshold may be reached, allowing an action potential to be generated that causes the muscle to contract. In order to produce sufficient depolarization with limitations in time and the number of channels available, the channels have a low probability of opening when the ACh concentration is low, and a high probability of opening when ACh is present in saturating amounts (greater than about 50 μM; Dilger & Brett, Reference Dilger and Brett1990). This large difference in opening probability is achieved by utilizing the binding energies of two ACh molecules, one at each site.
Electrophysiological recordings at the single-channel level have yielded detailed quantitative information about how the channels respond to ACh and other agonists (Colquhoun & Sakmann, Reference Colquhoun and Sakmann1985), including the possible involvement of intermediate closed states (Lape et al. Reference Lape, Colquhoun and Sivilotti2008; Mukhtasimova et al. Reference Mukhtasimova, Lee, Wang and Sine2009). The recordings show that individual channels stay open for varying durations, yet invariably exhibit the same all-or-none response. Given the same driving force and ionic conditions, the open channel conducts ions at the same rate whatever concentration of ACh (or other agonist) is used. Even with no ACh present the channels may still open, but with a probability that is extremely low (Jackson, Reference Jackson1986). On the other hand, when ACh is present in saturating amounts, so that both binding sites are occupied, the channels have a probability of being open approaching unity (>90%; Colquhoun & Ogden, Reference Colquhoun and Ogden1988; Dilger & Brett, Reference Dilger and Brett1990; Sine et al. Reference Sine, Claudio and Sigworth1990). Only when ACh is present for an extended period (greater than about 20 ms), does this high efficacy diminish due to a significant fraction of the channels converting to a non-conducting, desensitized state. However, desensitization is of little consequence for normal neuromuscular transmission, where the channels receive continuous ACh exposure lasting at most a few milliseconds.
The ACh receptor therefore behaves as a typical allosteric regulatory protein (Auerbach, Reference Auerbach2012; Changeux & Edelstein, Reference Changeux and Edelstein2005; Monod et al. Reference Monod, Wyman and Changeux1965): having just two conformations normally at the synapse, one reflecting the closed, or resting state and the other the fully open, or active state, with ACh acting simply to shift the equilibrium between either form. A very large shift in the equilibrium ratio of open/closed channels from neither binding site being occupied to both binding sites being occupied (∼107-fold; Jackson, Reference Jackson1989) ensures that this protein functions as a near perfect on-off switch.
3. Torpedo postsynaptic membrane
3.1 Organization of ACh receptors at the synapse
Single receptors are easily resolved in electron images because of their high molecular weight (∼290 kDa), and could be seen in early freeze-fracture micrographs from Torpedo tissue, both at intact synapses and in isolated postsynaptic membrane fragments (Cartaud et al. Reference Cartaud, Benedetti, Sobel and Changeux1978; Heuser & Salpeter, Reference Heuser and Salpeter1979). The micrographs showed double rows of particles extending across the membrane surface. In quick-frozen, deep-etched postsynaptic membrane fragments, these double rows were often seen lying two abreast, giving rise to a tight-packing arrangement (Fig. 1 a). The freeze-fracture particles had a similar appearance to the ‘doughnut’ shapes made by ACh receptors in isolated postsynaptic membranes when imaged in the negative stain. Hence, there were no difficulties in identifying the freeze-fracture particles with the receptor protein itself.

Fig. 1. Rows of paired receptors (dimer ribbons) and their relationship to tubes. (a) Freeze-fracture, deep-etched image of the extracellular surface of a postsynaptic membrane fragment obtained from gently sheared Torpedo tissue, showing dimer ribbons partially packed side-by-side. (b)–(e) Negative stain images of ACh receptor-rich vesicles after stronger mechanical treatment, followed by incubation in a low salt solution. (b), (c) The mechanical treatment destroys the original regular packing of receptors, but with time the dimer ribbons and the side-by-side arrangements reappear. Tube formation ((d), bottom and (e)) appears to be driven by tight side-by-side packing of the dimer ribbons. Arrows in (c) and (d) identify loosely packed ribbons, which in (d) are continuous with those forming the tube. ((a) is from Heuser & Salpeter, Reference Heuser and Salpeter1979; (b)–(d) are from Brisson & Unwin, Reference Brisson and Unwin1984). Scale bars: 500 Å.
Further insight into the synaptic organization of receptors came from morphological studies of postsynaptic membrane vesicles incubated in low salt solution for extended periods of time (Brisson, Reference Brisson1980; Brisson & Unwin, Reference Brisson and Unwin1984). The mechanical treatment during isolation yielded outside-out vesicles with receptors densely packed, and apparently organized randomly over their surfaces. However, after incubation at 17 °C for several days aggregates consisting of paired molecules aligning linearly and forming double rows – dimer ribbons of receptors – could be seen extending over some vesicles (Fig. 1 b). Elongated vesicles and short poorly ordered tubes, made by loose side-by-side association of the dimer ribbons, could also be seen at this stage (Fig. 1 c). After further incubation, the side-by-side association became tighter, giving rise to more regular tubes (Figs 1 d and 1e). In addition, the tubes lengthened, presumably by recruitment of receptors from the pool already present in the same vesicle.
These morphological observations on isolated vesicles imply a close structural correspondence between the tubes, which are simply elongated protein–lipid vesicles, and the receptor-rich membrane as it exists in vivo. Given that tube formation is most likely driven by tight side-by-side association of the dimer ribbons, the packing of receptors in tubes may be equivalent to that at the Torpedo synapse in regions where the receptors are most tightly packed. It is notable that the radius of curvature of a typical tube (350–500 Å) is close to the maximum curvature of the postsynaptic membrane found at the crests of the junctional folds (e.g. Sealock et al. Reference Sealock, Wray and Froehner1984). Therefore, the region underlying the active zone may be the region most favoured for tight receptor packing: a situation that would ensure the maximum postsynaptic response.
The involvement of the receptor clustering protein, rapsyn, in influencing the organization of receptors at the synapse, or in tubes, was not evident from these studies, even though ultrastructural localization using monoclonal antibodies shows the two proteins to be co-extensive (Sealock et al. Reference Sealock, Wray and Froehner1984). Later studies of the tubes by helical reconstruction indicated that rapsyn, although present on their inner surfaces, does not create a regular network consistent with the tube surface lattice (Toyoshima & Unwin, Reference Toyoshima and Unwin1988). All the intracellular density except for that at the extreme intracellular end could be accounted for by the receptor itself (Miyazawa et al. Reference Miyazawa, Fujiyoshi, Stowell and Unwin1999). Thus it is unlikely that rapsyn participates directly in achieving the ordered close packing present either in tubes or at the synapse. However, it may well play a role in the initial stages, for example, to concentrate receptors at the synapse and facilitate their association into dimer ribbons.
3.2 Early structural studies of sheets and tubes
While the specific interactions involved in stabilizing the tubes have now become better understood as a result of higher-resolution structural analyses (see Section 4.5), it was recognized at this stage that a disulphide bridge made between the δ subunits of neighbouring receptors was most likely responsible for the observed pairing. Both ribbons and tubes dispersed completely after incubation with small amounts (1 mM) of dithithreitol – the conditions used for separating δ–δ subunit-linked dimers in biochemical studies (Chang & Bock, Reference Chang and Bock1977; Kubalek et al. Reference Kubalek, Ralston, Lindstrom and Unwin1987).
The presence of the δ–δ dimer restricted the number of possible receptor arrangements and, together with the morphological evidence, strongly suggested that the tubes are composed of pairs of oppositely oriented, δ–δ-linked molecules organized on a p2 surface lattice. Tubes assembled from pentameric polyoma virus capsids (Baker et al. Reference Baker, Caspar and Murakami1983) were a possible precedent for this arrangement. The clue to the capsid arrangement was the presence of weak diffraction spots between the major hexagonally positioned ones in computed diffraction patterns of the images. Diffraction patterns from images of the ACh-receptor tubes displayed the same features (Fig. 2 a), and the paired-receptor arrangement was confirmed by crystallographic analysis (Brisson & Unwin, Reference Brisson and Unwin1984). Other images of tubes or planar sheets, apparently showing all molecules facing in the same direction (Kistler & Stroud, Reference Kistler and Stroud1981; Ross et al. Reference Ross, Klymkowski, Agard and Stroud1977), or in various directions (Giersig et al. Reference Giersig, Kunath, Pribilla, Bandini and Hucho1989), are therefore not of the normal, synapse-related polymorphic form.
Fig. 2. Analysis of flattened tubes. (a) Fourier transform (upper) and surface lattice (lower), viewed from the outside of a tube. Transforms from typical tubes contain only a small number of independent h,k peaks, the amplitudes of which are proportional to the shown diameters. The weak peaks, for which k is odd (‘superlattice’ peaks) are associated with a doubling of the b dimension of the surface lattice (indicated by alternate rows of closed and open circles along the (0,1) lines). Typical values for the cell dimensions are: a=90 Å, b=162 Å, γ=118°. (b) Three-dimensional map at 25 Å resolution of receptor molecules in the surface lattice as they would appear viewed from the synaptic cleft. The dimer ribbon lies in a horizontal direction, and the centre-to-centre separation of receptors along the ribbon is 90 Å. The δ subunits of neighbouring receptors are labelled to indicate the location of the δ–δ disulphide bridge. Successive sheets are of sections parallel to the membrane plane separated by spaces corresponding to 5 Å; data from ice-embedded flattened tubes. (Fig. 10 gives more details of the packing and the subunit arrangement around the pentamer. (a) is from Brisson & Unwin, Reference Brisson and Unwin1984; (b) is adapted from Brisson & Unwin, Reference Brisson and Unwin1985).
3.3 Analyses of flattened tubes
Initial attempts to explore the three-dimensional (3D) structure of the ACh receptor from the ordered arrays were influenced by the development of an electron crystallographic method to determine structures from two-dimensional (2D) crystals of unstained specimens (Henderson & Unwin, Reference Henderson and Unwin1975). With this method the protein molecule itself is imaged, using a low electron dose to minimize radiation damage, and a statistically significant picture of the molecule is built up by averaging over the large number of identical copies present in the crystal lattice (Unwin & Henderson, Reference Unwin and Henderson1975). Then to obtain different views, data are recorded from the crystals tilted at various angles to the electron beam. Finally, the information from the different views is combined and used to calculate a 3D map. By this approach, it is possible to reveal the internal structure of a protein much in the same way as is done by X-ray crystallography of 3D crystals.
The wider tubes could be flattened readily on the carbon support film and analysed as pairs of superimposed planar sheets. Crystallographic analyses of the flattened tubes, tilted over a range of angles to obtain the different views, showed that the receptor has a pseudo-fivefold symmetric quaternary structure, with features extending for some distance on both sides of the membrane (Fig. 2 b; Brisson & Unwin, Reference Brisson and Unwin1985; Mitra et al. Reference Mitra, Mccarthy and Stroud1989). At the same time, attempts were made to assign the locations of individual subunits, by labelling with subunit-specific reagents (Kubalek et al. Reference Kubalek, Ralston, Lindstrom and Unwin1987). However, the resolution of these studies was limited, particularly in the direction normal to the membrane plane, by the small lateral dimensions of the arrays, by the inability to make them perfectly flat and by the restricted range of tilt views. Also, the reliability of the labelling was questionable given that the deduced location of β in-between the two α subunits conflicted with biochemical evidence that the γ subunit is in that location. Definitive subunit assignments could not be made until the resolution was sufficient to trace the polypeptide chains (see Sections 3.8, 4.5).
3.4 Helical families
A major breakthrough in the field of biological electron microscopy came about when Jacques Dubochet and colleagues developed a simple way of freezing specimens in a thin aqueous film so rapidly that ice crystals could not form and the water instead went into a solid amorphous state (Adrian et al. Reference Adrian, Dubochet, Lepault and Mcdowall1984; Dubochet et al. Reference Dubochet, Adrian, Chang, Homo, Lepault, Mcdowall and Schulz1988). The specimens on the microscope grid were plunge-frozen into liquid nitrogen-cooled ethane, and the use of a cold stage, operating at a temperature low enough to retain the amorphous ice (<−130 °C), enabled them to be viewed directly, without dehydration in the microscope vacuum. Particularly striking were images of viruses recorded from regions of the frozen film spanning holes in the carbon support (Adrian et al. Reference Adrian, Dubochet, Lepault and Mcdowall1984).
When ACh-receptor tubes were prepared and imaged in this way, the required low electron dose limited detail present in each molecule, but the protein was big enough to be contrasted well against the more weakly electron-scattering membrane lipids and amorphous ice. Wide tubes (as in Fig. 1 e), lying within the aqueous film and attached to the carbon support, were easily flattened and were analysed as such, assuming they approximated to superimposed planar sheets (Brisson & Unwin, Reference Brisson and Unwin1985; Unwin, Reference Unwin1996). However, narrower tubes (<850 Å diameter), when suspended over holes in the support film and hence surrounded by (frozen) water on all sides, retained their circular cross-section (Fig. 3). Under these conditions, the surface lattice formed a regular helix, making it possible to conduct an analysis by the Fourier method of helical image reconstruction (DeRosier & Klug, Reference Derosier and Klug1968; DeRosier & Moore, Reference Derosier and Moore1970; Toyoshima & Unwin, Reference Toyoshima and Unwin1988, Reference Toyoshima and Unwin1990).
Fig. 3. Electron images of ACh-receptor tubes in amorphous ice, spanning holes in the carbon support film. The three tubes shown belong to different helical families and have different diameters, characteristic of the particular family. In each example, the two indices in the upper right-hand corners denote the start numbers of the two principal helices, deduced from Fourier transforms of the images (see Fig. 4), (from Toyoshima & Unwin, 1990). Scale bar: 1000 Å.
A fundamental property of a helix is that it presents a complete set of equally spaced views of a molecule, making it unnecessary to tilt the specimen to obtain the information needed to calculate a 3D structure, as was necessary with the crystallographic method based on flattened tubes. The resulting simplification in data collection and the potential for greater resolution from an unperturbed, fully symmetric structure meant that it was now preferable to examine the structure of the receptor in tubes over holes, treating them as helical objects.
A simple way of representing the surface lattice of a helical tube is to imagine it drawn on a cylindrical surface, which is then cut open and laid out flat (Fig. 4 a). The lattice is built from dimers of receptors (linked circles in figure), arranged with p2 plane group symmetry, so that the dimers run obliquely upwards from left to right. Lines following a given direction represent a set of helices and may be characterized by (h,k) indices, based on the a and b vectors of the unit cell. A key parameter for defining the surface lattice is the number of lines (helices) required to make up the circumference. In Fig. 4 a, the start numbers for the two principal sets of lines corresponding to the unit cell vectors, (1,0) and (0,1), are 16 and 6, respectively. Taking the direction (handedness) into account, this surface lattice is referred to as belonging to the (−16,6) helical family.
Fig. 4. The helical p2 surface lattice and 3D image reconstruction. (a) The surface lattice consists of a regular array of receptor dimers (linked circles) arranged on a cylindrical surface. The diagram is made by opening the cylinder and viewing it from the outside. The vertical axis is parallel to the tube axis and the horizontal axis shows the azimuthal angle around this axis; a line on the surface lattice corresponds to a helix. The lattice can be characterized by the numbers (start numbers) of the two principal lines (helices) required to fill 360° of azimuth. In this example, a 16-start left-handed helix and a 6-start right-handed helix, denoted by (1, 0) and (0, 1), respectively, are the principal helices; the lattice is therefore of the (−16, 6) family. Unit cell vectors, a and b, are indicated. (b) The ribbons of receptor dimers, lying along the a-direction in (a), form helices having slightly different pitches depending on the helical family and the exact dimensions of the unit cell. (c) Surface representation reconstructed from images of (−16, 6) tubes embedded in ice. The darker shading highlights the basic helix formed by a dimer ribbon of receptors.
ACh-receptor tubes form many different helical families, each having a distinct diameter, depending on the start numbers of the two principal helices (Toyoshima & Unwin, Reference Toyoshima and Unwin1990). The narrower tubes, which can be imaged in thinner films of ice with minimal flattening, are the most suitable kind for structural studies. Of these, the most frequently encountered belong to the (−15, 5), (−17,5), (−16,6), (−15,7) and (−18,6) families. They have mean diameters of 710, 760, 770, 790 and 830 Å, respectively, with some intra-family variation due to slight differences in unit cell dimensions of the surface lattice. The basic helix, corresponding to the dimer ribbon (Fig. 4 b; shaded helix in Fig. 4 c), has a slightly different pitch, depending on the family, again with some intra-family variation.
3.5 Image reconstruction from well-ordered tubes
Tubes of the different helical families yield similar diffraction patterns, with layer-lines in slightly different positions depending on the family in question. The diffraction patterns from single tubes do not show layer-lines extending beyond about 30 Å resolution, indicating a significant degree of disorder (see Section 3.6). However, in initial attempts to elucidate the 3D structure of the ACh receptor by helical reconstruction, a resolution of ∼17 Å was able to be attained simply by averaging information from several exceptionally well-ordered tubes in the same family (Toyoshima & Unwin, Reference Toyoshima and Unwin1988, Reference Toyoshima and Unwin1990). By avoiding the technical limitations associated with analyses of flattened tubes, the helical method revealed more accurately the shape of the receptor and the central ion pathway (Fig. 4 c).
In cross-section, the ion path through the receptor was now resolved into a narrow pore spanning both leaflets of the lipid bilayer and framed on either side by large extracellular and intracellular vestibules (Fig. 5 a). By averaging data from yet more (26) well-ordered tubes, it then became possible to extend the resolution to ∼9 Å and hence reveal even finer detail (Unwin, Reference Unwin1993), which could be linked tentatively with results from biochemical and mutation-combined-with-electrophysiological studies. For example, one could observe in the 3D map bent rods of density shaping the pore (Fig. 5 b) and tentatively align these with the amino-acid sequence corresponding to the putative pore-lining helix, M2 (Fig. 5 c; Giraudat et al. Reference Giraudat, Dennis, Heidmann, Chang and Changeux1986; Hucho et al. Reference Hucho, Oberthur and Lottspeich1986; Imoto et al. Reference Imoto, Methfessel, Sakmann, Mishina, Mori, Konno, Fukuda, Kurasaki, Bujo, Fujita and Numa1986; Leonard et al. Reference Leonard, Labarca, Charnet, Davidson and Lester1988). This alignment tentatively suggested that a universally conserved leucine residue (L251 of the α subunit), near the middle of the membrane, might be involved in forming the gate of the channel. However, the accuracy of the alignment was obviously limited, given the weak density features, and simply stressed the fact that a higher-resolution framework would be needed to integrate in a useful way existing biochemical and electrophysiological data.
Fig. 5. Structures obtained by averaging data from images of ice-embedded tubes spanning holes in the carbon support film. (a) Cross-section normal to the tube axis showing the extracellular (outside) and intracellular (inside) portions of the receptors projecting from the phospholipid headgroup regions of the membrane (pair of concentric rings of density, 30 Å apart). A single receptor, cut centrally, is identified by the vertical arrow. (b) A single receptor in profile with positions of transmembrane rods and estimated limits of the lipid bilayer (dotted lines, 30 Å apart) superimposed. (c) α-helical net plot giving a tentative alignment of the amino acid sequence along the M2 pore-lining helix (Torpedo α subunit) with the densities in the map. The alignment suggests that a highly conserved leucine residue (L251; asterisk) lies near the middle of the membrane at the narrowest part of the pore. ((b) and (c) from Unwin, Reference Unwin1993).
3.6 Correction of image distortions
Further improvement of the helical method depended on finding a way to minimize the loss of signal due to crystalline disorder: a property which, in turn, is determined by the inherent flexibility of the tubes and other factors, such as heterogeneity of the endogenous lipids. Disorder degrades the diffraction patterns by causing regions of a tube to be displaced from their exact lattice positions, leading to a progressively more severe weakening of diffraction intensities at higher resolution. Displacements of a few Angstroms are easily generated by only slight bending in the image plane, tilting (bending away from the image plane), variations in twist around the tube axis, changes in unit cell dimensions due to slight tension or compression of the tube or to changes in lipid composition, and by shearing (so that the layer-lines in diffraction patterns no longer lie exactly perpendicular to the tube axis).
In the case of 2D purple membrane crystals, the correction of displacements away from the strict lattice positions had been shown to restore a large fraction of the signal (Henderson et al. Reference Henderson, Baldwin, Downing, Lepault and Zemlin1986), allowing ultimately the atomic details of bacteriorhodopsin to be revealed. This success with a 2D crystal inspired development of an analogous method for improving the analysis of ACh-receptor tubes (Beroukhim & Unwin, Reference Beroukhim and Unwin1997). The tube image is divided into short segments (Fig. 6 a), each of which is independently aligned, in reciprocal space, by cross-correlation against a reference structure. The variations in the different parameters are then mapped along the length of the tube (Fig. 6 b), and the tube image is reconstructed from the individual segments with the measured misalignments corrected. Thus each short segment is treated as a perfect helix, and any small displacements caused by distortions within the segment are neglected. By using short enough segments, all displacements (including those within segments that are not corrected) are, in principle, reduced to the point they no longer have a significant effect. In practice, there is a limit on the shortness of the segment that can be used, since the information content lessens as the length decreases, leading to increasing alignment errors.
Fig. 6. Measurement and correction of distortions. (a) Electron micrograph of an ice-embedded tube ((−15, 7) helical family), recorded at liquid helium temperatures using a 300 kV field emission microscope (defocus: 17 400 Å). The contrast is low due to the high accelerating voltage, but the low imaging temperature and the high stability of the microscope ensures that the finest details are present. The boxed region shows a typical-sized segment which is scanned along the length of the tube to measure the distortions present. (b) Plots showing the variation of several parameters (tube axis position; bend angle; tilt angle; apparent repeat length) along the length of the tube, determined from the segments (Beroukhim & Unwin, Reference Beroukhim and Unwin1997). A complete 3D alignment (defined by eight parameters) is obtained from each segment and the aligned segments are added to reconstruct a whole helical repeat. Subsequently, whole repeats along the length of the tube are averaged together. The same procedure, using overlapping segments, leads to a significant improvement in signal-to-noise ratio at higher resolution (Fisher et al. Reference Fisher, Ward, Milligan, Unwin, Potter and Carragher2011; Unwin & Fujiyoshi, Reference Unwin and Fujiyoshi2012). As is typical, the tube shown bends only slightly in-plane (bend angle), but bends more substantially out-of-plane (tilt angle) and varies in apparent helical repeat (with corresponding variations in unit cell dimensions of the surface lattice).
The limited diffraction from the ACh-receptor tubes can be readily understood, given the extent of the distortions present. Even when a tube appears straight and regular, by eye (as in Fig. 6 a), it can be quite badly bent out-of-plane or variable in terms of unit cell dimensions (see plots of tilt angle, apparent repeat, Fig. 6 b). In most cases, about half of the associated signal loss arises from effects that are correctable in the image plane (bending, changes in unit cell dimensions, shear) and about half from effects requiring out-of-plane correction (variations in tilt and twist around the tube axis). Preliminary tests using a segment size similar to that in Fig. 6 a showed that such losses could be restored almost completely by realignment of the segments to reconstruct a more perfect helix (Beroukhim & Unwin, Reference Beroukhim and Unwin1997). However, the resolution attainable then became limited by the electron-optical quality of the images. Clearly, full benefit from the distortion corrections could only be achieved by making use of an ultra-high performance electron microscope.
The effectiveness of this segmental Fourier method for measuring and correcting distortions in tubes stems from the fact that helical symmetry confers the ability to determine accurately and correct the distortion-induced displacements in all three dimensions. The analysis is not confined to displacements in the image plane. Furthermore, the method enables critical assessment of the data (e.g. extent of preservation of helical symmetry and of twofold symmetry perpendicular to the tube axis), and the ready incorporation of corrections for the focus changes at different levels in the structure (DeRosier, Reference Derosier2000). An alternative real-space method has also been developed that avoids the Fourier formulation by treating segments as a string of single particles (Egelman, Reference Egelman2007). Both methods have given similar results when applied to other tubular membrane crystals (Coudray et al. Reference Coudray, Valvo, Hu, Lasala, Kim, Vink, Zhou, Provasi, Filizola, Tao, Fang, Penczenk, Ubarretxena-Belandia and Stokes2013; Pomfret et al. Reference Pomfret, Rice and Stokes2007).
3.7 Microscopy at liquid helium temperatures
A modern electron microscope, equipped with a high-voltage field-emission gun, enables images of relatively stable materials, such as semiconductors and metals, to be recorded routinely with near-perfect contrast transfer out to a resolution of about 3 Å. However, a radiation-sensitive biological specimen, maintained in a thin film of amorphous ice, is another matter. The likelihood of movement produced by a short pulse of high-energy electrons (beam-induced movement) or, for example, small temperature gradients (drift), makes retention of the precise electron-optical signal much more difficult to achieve.
At the time the distortion–correction method was being developed, the symmetrical, top-entry liquid helium-cooled stage, designed by Fujiyoshi et al. (Reference Fujiyoshi, Mizuasaki, Morikawa, Yamagishi, Aoki, Kihara and Harada1991), offered the greatest potential for recording the needed high-quality images of tubes. In experiments leading to atomic models from 2D crystals (Fujiyoshi, Reference Fujiyoshi1998; Kuhlbrandt et al. Reference Kuhlbrandt, Wang and Fujiyoshi1994), this stage had demonstrated exceptional drift-stability compared with standard side-entry cold stages, and the overall microscope design was well suited for extensive grid-searching and rapid specimen exchange. Beam-induced movement becomes a more serious problem when recording images at liquid helium temperature (4 K), because of the greatly reduced conductivity of the specimen and consequent build-up of electric charge. However, we found that such movement could largely be avoided by the use of more conductive pre-irradiated carbon grids and incorporation of a clean, gold-coated objective aperture (Miyazawa et al. Reference Miyazawa, Fujiyoshi, Stowell and Unwin1999).
3.8 Refined 4 Å structure
The better quality images obtained with the liquid helium-cooled stage, when combined with distortion correction, led to significant improvements. A resolution of 4 Å was achieved after merging data from images of tubes in four helical families (equivalent to ∼106 molecules), recorded with the stage. This was sufficient, in the first instance, to trace all five polypeptide chains through the 3D densities in the α-helical membrane-spanning domain of the receptor (Miyazawa et al. Reference Miyazawa, Fujiyoshi and Unwin2003). Later, a refined atomic model was derived using standard crystallographic methods (Unwin, Reference Unwin2005), and helped by knowledge of the 2.7 Å X-ray structure of a related soluble ACh-binding protein (AChBP; Brejc et al. Reference Brejc, Van Dijk, Klaassen, Schurmanns, Van Der Oost, Smit and Sixma2001). This model did not allow a full chemical interpretation of the structure, because of the modest resolution, but nevertheless provided a detailed representation of the complex architecture of the receptor in all three domains. The model included 80% of the 2335 amino acids comprising the whole assembly, with most of the missing residues being in the long, apparently disordered (M3–M4) intracellular loop.
AChBP is a water-soluble homopentameric protein from snail, the protomer of which has similarity (20% amino acid identity) with extracellular part of the Torpedo α subunit. It was key to chain tracing in the extracellular domain because of the limited quality of the density map and the difficulty in interpreting finer, non-α-helical features. AChBP was also important in ensuring correct assignment of the subunits around the pentamer. The X-ray structure confirmed the results of biochemical experiments identifying ACh-binding residues (Grutter & Changeux, Reference Grutter and Changeux2001), and corroborated the inference that γ is the single subunit between the two α subunits (Karlin, Reference Karlin1993; Sine et al. Reference Sine, Kreienkamp, Bren, Maeda and Taylor1995). Only with this assignment were specific residues shown to influence ACh binding to the receptor, and equivalent residues in AChBP, clustered in a compatible way. Moreover, the handedness apparent in AChBP clarified the spatial order of αγ and αδ, the α subunits shaping the ACh-binding sites at the interfaces with γ and δ, respectively. Therefore, once the location of the δ subunit was established next to one of the radial twofold axes (see Section 4.5), the assignments of αδ, γ, αγ and the remaining β subunit automatically followed.
4. Atomic model of the closed channel
4.1 Architecture and fold
The four polypeptide chains of the receptor are similar in size and have the same 3D fold (Fig. 7 a). They form ∼160 Å long rod-shaped subunits, which lie almost perpendicular to the membrane and are split into three structurally distinct parts: an N-terminal extracellular part organized around a β-sandwich core, a membrane-spanning part composed of four α-helices and an intracellular part containing one α-helix. The extracellular part has the most complex organization. The two β-sheets composing the β-sandwich are made from six strands (inner sheet: green) and four strands (outer sheet: pink), and are joined through the Cys-loop disulphide bridge. Several of the loops connecting β-strands are critical for ACh receptor function. These include: the Cys loop, overlying the outer membrane-spanning helices; the loops A, B and C, which in the α subunits are involved in shaping the ACh-binding site; and the β1/β2 loop, connecting the inner β-sheet to the inner, pore-lining helix M2. Also specific to the α subunits is a loop near the N-terminal, which forms the main immunogenic region (MIR), the region where antibodies bind in the auto-immune disease, myasthenia gravis (Tzartos & Lindstrom, Reference Tzartos and Lindstrom1980). In the membrane-spanning part, the helices M1–M4 have a simple arrangement but splay apart towards the extracellular end, where they extend into solvent. The pore-lining helix M2, in the closed channel, is bent inward towards the lumen of the pore, but not as sharply as lower resolution analyses had suggested. The intracellular part is composed mainly of the stretch of sequence between M3 and M4, and includes a curved helix (MA), which precedes M4. As mentioned above, most of the rest of M3–M4 is disordered and not seen in the structure.
Fig. 7. Architecture and fold of the ACh receptor (closed-channel form (2BG9)), illustrated with ribbon diagrams. (a) The αγ subunit viewed from the side, with the central axis of the receptor on the right; the outer and inner sheets composing the β-sandwich core are in pink and green, respectively. The loops A, B and C harbour ACh-binding residues (see Fig. 8). (b) Whole assembly, as viewed from the synaptic cleft, with individual subunits identified; the C loop of αδ (broken trace) is not resolved in the density map, whereas the C loop of αγ is clearly seen, presumably because its flexibility is restricted by interactions with the equivalent loop on a neighbouring receptor. (c) Whole assembly viewed from the side with the αγ and γ subunits in foreground. Also shown in the figures in red are the side-chains of the ACh-binding residue αW149 on the B loop and αV46 on the β1/β2 loop. The position of the membrane is indicated by the horizontal bars.
The individual subunits arrange side-by-side in a ring, forming a pseudo-fivefold symmetric assembly that is ∼160 Å long and up to 80 Å in diameter (Fig. 7 b). The fold of the subunits partitions this assembly naturally into three functionally distinct domains (Fig. 7 c): an extracellular domain, which contains the two ACh-binding sites; a membrane-spanning domain, which contains the gate of the channel; and an intracellular domain: an inverted cone of five α-helices, which interacts with the clustering protein, rapsyn, at its base. The ion path running along the central axis is similarly divided, and consists of a narrow pore across the membrane, framed on the extracellular side by a long, ∼20 Å diameter cylindrical vestibule, and on the intracellular side by a shorter conical-shaped vestibule opening through five side-windows into the interior of the cell.
The two ACh-binding sites are about 40 Å from the membrane, and reside principally in the α subunits at the interfaces with neighbouring γ and δ. The entrances to these sites are framed by the C loop and a coordinating tryptophan residue, W149, on the B loop of α (Fig. 7 b). As will be described in Section 4.3, the gate of the channel is located near the middle of the membrane-spanning pore, more than 50 Å away from the binding sites (Fig. 7 c). Thus the ACh-triggered conformational change to open the channel extends over a remarkably long distance given that the transition is so fast.
The ACh receptor is a cation-selective ion channel, and when open allows only small cations (Na+, K+ and some Ca2+) to permeate. A function of the extracellular and intracellular vestibules is to enhance the selectivity of cations over anions. There is an excess of negatively charged groups, creating cation-stabilizing environments, on the inner walls of both vestibules. Rings of negatively charged side-chains are also located strategically at the ends of the pore-lining helices (Imoto et al. Reference Imoto, Busch, Sakmann, Mishina, Konno, Nakai, Bujo, Fukuda and Numa1988), and on the helices forming the intracellular domain, to concentrate cations near the entrances of the narrow pore. The ‘intermediate ring’ of negative charge at the intracellular end of the pore appears to play a particularly important role. Unlike the case of ion-specific channels, such as the potassium channel (Doyle et al. Reference Doyle, Morais Cabral, Pfuetzner, Kuo, Gulbis, Cohen, Chait and Mackinnon1998), the pore itself does not play a major role in ion discrimination but restricts the size of ion that can pass through.
4.2 ACh-binding sites
The detailed structure of the ACh-binding site in either αγ or αδ is not yet known. However, the ACh-binding site in AChBP shares the same core structure, since the key coordinating amino acids are conserved between the two proteins. The canonical site is an aromatic cleft shaped largely by amino acid residues on the C loop and on the body of the α subunit. Figure 8 shows how carbamylcholine (a close analogue of ACh) is coordinated with surrounding side-chains in the principal α-subunit-equivalent side of the protomer of AChBP (Celie et al. Reference Celie, Van Rossum-Fikkert, Van Dijk, Brejc, Smit and Sixma2004). One conserved tryptophan on the B loop (equivalent to W149 in Fig. 7), two conserved tyrosines on loops A and C (equivalent to αY93 and αY190), as well as one of the pair of conserved cysteines on the C loop (equivalent to αC192) contact the bound molecule. The most important stabilizing influence is a cation-π interaction between the tryptophan side-chain and the cationic quaternary nitrogen of the ligand (Zhong et al. Reference Zhong, Gallivan, Zhang, Li, Lester and Dougherty1998), but the carbonyl group of the tryptophan and another aromatic side-chain on the C loop (αY198) are involved as well. Several more variable residues on the adjacent AChBP protomer (equivalent to the γ or δ subunit of the receptor) also participate in the interactions, consistent with biochemical studies showing that residues from the γ and δ subunits modulate the properties of the two binding sites (Karlin, Reference Karlin2002; Sine, Reference Sine2002; Xie & Cohen, Reference Xie and Cohen2001).
Fig. 8. ACh-binding site of AChBP in the presence of the ACh analogue, carbamylcholine (1UV6; Celie et al. Reference Celie, Van Rossum-Fikkert, Van Dijk, Brejc, Smit and Sixma2004). The view of the whole pentamer (left) is equivalent to that of the receptor in Fig. 7b, with a binding site region identified by a square. The enlargement of the square (right) shows the principal ‘α subunit’ component of the binding site, including the arrangement of conserved aromatic side-chains and a conserved cysteine residue, which co-oordinate with the bound ligand. The tryptophan on the B loop, the tyrosines on the A and C loops, and the cysteine on the C loop are equivalent, respectively to αW149, αY93, αY190 and αC192 of the receptor. (ACh differs from carbamylcholine only by the replacement of the NH2 group with a methyl group).
The AChBP conformation in Fig. 8 is thought to resemble the fully coordinated, agonist-stabilized desensitized conformation of the receptor (Grutter & Changeux, Reference Grutter and Changeux2001). Nevertheless, it is interesting to compare this conformation (Fig. 8) with that of the closed channel (Fig. 7 b), as one might expect the open channel to be partially coordinated with a conformation between the two extremes. An obvious difference between the two structures lies in the orientation of the C loop. This loop bends inward to coordinate with the bound carbamylcholine in Fig. 8, but projects outward almost tangentially in Fig. 7 b. Thus residues in the binding site of the receptor must contract around the bound ACh molecule, as studies with AChBP have suggested (Gao et al. Reference Gao, Bren, Burghardt, Hansen, Henchman, Taylor, Mccammon and Sine2005), but access to the binding site is enhanced when it is empty. A description of the actual movement observed in this region upon channel opening is given in Section 5.3.
The structural differences between the two binding sites of the receptor are reflected in their different affinities for ACh (and for other ligands). These differences are important in terms of the activation mechanism, a central concept of allosteric regulation being that agonists bind to the inactive state of a protein (closed channel) less tightly than to the active state (open channel), and that this difference in binding energy is used to drive the activation event. In the case of the Torpedo receptor, both electrophysiological and biochemical studies have indicated that the closed-state affinity of αγ for ACh (∼100 μM) is less by ∼100-fold than that of αδ (Andreeva et al. Reference Andreeva, Nirthanan, Cohen and Pedersen2006; Sine et al. Reference Sine, Claudio and Sigworth1990), whereas the open-state affinities are more nearly equal and in the nanomolar range (Jackson, Reference Jackson1988). Therefore, αγ would be expected to make the greater energetic contribution to the conformational change driving channel opening. It is thought that the lower affinity of one site facilitates rapid termination of the synaptic response, whereas the higher affinity of the other site may accelerate activation (Jackson, Reference Jackson1989). The existence of a second binding site may also be considered the energetic cost of perfecting a structural switch that is capable of driving such a large change in the channel-gating equilibrium.
4.3 Membrane-spanning pore
The α-helical segments of the five subunits arrange regularly in the membrane, forming concentric rings around a tapered, water-filled pore (Fig. 9 a). There is an inner ring of helices (M2) lining the pore, and an outer ring of helices (M1, M3 and M4) facing the lipids. Both sets of helices splay apart to some extent towards the extracellular side, but otherwise are organized rather differently. In the closed channel, the inner M2 helices bend inward and only make significant side-to-side contacts near the middle of the bilayer and in the intracellular leaflet (Fig. 9 b). The outer M1 and M3 helices make tighter side-to-side contacts and twist around each other as in a regular left-handed coil. This difference in packing leads to a slight separation between the inner and outer inner rings, especially in the extracellular portion of the membrane domain. The outermost M4 helices make limited contact with the rest of the protein, but interact extensively with the lipids.
Fig. 9. Membrane-spanning structure of closed channel (2BG9). (a) View down the axis of the receptor (extracellular side uppermost) showing the symmetrical pentagonal arrangement made by the ∼40 Å long transmembrane α-helices. Each subunit contributes four helices, M1-M4, which are splayed apart at their extracellular ends, but come closer together towards the intracellular membrane surface, creating a tapered central pore. The bent pore-lining M2 helices are somewhat separated from the ring of M1/M3 helices, most especially in the extracellular portion of the bilayer. The M2 helices only make significant contacts with each other at the middle of the membrane and in the intracellular leaflet of the bilayer. At least two rings of hydrophobic side-chains (indicated by the yellow sticks, at positions 9′ and 13′) project into the pore from the encircling helices, to create a barrier for ion permeation near the middle of the membrane. The arrows indicate the directions in which αγM2 and δM2 flex to straighten when the channel opens. (b) Side view with the front two subunits removed, showing the tapered pore profile made by the bent M2 helices of the αγ and δ subunits. The pore narrows from the extracellular side (uppermost) to become most constricting near the middle of the bilayer (asterisk), where the large hydrophobic residues are located. The density contours superimposed on the atomic model are from the 6-Å closed-class structure (see Section 5), i.e. using data from different helical families than were used to derive the model. The indicated curvature and positions of the helices are the same in either case. The locations of specific pore-facing amino acid residues on αγM2 (at positions: −1′ (E), 2′ (T), 9′ (L), 13′ (V) and 20′ (E)) are shown. The pink contours indicate the locations of the rings of density associated with the phospholipid headgroup regions of the lipid bilayer (see Fig. 5a).
The water-filled space between the inner and outer rings seems crucial to the channel-opening mechanism (Section 5.5), since it would allow the pore-lining helices some freedom of movement to reshape the pore independently of the outer wall, where the M1 and M3 helices are more tightly packed. In the neuronal α7 nicotinic ACh receptor (which shares 36% sequence identity with the Torpedo α subunit), synthetic compounds forming positive allosteric modulators and allosteric agonists are able to bind in this interstitial space and potentiate or trigger channel opening. The location in α7 of most compounds is intra-subunit (Dacosta et al. Reference Dacosta, Free, Corradi, Bouzat and Sine2011; Gill et al. Reference Gill, Savolainen, Young, Zwart, Sher and Millar2011; Young et al. Reference Young, Zwart, Walker, Sher and Millar2008), but in the case of ivermectin (Krause et al. Reference Krause, Buisson, Bertrand, Corringer, Galzi, Changeux and Bertrand1998), the structure of a GluCl–ivermectin complex (Hibbs & Gouaux, Reference Hibbs and Gouaux2011) suggests that an inter-subunit location would be involved. Other members of the Cys-loop superfamily, notably glycine and GABAA receptors, harbour specific binding sites for alcohols, small neurosteroids and volatile anaesthetics in the same region (Hosie et al. Reference Hosie, Wilkins, Da Silva and Smart2006; Li et al. Reference Li, Chiara, Sawyer, Husain, Olsen and Cohen2006; Mascia et al. Reference Mascia, Trudell and Harris2000).
The phospholipid headgroup regions of the membrane are visible in the images (Fig. 5 a), and therefore specify how the helices, and the central pore, are positioned relative to the hydrophobic core of the lipid bilayer. The helices extend up to about two turns beyond the headgroups (pink contours in Fig. 9 b) on the extracellular side, but terminate fairly uniformly at, or slightly before the headgroups on the intracellular side. The pore is narrowest where it passes through the hydrophobic core of the bilayer, and widens at the extracellular end. The precise profile of the pore is determined by the paths taken by the encircling helices and by the side-chains projecting from them. In the closed channel, the pore-lining helices bend slightly, as is apparent in Fig. 9 b both from the atomic model and (superimposed) a lower-resolution surface representation obtained from an independent analysis (Unwin & Fujiyoshi, Reference Unwin and Fujiyoshi2012; see Section 5). The effect of the bending is to constrict the pore most near the middle of the membrane (asterisk, Fig. 9 b), and to maintain a narrow diameter thereafter until the intracellular entrance is reached.
A high-resolution atomic structure is not yet available to indicate the exact conformations and positions of individual side-chains lining the pore, but the details revealed in the crystal structures of related homomeric channels (Bocquet et al. Reference Bocquet, Nury, Baaden, Le Poupon, Changeux, Delarue and Corringer2009; Hibbs & Gouaux, Reference Hibbs and Gouaux2011; Hilf & Dutzler, Reference Hilf and Dutzler2008) are similar to those of the atomic model. Figure 9 b shows the locations of some key amino acids affecting ion transport, based on this model. The glutamate residues (αE241 and αE262; positions − 1′ and 20′), in the intracellular and extracellular phospholipid headgroup regions, are components of the intermediate and extracellular rings of charge affecting cation conductance through the open pore (Imoto et al. Reference Imoto, Busch, Sakmann, Mishina, Konno, Nakai, Bujo, Fukuda and Numa1988). The threonine (αT244; position 2′) is in the most constricting region when the channel is open (Imoto et al. Reference Imoto, Konno, Nakai, Wang, Mishina and Numa1991; Villarroel et al. Reference Villarroel, Herlitze, Koenen and Sakmann1991). The two hydrophobic residues (αL251 and αV255; positions 9′ and 13′) are components of adjacent rings of highly conserved leucines and valines forming a tight hydrophobic girdle around the closed pore (Blanton et al. Reference Blanton, Dangott, Raja, Lala and Cohen1998; White & Cohen, Reference White and Cohen1992).
4.4 Hydrophobic gate
The girdle formed by leucine and valine side-chains (positions 9′ and 13′) was identified by photolabelling experiments using a small uncharged photoactivatable compound (3-(trifluoromethyl)-3-(m-[125I]iodophenyl)diazarine). The compound bound efficiently to these side-chains on βM2 and δM2 only in the absence of agonist, suggesting that the pore-lining helices came together to form compact hydrophobic binding environment only when the pore was closed; otherwise, in the presence of agonist, the helices were more splayed apart (White & Cohen, Reference White and Cohen1992). This biochemical evidence was interpreted to indicate that the hydrophobic girdle forms all, or part of, the gate. In the closed-channel structure, the rings of leucine and valine side-chains are at the precise level where the pore becomes most constricted (Fig. 9 b; Miyazawa et al. Reference Miyazawa, Fujiyoshi and Unwin2003). They are also in the region where the pore widens most substantially when the channel opens (see Sections 5.3 and 5.5). The structural details therefore agree with the biochemical interpretation, and cannot be reconciled with other interpretations that place the gate at the intracellular membrane surface (e.g. Paas et al. Reference Paas, Gibor, Grailhe, Savatier-Duclert, Dufresne, Sunesen, De Carvalho, Changeux and Attali2005; Wilson & Karlin, Reference Wilson and Karlin1998).
Originally, it was thought that the gate might be an occluding barrier to ion permeation formed by the conserved leucine side-chains projecting inward towards the axis of the pore (Unwin, Reference Unwin1993; Fig. 5). However, the higher-resolution structure shows that this is not the case: there is a hole of 3.0–3.5 Å radius over a distance of about 8 Å along the axis where the hydrophobic girdle is located (Miyazawa et al. Reference Miyazawa, Fujiyoshi and Unwin2003). Although this radius is larger than that of a Na+ or K+ ion, the region contains no polar groups that could stabilize it electrostatically. Hence, the ion cannot readily shed its hydration shell and, in effect, becomes too large to pass through (Beckstein et al. Reference Beckstein, Biggin and Sansom2001; Unwin, Reference Unwin2000). The gate therefore works as an energy barrier, rather than a physical barrier, in preventing ion permeation across the membrane.
Interestingly the mechanosensitive ion channel, MscL, appears to have a similar hydrophobic gate (Chang et al. Reference Chang, Spencer, Lee, Barclay and Rees1998). This channel forms a homopentamer, but is otherwise unrelated to members of the Cys-loop ion channel superfamily. The gate of MscL is also a constriction, rather than an occlusion, made by adjacent rings of leucine and valine side-chains projecting symmetrically from five pore-lining α-helical segments (Birkner et al. Reference Birkner, Poolman and Koçer2012). The LxxxV sequence motif is common to both channels, suggesting that the exposed aliphatic leucine-next-to-valine side-chains may be particularly suited to forming the required functionally robust, but unstable permeation barrier.
4.5 Receptor–receptor interactions
The density maps from four helical families ((−15,7), (−17,5), (−16,6) and (−18,6)) were used to obtain the refined atomic model of the ACh receptor. Docking of this model into each of the four maps allows a more thorough examination of the interactions affecting receptor packing in the p2 surface lattice (Fig. 10). In fact, the packing is invariant between families, except for very minor adjustments to take account of differences in tube curvature. In each family, the receptors come closest to one other at the two radial twofold axes (asterisks, Fig. 10). The intersubunit disulphide bridge between the penultimate δ-Cys-500 residues of neighbouring receptors (DiPaola et al. Reference Dipaola, Czajkowski and Karlin1989) lies near the membrane at one such axis (blue asterisk). The C loops projecting from the αγ subunits of neighbouring receptors lie about 40 Å from the membrane at the other (red asterisk). Apparently the pair of C loops, as with the covalent δ–δ link, make specific contacts because their separation is constant, i.e. independent of the helical family in question. Both these contacts seem to act as swivel points about which the receptors tilt in order to allow the different curvature associated with each family. Flexure of this kind might be involved in facilitating changes in curvature at the crests of the junctional folds.
Fig. 10. Packing of receptors and subunit arrangement on the surface of a tube, viewed parallel with (upper) and normal to (lower) the plane of the membrane. Individual molecules come closest to each other at the two unique radial twofold axes (asterisks). A disulphide bridge between cysteine residues of neighbouring δ subunits lies at one such axis (blue asterisk); the C loops of neighbouring α subunits (αγ) lie at the other (red asterisk). The angles to the tube axis made by the dimer ribbons (identified by the pair of obliquely sloping broken lines in lower panel) range from 64 to 72°, depending on the helical family ((−16, 6) family in this example). The pair of arrows in the bottom panel indicates the plane including both twofold axes, and hence the orientation of the slab through the structure shown in the top panel.
4.6 Comparison with related proteins
Some features of the atomic model of the ACh receptor have been confirmed by X-ray structures of related proteins or domains. For example, in a 1.94 Å X-ray structure of the extracellular domain of the mouse α1 subunit bound to α-bungarotoxin (Dellisanti et al. Reference Dellisanti, Yao, Stroud, Wang and Chen2007), the β-sheet core superimposes well with the corresponding region of the model (Fig. 11 a). As would be expected, there is similar agreement with the β-sheet core structure of AChBP (Fig. 11 b). The homomeric anion-selective Cys-loop receptor, GluCl (Hibbs & Gouaux, Reference Hibbs and Gouaux2011), as well as the more distantly related homomeric prokaryotic ion channels (Bocquet et al. Reference Bocquet, Nury, Baaden, Le Poupon, Changeux, Delarue and Corringer2009; Hilf & Dutzler, Reference Hilf and Dutzler2008), share the same overall fold in the extracellular and membrane domains (Figs 11 c and 11d).
Fig. 11. Ribbon diagrams of the ACh receptor and related proteins. (a) Superposition of the mouse α1 subunit (2QC1) with the αγ subunit of the ACh receptor (2BG9). (b) AChBP (1I9B). (c) GluCl (3RIA). (d) Superposition of GluCl (open channel, stabilized by ivermectin (3RIA)) with the ACh receptor (closed channel (2BG9)) at the level of the gate. Related proteins are in gold; the ACh receptor is in grey.
In detail, there are some interesting differences between the homomeric ion channels and the ACh receptor. For example, the MA portion of the intracellular vestibule is missing from the homomeric channels. Yet it plays a significant role in the more highly evolved Cys-loop receptors, where it may act to control the single-channel channel conductance (Kelley et al. Reference Kelley, Dunlop, Kirkness, Lambert and Peters2003), or to filter out ions of the wrong size and charge (Hales et al. Reference Hales, Dunlop, Deeb, Carland, Kelley, Lambert and Peters2006; Song & Corry, Reference Song and Corry2009; Unwin, Reference Unwin2005). Presumably, the required conduction properties can be achieved equally, if not more efficiently by incorporating a framework that enables additional weak ion–protein interactions away from the narrow intracellular entrance of the pore.
Another difference is apparent in Fig. 11 d, comparing the helices encircling the open pore of GluCl (stabilized with ivermectin) with the closed pore of the ACh receptor, at the level of the gate. While the open pore of GluCl is indeed slightly wider at this level (based on Cα positions), the packing of its helices, like those of the prokaryotic ion channels (Brannigan et al. Reference Brannigan, Hénin, Law, Eckenhoff and Klein2008), is more compact. The looser helix packing of the ACh receptor, where the membrane is present, possibly reflects the fact that native lipids such as cholesterol, not only play a critical functional role (Brannigan et al. Reference Brannigan, Hénin, Law, Eckenhoff and Klein2008; Dacosta et al. Reference Dacosta, Medaglia, Lavigne, Wang, Carswell and Baenziger2009; Dalziel et al. Reference Dalziel, Rollins and Mcnamee1980), but also are needed to retain the exact transmembrane architecture.
5. Transition to open-channel form
5.1 Introduction
A description of the transient open-channel form of the receptor is fundamental to our understanding of how the protein works. What are the structural changes in the extracellular domain producing this form? How do they couple with the membrane domain to bring about a change in the α-helices encircling the pore? What are the helix motions that open the pore and let ions through? These questions have been probed in varying depth by a range of biophysical techniques. For example, rate-equilibrium free-energy relationship analysis has suggested that gating of the channel occurs by means of a ‘conformational wave’ that proceeds from the extracellular domain to the region of the gate (Auerbach, Reference Auerbach2010; Mitra et al. Reference Mitra, Cymes and Auerbach2005). Mutation analysis defining inter-residue couplings has identified a principal pathway linking ACh binding to opening of the pore (Lee & Sine, Reference Lee and Sine2005). Electrophysiological measurement of the extent of current block by substituted lysine residues has suggested that channel opening involves a subtle rearrangement of helices around the pore (Cymes et al. Reference Cymes, Ni and Grosman2005; Cymes & Grosman, Reference Cymes and Grosman2008). Molecular dynamics simulations based on the structure of the closed channel have illuminated the nature of the displacements required to achieve a fully conducting state (Corry, Reference Corry2006; Wang et al. Reference Wang, Cheng, Taylor, Mccammon and Sine2008, Reference Wang, Toghraee, Papke, Cheng, Mccammon, Ravaioli and Sine2009).
However, the ACh receptor is a complex heteromeric assembly and the actual conformational change must entail a range of finely balanced movements, distributed intricately in three dimensions. The magnitudes and directions of such movements cannot be defined in detail by the biophysical techniques, and cannot be predicted fully by even the most accurate energy calculations. They can only be established unequivocally by comparing directly solved structures of the same protein in the physiological closed- and open-channel forms. As will be shown later, these two forms differ in most regions by less than an Angstrom, and the changes relating them are asymmetric. Thus gating models based on comparisons of X-ray crystal structures of homomeric prokaryotic receptors, engineered from different polypeptide chains (Bocquet et al. Reference Bocquet, Nury, Baaden, Le Poupon, Changeux, Delarue and Corringer2009; Hilf & Dutzler, Reference Hilf and Dutzler2009), are of limited validity and do not relate straightforwardly to the conformational change occurring in the ACh receptor.
Electron microscopy of the tubes in principle provides an ideal way to investigate this conformational change, since the receptors in the surface lattice of a tube can be trapped in the open-channel form (Berriman & Unwin, Reference Berriman and Unwin1994), as well as in the close-channel form, and compared. This method avoids the concerns about relevance of the X-ray structures, and moreover eliminates possible ambiguity about the actual functional state. In preliminary experiments imaging the trapped open channels (Unwin, Reference Unwin1995), the resolution was insufficient to resolve individual membrane helices and the membrane domain was incorrectly assumed to be fivefold symmetric. However more recently, helped by technical developments (Sections 3.6 and 3.7), it has been possible to improve the resolution and to define the gating motions of the individual helices with demonstrated precision (Unwin & Fujiyoshi, Reference Unwin and Fujiyoshi2012). The following paragraphs give a brief description of the trapping experiments and of the indicated conformational change, providing a background for later discussion about how the channel works.
5.2 Spray-freeze-trapping experiments
The time-resolved aspect of trapping the open-channel form can be accomplished in several different ways (e.g. Lu et al. Reference Lu, Shaikh, Barnard, Meng, Mohamed, Yassin, Mannella, Agrawal, Lu and Wagenknecht2009; Ménétret et al. Reference Ménétret, Hofmann, Schröder, Rapp and Goody1991; White et al. Reference White, Thirumurugan, Walker and Trinick2003). Our work made use of an ACh-containing spray, to mimic synaptic activation, together with rapid freezing, to arrest the reaction before a significant number of receptors would convert to a desensitized state. In the experimental setup (Fig. 12 a), a solution containing the tubes is applied to a microscope grid, held in tweezers, and blotted so that only a thin aqueous film remains. The grid is then plunged by free-fall into liquid-nitrogen-cooled ethane. Just before it hits the ethane surface, the grid is intercepted by a fine spray of ACh droplets (∼1 μm diameter; 200 mM ACh) containing ferritin marker particles, which spread over and mix with the contents of the aqueous film. The distance between the spray nozzle and the ethane surface is made short, so that the maximum reaction time is brief (∼10 ms). Tubes that have actually been exposed to ACh (in estimated local concentrations of 1–5 mM) are identified in the images by the presence of nearby ferritin particles (Fig. 12 b).
Fig. 12. Spray-freeze trapping. (a) The grid-supported solution containing tubes is allowed to drop by free-fall into liquid-nitrogen-cooled liquid ethane and is sprayed with an ACh solution containing ferritin marker particles just before it hits the ethane surface. The atomiser spray produces a pulse of concentrated ∼1 μm diameter droplets, and the reaction time is made brief by having the level of the nozzle only a short distance above the ethane surface. The distances d 1 and d 2 are typically 4 and 0.8 cm, respectively. (b) Tubes that have interacted with ACh can be identified from the images by the presence of nearby ferritin particles (arrow). (c) Mixing of a 1 μm diameter spray droplet, containing ACh and ferritin, with the grid-supported aqueous film 10 ms after impact: ACh (reddish colour) spreads beyond the ferritin (dots) within the zone of coalescence (inner dashed circle), reaching some tubes by diffusion. ACh receptors in tubes located in this outer ‘diffusion zone’ may have received saturating amounts of ACh for up to ∼2 ms and so would also have a high probability of being open. These tubes can be identified a posteriori by correlation against closed-channel and open-channel reference structures. ((a) is from Berriman & Unwin (Reference Berriman and Unwin1994); (c) is from Unwin & Fujiyoshi (Reference Unwin and Fujiyoshi2012)).
A later refinement of this technique made use of tubes located in a ‘diffusion zone’ extending beyond the ferritin-delineated edge of the coalescing droplet (Fig. 12 c), where the reaction times were even shorter (0–2 ms) and the ACh concentration lower than 1 mM (Unwin & Fujiyoshi, Reference Unwin and Fujiyoshi2012). Although the ACh concentration is falling off in this region, it can still be above the saturation level required to ensure a high probability of channels being open. Moreover, in the absence of ‘labelling’ by ferritin particles, it was possible to determine functional state a posteriori by comparing the (average) structure of the receptor in each tube with closed- and open-channel reference structures. The sorting procedure led to roughly equal numbers of ‘closed’ and ‘open’ class images, and comparison of the two equal-quality 6.2 Å density maps obtained from these images allowed accurate measurement of small changes throughout the structure. About 80% of the open-channel images used in this analysis were from tubes located in the diffusion zone. In terms both of ACh concentration and of timescale, therefore, the experimental conditions recapitulated almost perfectly the activation conditions existing at the neuromuscular junction (Kuffler & Yoshikami, Reference Kuffler and Yoshikami1975).
5.3 Signature changes at binding site and gate
The ACh-coordinating amino acid side-chains on the C loop and the loops A and B of the αγ subunit are far apart in the closed-channel form of the receptor (Figs 7 a and 7b), indicating that a localized disturbance, involving closure of the C loop, must occur upon activation. Indeed, the open-class structure in this region (Fig. 13 b) shows the C loop to have moved inward by about 2 Å, compared with its orientation in the closed-class structure (Fig. 13 a), bringing it nearer to residues, such as W149 on the B loop, on the other side of the binding site. However, the C loop has not moved as far inward as its counterpart in ‘desensitized’ AChBP (green trace, Fig. 13 b; see also Fig. 8). The orientation shown in Fig. 13 b therefore suggests that the open state may correspond to a partially co-ordinated intermediate conformation.
Fig. 13. Closure of C loop and widening of pore on activation by ACh. (a) and (b) show the C-loop conformations of αγ in the closed- and open-channel forms, respectively, in equivalent slabs through the two 6 Å density maps. The view is similar to that of Fig. 7b, from the synaptic cleft, and is adjacent to a radial twofold axis (cross) in the tubular surface lattice. Also shown are the superimposed Cα backbones from the atomic model of the closed channel (black (2BG9)) and, in (b), loop C from the aligned AChBP complexed with carbamylcholine (green (1UV6); Celie et al. Reference Celie, Van Rossum-Fikkert, Van Dijk, Brejc, Smit and Sixma2004). (c) and (d) show the pore profiles made by the helices, αγM2 and δM2 in the closed- and open-channel forms, respectively, in equivalent near-axial slabs through the two 6 Å density maps. The fitted Cα backbones (black, closed; red, open) are superimposed. These helices widen the pore by straightening (arrows) in response to ACh binding, as indicated for αγM2 by comparison with the overlaid broken line. The straightening of δM2 is not fully evident in this figure because it occurs predominantly in a direction tangential to the axis of the pore (see Figs. 9a, 15). The vertical yellow bar in (c) identifies the location of the gate, near the middle of the membrane (grey bars). Extracellular side is uppermost. The net spacing corresponds to 1 Å. Contours are at 1σ. (From Unwin & Fujiyoshi, Reference Unwin and Fujiyoshi2012).
Another defining signature of an opened channel is an increase in dimensions of the pore (and/or increase in polarity) in the vicinity of the gate. In fact, the open-class pore widens maximally at the gate, and in the adjoining extracellular region, according to comparisons between the two 6-Å structures. Figures 13 c and 13d show the alternative profiles shaped by the oppositely facing pore-lining helices of the αγ and δ subunits. In the closed channel configuration (Fig. 13 c) both αγM2 and δM2 bow inward towards the gate (yellow bar), whereas in the open-channel configuration (Fig. 13 d) they no longer bow inward, but follow straight paths (indicated for αγM2 by the broken line). The pore develops a uniformly tapering profile as a result of the helix straightening and, in this cross-section, widens by almost 2 Å at the gate (see also Section 5.5).
5.4 Conformational change driving channel opening
The extended conformational change linking the disturbance at the C loop (Figs 13α and 13b) to the helix gating motion (Figs 13 c and 13d) occurs by mechanically distinct movements in the extracellular and membrane domains. The main movements are a structural rearrangement involving the αγ and β subunits in the extracellular domain, and a rigid-body tilting of the β subunit in the membrane domain. The αγ subunit, which contains the low-affinity ACh-binding site, is the main driver of the conformational change, whereas the β subunit is the main partner transmitting the action to the membrane.
Figure 14 shows essential details of this mechanism. When the C loop of αγ is drawn in towards the binding pocket it brings the outer sheet with it, rotating the sheet in the plane of the membrane (curved arrow and pink shading, Fig. 14 a) and tilting it slightly in a perpendicular direction. To accommodate this movement, the inner sheet (green shading) is pushed towards the β subunit (yellow arrow). The β subunit responds with a rocking motion (Fig. 14 b): the extracellular part moves outward, tilting slightly to maximize displacement near the membrane surface, whereas the membrane part tilts by ∼2° in the opposite sense, in the same direction. That is, the two parts of β move as rigid bodies, flexing about their shared interface.
Fig. 14. Conformational change driving channel opening. (a) Slab showing fitted Cα backbones (black, closed; red, open) through the closed- and open-class density maps at the level of the ACh-binding site in αγ. Closure of the C loop around the bound ACh is accompanied by rotation (curved arrow) of the outer sheet (pink shading). The displacement of the outer sheet is accommodated by a readjustment of the inner sheet (green shading), resulting in outward displacement of the extracellular part of β (yellow arrow). Individual β-strands, and W149, are also identified in the figure. These changes are indicated in the sketch of the whole cross-section on the right. (b) The outward displacement of the extracellular part of β (upper black arrow), driven by αγ (yellow arrow), is coupled to tilting of the membrane part (lower black arrow), affecting the stability of the hydrophobic gate. The sketch on the right indicates that both components move from their closed-channel positions (broken lines) as rigid bodies. (Adapted from Unwin & Fujiyoshi, Reference Unwin and Fujiyoshi2012).
The rearrangement within the αγ subunit is best described as a small readjustment of the inner and outer sheets composing the β-sandwich, and may be contrasted, for example, with the larger ‘clamshell’ closing movement around the bound agonist, as occurs in glutamate receptors (Gouaux, Reference Gouaux2004). The β-sheet core of αγ in fact becomes more similar to the β-sheet cores of the non-α subunits as a result of this transition, and the whole extracellular domain becomes more pentagonally symmetrical. Thus, the outer and inner β-sheets of αγ must be initially in a ‘distorted’ or sub-optimal configuration, which the ACh-induced readjustment helps to relieve, thereby increasing the affinity of the binding site. The rearrangement in αδ (and corresponding change in affinity) is smaller than in αγ, but appears to contribute in pushing β outward, using the extracellular part of γ to transmit a small displacement in the same sense around the ring.
The coordinated rocking motion of the β subunit must be the primary means by which the effect of ACh binding is coupled to opening of the gate, because β is the only subunit where matching displacements are found on either side of the domain interface. Other membrane proteins use a similar mechanism for communicating between ligand-binding and membrane domains (Bublitz et al. Reference Bublitz, Poulsen, Morth and Nissen2010; Locher, Reference Locher2009). Nevertheless mutation-combined-with-electrophysiology experiments have shown that inter-domain contacts in the remaining subunits also play significant roles (Jha et al. Reference Jha, Cadugan, Purohit and Auerbach2007; Lee et al. Reference Lee, Free and Sine2009; Xiu et al. Reference Xiu, Hanek, Wang, Lester and Dougherty2005). Most notable is an interaction between the inner β-sheet of αγ and the end of the helix αγM2 through a valine side-chain, V46, projecting from the tight β1/β2 loop (Fig. 7 a; Hanek et al. Reference Hanek, Lester and Dougherty2008; Lee & Sine, Reference Lee and Sine2005; Miyazawa et al. Reference Miyazawa, Fujiyoshi and Unwin2003). Clearly any displacement of the inner sheet is likely to affect the flexing of αγM2, and hence the stability of the gate.
5.5 Asymmetric gating by the membrane helices
The pore-lining M2 helices of the αγ, δ and β subunits, through flexing (αγ, δ; Figs 13 c and 13d) and tilting (β; Fig. 14 b), bring about the major changes creating an open pore, whereas the pore-lining helices of αδ, and γ do not change appreciably. As Fig. 15 shows the M2 motions, and degree of participation of M1, M3 and M4, are different with each subunit. In the case of αγ, the straightening of αγM2 occurs independently of the other three helices, producing a maximum displacement of 1.5 Å (arrow, Fig. 15 a) approximately towards αγM3, thereby contracting the interstitial space. In the case of δ, the straightening of δM2 also occurs nearly independently of the other three helices, but the displacements are in a direction between βM1 and δM3, i.e. away from αδM2 (arrow, Fig. 15 b). As a result, a 1 to 2-Å wide crevice (blue wedge, Fig. 15 b) is created in between δM2 and αδM2. In the case of β, the tilting of βM2 occurs along with the other helices as part of a rigid domain (Fig. 15 c). Since the relevant helix motions are not equivalent relative to the pore axis (radial for αγM2 and βM2, near-tangential for δM2; Fig. 15 d), the pore becomes markedly more asymmetric in the open-channel form.
Fig. 15. Pore opening is mediated principally by the αγ, δ and β subunits. Different movements are involved, depending on the subunit in question, as shown by superpositions of the Cα backbones fitted to the closed-class (grey) and open-class (red) density maps. In (a) αγM2 converts from a bent conformation to a straight conformation by flexing by up to 1.5 Å towards αγM3 (arrow); this is in a radial direction with respect to the axis of the pore. The remaining helices (M1, M3 and M4) retain approximately fixed positions. In (b) δM2 converts from a bent conformation to a straight conformation by flexing to a similar degree as αγM2, but towards the space between βM1 and δM3. This flexure therefore has a tangential component with respect to the pore axis, exposing a crevice (arrow, blue wedge) between δM2 and αδM2. The view shown is from inside the pore. In (c) all four helices of β tilt equally by ∼2°, as indicated by the pairs of overlaid lines. (d) Shows the directions of movement of the pore-lining M2 helices in each example, relative to the axis of the pore (see also Fig. 9a). ((a)–(c) from Unwin & Fujiyoshi, Reference Unwin and Fujiyoshi2012).
It is notable that the maximum displacement of αγM2 is towards the space where, in the neuronal α7 nicotinic ACh receptor, the intra-subunit allosteric modulators appear to bind, whereas the maximum displacement of δM2 is towards the space where inter-subunit allosteric modulators appear to bind (arrows, Fig. 9 a). The interactions of allosteric modulators with α7 therefore draw attention to the relevance of the interstitial spaces for normal neuromuscular transmission, which must be to provide room for the pore-lining helices to flex when the channel opens.
How does the conformational change transmitted primarily through the β subunit bring about this change in helix configuration around the pore? We have argued that the pore-lining helices bending inward to form the hydrophobic gate, and somewhat separated from the outer protein wall, are held together symmetrically by a small number of side-to-side contacts involving the same conserved amino acid side-chains, and that this organization makes the gate intrinsically unstable, since its integrity would depend on equal sets of interactions between each component of the ring (Miyazawa et al. Reference Miyazawa, Fujiyoshi and Unwin2003). Hence, when a disturbance is introduced, as through the tilting of βM2, at least one set of interactions is eliminated and the whole gate assembly should break up. The non-equivalent displacements of the αγM2 and δM2 helices on either side of βM2 (Fig. 15 d), are explained simply on this basis since the interstitial space available to the now freed helices would be different, depending on the exact locations and side-chain compositions of the neighbouring helices forming the outer wall.
The argument that the integrity of the gate depends on equal side-to-side interactions is supported by the fact that it can be weakened by minor perturbations in multiple ways. For example, in oocyte expression studies, mutation of the leucine (position 9′) to serine or threonine, in any of the subunits, increases the opening sensitivity of the channel by roughly equal amounts (Filatov & White, Reference Filatov and White1995; Labarca et al. Reference Labarca, Nowak, Zhang, Tang, Deshpande and Lester1995). Mutation of the valine (position 13′) to alanine, in any of the subunits, gives similar results (Moriconi et al. Reference Moriconi, Di Castro, Fucile, Eusebi and Grassi2010). Likewise, a small molecule that binds in the interstitial space behind αγM2 should be able open the channel using contraction around the bound ligand, rather than tilting of a subunit, to destabilize the gate. This seems to be the mechanism of activation of the neuronal α7 nicotinic receptor by the allosteric agonist, 4BP-TQS (Gill et al. Reference Gill, Savolainen, Young, Zwart, Sher and Millar2011).
Elimination of the central hydrophobic constriction through flexing and tilting of the pore-lining helices transfers the narrowest part of the pore to a region close the intracellular membrane surface, where polar residues projecting inward (such as αT244; position 2′) would keep the ions solvated. Although the pore has widened by only a small amount at the level of the constriction (about 1 Å, as sensed by a sphere), the unequal helix motions would have exposed buried polar groups in spaces where the helices have become separated (Fig. 15 b) and made room here, and in the lumen, for additional water molecules, thereby strongly perturbing the hydrophobic character of this region. Indeed, the polar crevice opened up by δM2 may have some analogy with the ‘guide-rail’ of exposed polar carbonyl groups in aquaporins which enables water transport at very high rates (∼3×106 molecules ms−1) through an otherwise largely hydrophobic and much narrower pore (∼3 Å diameter; Tani et al. Reference Tani, Mitsuma, Hiroaki, Kamegawa, Nishikawa, Tanimura and Fujiyoshi2009). This analogy, at the very least, stresses the importance of the exact surface chemistry.
Unfortunately, it is not possible to evaluate quantitatively the effect of these helix displacements on ion transport rates. The change to the pore has not been resolved in atomic detail, and may incur additional changes in side-chain conformation. Experimental limitations – the inability to record images where all channels are open – may mean that the gating motions indicated in Fig. 15 have been slightly underestimated. However, given that the lumen of the closed pore at the gate is already quite wide (see Fig. 11 d), the smallness of the change should not be surprising. It is entirely consistent with molecular dynamics simulations of hydrophobic gating (Beckstein & Sansom, Reference Beckstein and Sansom2004, Reference Beckstein and Sansom2006), indicating that only minor readjustments are sufficient to achieve the full flow of hydrated ions.
5.6 Allosteric mechanism
These electron crystallographic experiments on the Torpedo receptor help to define the structural mechanism underlying the activation event at the synapse. As they show, opening of the channel takes place by a concerted conformational change, involving a number of small inter-dependent movements. All five subunits participate, but three of them (αγ, β and δ) play a dominant role. By focusing on just these three subunits, the conformational change can be simplified into its principal elements. These are depicted in Fig. 16 a. The ACh molecule, on coordinating with residues in the αγ-binding site, draws the C-loop inward (red arrow). Rotation of the C loop brings the outer β-sheet with it, and the inner sheet readjusts by moving in the direction of the β subunit (green arrow). The β subunit is thereby pushed outward, and moves by displacement of its extracellular part and tilting of its membrane part (yellow arrow). Tilting of the membrane part of β disrupts the gate, freeing the pore-lining helices, which consequently straighten.
Fig. 16. Mechanism of switching between closed and open states. (a) ACh on entering the two binding sites induces a concerted conformational change, in which the αγ, β and δ subunits, shown here, play the principal roles. The αγ subunit undergoes a small β-sheet rearrangement, driving the conformational change; the β subunit communicates the rearrangement in αγ to the gate; and the pore-lining helix of δ (along with that of αγ) straightens, altering the size, shape and polarity of the pore. Arrows denote the movements linking the binding site in αγ to the gate; red arrow: inward displacement of C loop; green arrow: displacement of inner β-sheet of αγ towards β subunit; yellow arrow: outward displacement and tilting of β subunit; black arrows: straightening of the pore-lining helices of αγ and δ in radial and near-tangential directions, affecting the gate. Outer and inner β-sheets of αγ are in pink and green, respectively; pore-lining M2 helices of αγ and δ are in blue; gate, yellow bar. (b) Shows the alternative pore configurations before (closed) and after (open) the transition. Although the lumen of the closed pore is wider than the diameter of a Na+ ion (cyan sphere) at the gate (yellow bar), the pore-lining hydrophobic side-chains (yellow spheres) present an energy barrier to ion flow through the pore. The open pore is wider and more polar at the gate, due mainly to the straightening of αγM2 and δM2, allowing ions to pass through.
Figure 16 b compares the αγM2 and δM2 helix configurations before (closed) and after (open) the conformational change to illustrate how the straightening of these helices increases the diameter (and polarity) of the pore in the region of the gate. Once straightening has occurred, the gate region ceases to function as a permeation barrier and the ions readily pass through.
Small changes in symmetry accompany this conformational change, providing further insight into the allosteric mechanism. As mentioned earlier, the packing of β-sheet becomes more regular when the channel is open, strengthening the fivefold symmetry of the extracellular domain. In contrast, the pore-lining helices organize less symmetrically when the channel is open. The weaker initial symmetry of the extracellular domain can be explained by assuming that the β-sheet arrangements in two α subunits (mainly αγ) are ‘distorted’ by their interactions with neighbouring subunits. Coordination of ACh with amino acids in the binding pockets then (partially) overcomes these distortions. This allows more optimal arrangements of their β-sheets, like those in the relatively stable and rigid non-α subunits – hence promoting more symmetrical packing. The reduction in symmetry around the pore can be explained by assuming the bent pore-lining helices are ‘distorted’ by the equal side-to-side interactions needed to form the gate – interactions, which are eliminated when the channel opens.
The structure of the closed channel therefore appears to be perturbed in both domains in a way that would not happen if the interactions between neighbouring subunits were not present. Since the binding of ACh acts to reduce these perturbations in both domains, it shifts the whole structure from a ‘tense’ towards a more ‘relaxed’ state. A parallel can therefore be drawn between the ACh receptor and soluble protein complexes or regulatory enzymes having similar allosteric properties (Monod et al. Reference Monod, Wyman and Changeux1965). A well-studied example is haemoglobin, which converts from a low-affinity tense state to a high-affinity relaxed state when oxygen binds (Perutz, Reference Perutz1989). The closed-channel form of the receptor would be equivalent to the low-affinity tense state of haemoglobin, and the open-channel form would be more nearly equivalent to the high-affinity relaxed state of haemoglobin (although the desensitized form is presumably the most equivalent).
One would expect the switch from a low-affinity tense state to a higher affinity, more relaxed state to underlie the gating process in other members of the Cys-loop receptor superfamily, because of their shared molecular design, implying conservation of function. Functional conservation has been demonstrated convincingly by the creation of chimaeric receptors that combine distinct regions of different superfamily members to build new receptors having the functional properties of both (Eiselé et al. Reference Eiselé, Bertrand, Galzi, Devillers-Thiéry, Changeux and Bertrand1993; Grutter et al. Reference Grutter, De Carvalho, Dufresne, Taly, Edelstein and Changeux2005). On the other hand, less basic aspects of the mechanism, such as the degree and nature of participation by each subunit, must certainly differ from one superfamily member to the next. In the case of homomeric channels, the existence of a tense closed state implies some degree of departure from fivefold symmetry, since a perturbed β-sheet arrangement would not be distributed equally among the subunits. So far, only symmetrical ‘closed state’ prokaryotic pentamers have been studied by X-rays (Hilf & Dutzler, Reference Hilf and Dutzler2008; Prevost et al. Reference Prevost, Sauguet, Nury, Van Renterghem, Huon, Poltevin, Baaden, Delarue and Corringer2012), but how closely the crystal structures of these proteins represent the physiological membrane-bound form remains to be seen (Gonzalez-Gutierrez & Grosman, Reference Gonzalez-Gutierrez and Grosman2010; Gonzalez-Gutierrez et al. Reference Gonzalez-Gutierrez, Lukk, Agarwal, Papke, Nair and Grosman2012; Zimmermann & Dutzler, Reference Zimmermann and Dutzler2011). The requirement for well-diffracting crystals, and detergent interactions with the gate, may make X-ray structures of the genuine closed (or resting) state difficult to achieve.
Another process that may be widely implemented is the tilting, or rocking about the extracellular-membrane domain interface (Fig. 14 b), to communicate the effect of ligand binding to the gate. As described here, ACh binding to αγ drives a rearrangement of the pair of β-sheets, an action that pushes the extracellular part of β outward and is accommodated by tilting of its membrane part. However alternatively, the rearrangement can be seen as changing the shape of the extracellular part of the αγ subunit, producing a small circumferential expansion (sketched in Fig. 14 a, right). Presumably in other superfamily members similar expansions occur when the ligand binds, and these also must be accommodated somehow in the membrane domain. A tilting action would be favoured over other possibilities because the membrane helices only pack together tightly at the intracellular membrane surface, where the resulting disturbance would be least. Whether the tilting is by means of a partner (equivalent to the β subunit) or distributed among several subunits, or by the ligand-binding subunit alone, any such action could be sufficient to destabilize a gate having the properties described in Section 4.4.
Finally, these structural studies of Torpedo postsynaptic membranes have shown us that the ACh receptor is designed to achieve its rapid gating action through small concerted movements of the subunits. Given the functional conservation, demonstrated in the studies using chimaeric receptors, other members of the Cys-loop superfamily must work by means of similar small concerted movements. In other words, the gating is invariably accomplished by means of small displacements around the pore. Since hydrophobic gating requires the least displacements to convert a closed pore into one that is fully open, it seems likely that gates of the kind used by the ACh receptor will be a common, if not a universal theme among members of this ion channel superfamily.
6. Conclusions
The ACh receptor at the neuromuscular junction has been probed intensively over the last few decades by a wide range of experimental approaches. Recently, the knowledge derived from X-ray structures of related proteins, and studies combining mutagenesis with measurements of receptor function have yielded some important new insights. Electron microscopy has complemented these approaches by providing a direct structural picture of the protein functioning in its natural membrane setting. Together the researches have led to a broad understanding of this model ligand-gated ion channel, and defined some key principles by which it transduces a chemical signal into an electrical signal with such high efficiency and speed.
The experiments on Torpedo membranes, highlighted in this review, show that the conformational change to open the channel takes place by means of small concerted movements of the subunits. The displacements linking the binding sites in the extracellular domain to the gate in the membrane domain are too small to necessitate significant readjustment of non-covalent bonds, which may slow down a conformational change. The whole transition can be explained in terms of an allosteric mechanism in which the binding of ACh relieves pre-existing tense structures in both domains.
In the membrane, the pore-lining helices have alternative ‘closed’ and ‘open’ configurations of nearly equal stability, allowing the controlled movements associated with the conformational change simply to tip the balance from the one to the other. In the closed configuration, the pore-lining helices bend inward making a constricting hydrophobic girdle. This girdle forms an energy barrier to ion permeation across the membrane, and so functions as the gate of the channel. The gate is destabilized by the conformational change, and helix straightening makes the pore wider and more polar, allowing the ions to pass readily through.
The basic allosteric mechanism, the means of communication between the binding sites and gate, and hydrophobic gating are properties of the ACh receptor that are likely to be shared by other members of the Cys-loop ion channel superfamily.
7. Acknowledgements
I would like to thank colleagues at the MRC Laboratory, Kyoto University and the Scripps Research Institute for many helpful discussions, and Richard Henderson for comments on the manuscript. I thank John Heuser for Fig 1a. The author's research is supported by grants from the US National Institutes of Health (grant no. GM61941), the Medical Research Council (MRC file reference number U105184294), and by funds from the Louis-Jeantet Foundation.

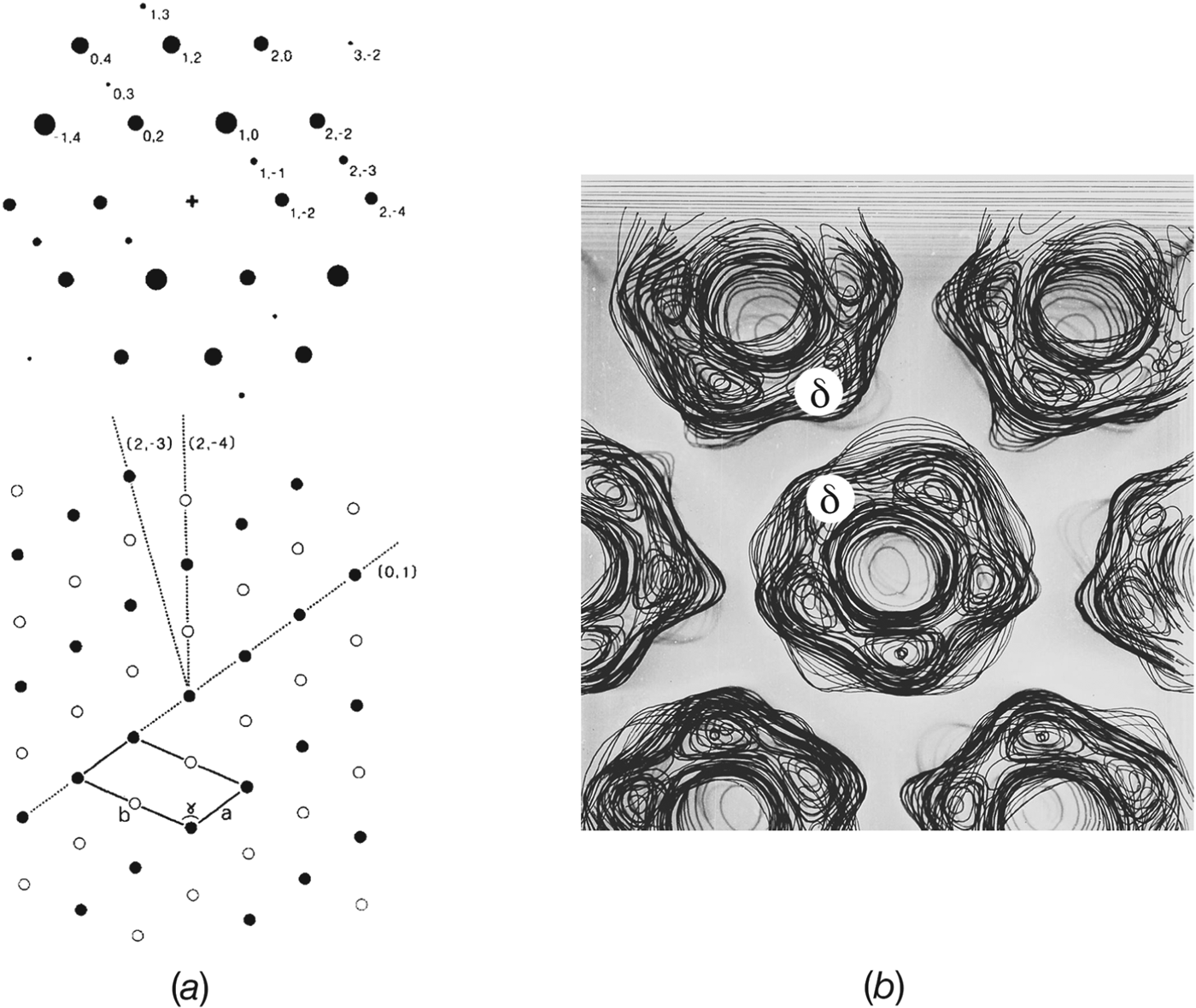

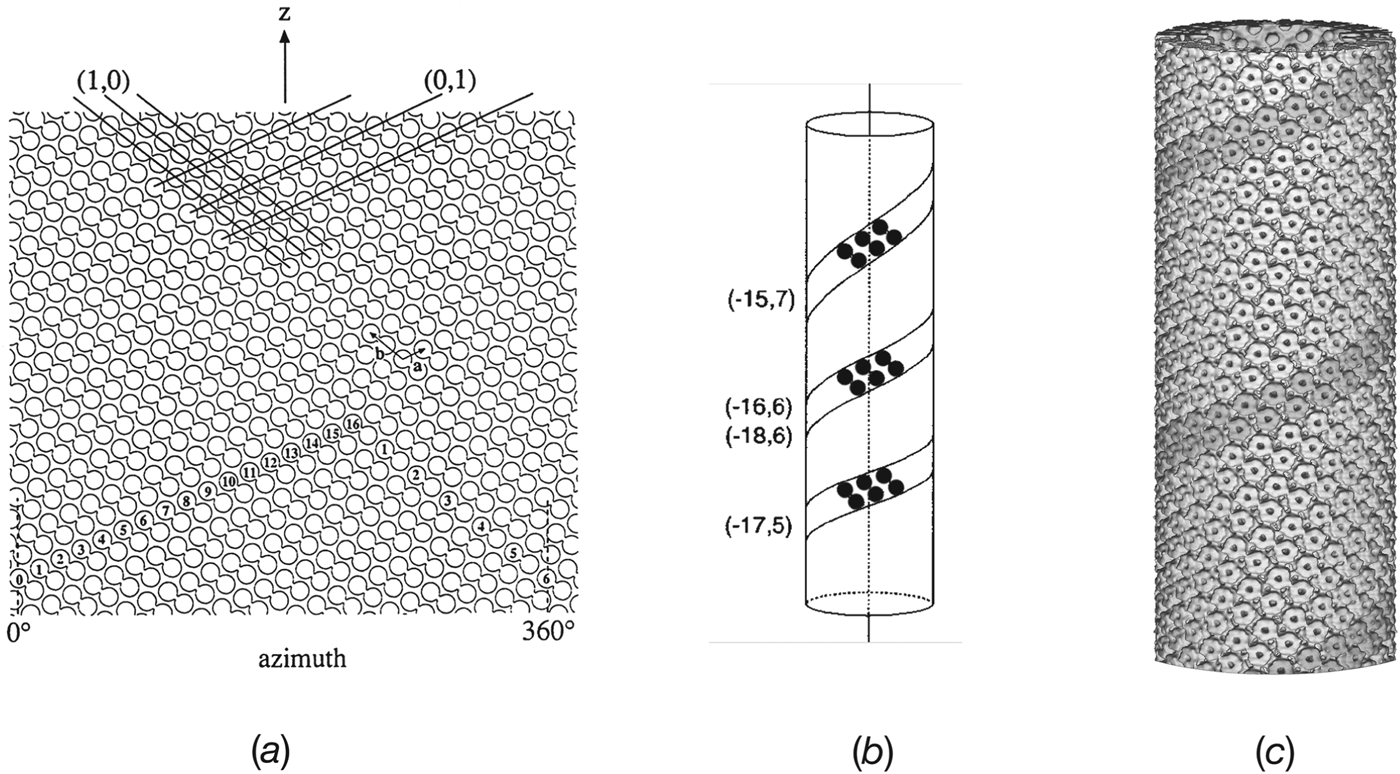
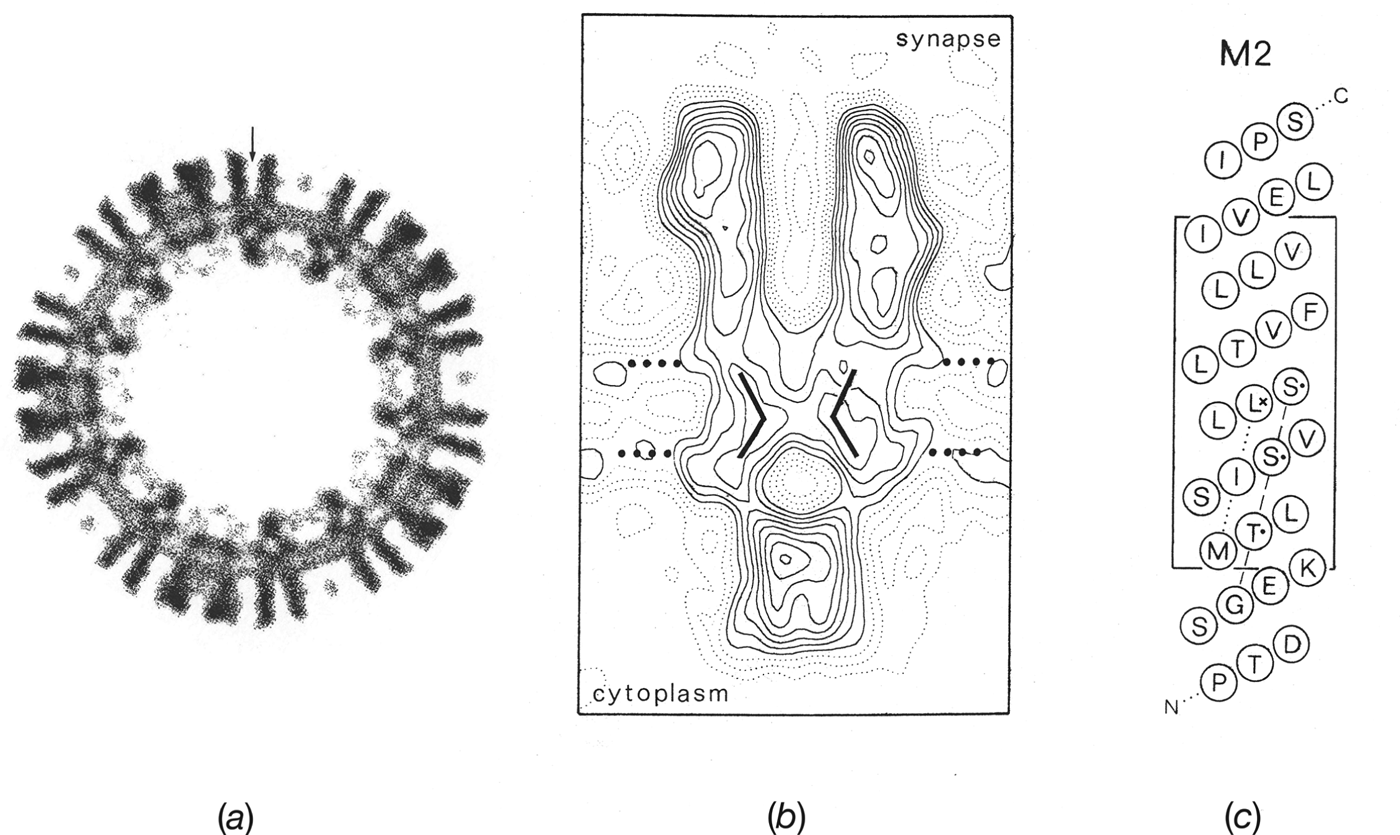
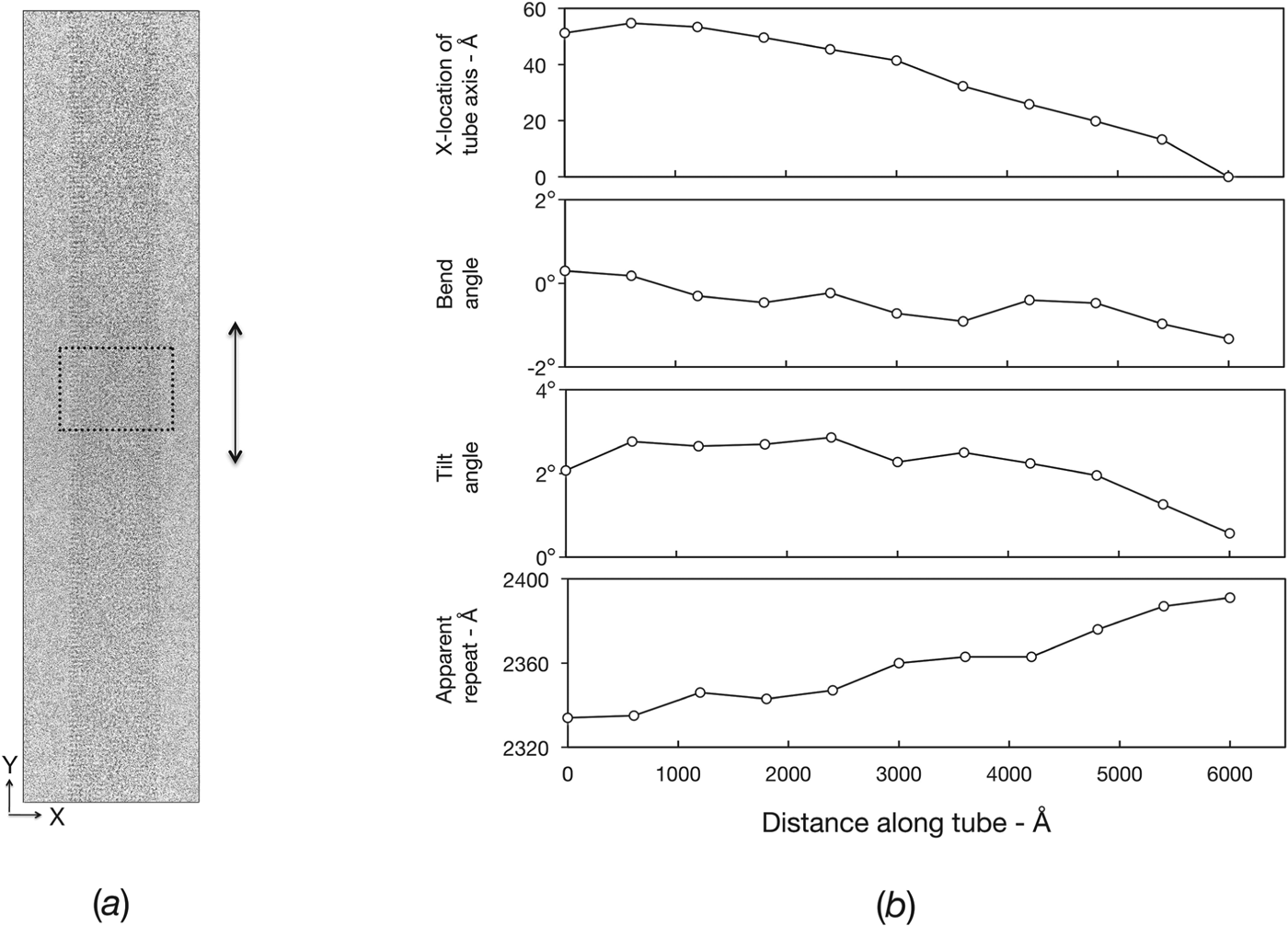


 Open access
Open access