


Introduction
CRISPR (clustered regularly interspaced short palindromic repeat)–Cas9 is a bacterial adaptive immune system, which has revolutionized life sciences through the introduction of a facile genome editing technology (Doudna and Charpentier, Reference Doudna and Charpentier2014; Jinek et al., Reference Jinek, Chylinski, Fonfara, Hauer, Doudna and Charpentier2012). In this system, the endonuclease Cas9 can be programmed with single-guide RNAs to site-specifically recognize and cleave any DNA sequence bearing a protospacer adjacent motif (PAM) sequence, which serves as a key recognition element across the genome. This enables genetic engineering of biological systems with unprecedented efficiency, resulting in transformative applications in the life sciences, including the fields of medicine and biotechnology.
During CRISPR–Cas9 activation, the DNA binds Cas9 by matching the guide RNA with one strand (the target strand, TS) and forming an RNA:DNA hybrid, while the non-target strand (NTS) is displaced. Two nuclease domains, HNH and RuvC, catalyze the cleavage of the TS and NTS, respectively. The Cas9 protein comprises a recognition (REC) lobe, which mediates the nucleic acid binding through three recognition domains (REC1–3), and a nuclease lobe including the RuvC and HNH catalytic cores (Fig. 1). At the protein C-terminus, a domain structurally similar to type II topoisomerase constitutes the PAM interacting (PI) region (Jiang and Doudna, Reference Jiang and Doudna2017; Nishimasu and Nureki, Reference Nishimasu and Nureki2017). Structural and biophysical studies have revealed that relevant conformational changes occur upon binding of the nucleic acids (Chen and Doudna, Reference Chen and Doudna2017). Specifically, RNA binding primes the protein for subsequent DNA binding (Jiang et al., Reference Jiang, Zhou, Ma, Gressel and Doudna2015), while the dynamics of the HNH exert conformational control over Cas9 nuclease activity (Sternberg et al., Reference Sternberg, Lafrance, Kaplan and Doudna2015). Upon DNA binding, the HNH domain undergoes a structural transition from an inactivated state, in which the catalysis is hampered, to an activated state prone for the cleavage of the TS. While the inactivated state has been well characterized via X-ray crystallography (Anders et al., Reference Anders, Niewoehner, Duerst and Jinek2014; Nishimasu et al., Reference Nishimasu, Ran, Hsu, Konermann, Shehata, Dohmae, Ishitani, Zhang and Nureki2014), high-resolution data for the activated state are missing. Indeed, the most complete X-ray structure of Cas9 from Streptococcus pyogenes in complex with the nucleic acids captured a pre-activated state of the system, with the HNH catalytic H840 located ~19.4 Å from the cleavage site on the TS (Fig. 1b) (Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016). Clearly, the activation of the system toward catalysis requires a further conformational change of the HNH domain (Fig. 1b, lower panel). Very recently, a novel cryo-EM structure has been reported at a 5.2 Å resolution (EMD-8236) (Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017), in which the HNH domain approaches REC1. The difficulty in experimentally capturing the active conformation of CRISPR–Cas9 and the associated conformational transitions reflects the ‘striking flexibility’ of the protein, as arising by the interplay with the nucleic acids, during recognition, association, and cleavage (Palermo et al., Reference Palermo, Miao, Walker, Jinek and Mccammon2016; Sternberg et al., Reference Sternberg, Lafrance, Kaplan and Doudna2015). As a support to the structural characterizations, extensive experimental efforts, including Forster Resonance Energy Transfer (FRET) and structural comparisons with homologous systems, identified the conformational requirements of a catalytically competent Cas9 (Chen and Doudna, Reference Chen and Doudna2017). In an initial study, Sternberg et al. used bulk FRET to reveal that the conformational dynamics of the HNH domain controls DNA cleavage (Sternberg et al., Reference Sternberg, Lafrance, Kaplan and Doudna2015). Subsequently, single-molecule FRET (smFRET) experiments characterized the conformational features of the activated HNH docked at the cleavage site (Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017), and also revealed that the high flexibility of the REC lobe facilitates the activation of the HNH domain (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Osuka et al., Reference Osuka, Isomura, Kajimoto, Komori, Nishimasu, Shima, Nureki and Uemura2018). Moreover, computational studies have contributed in understanding the conformational dynamics of HNH from the apo form of Cas9 up to the DNA-bound state (Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017; Palermo et al., Reference Palermo, Miao, Walker, Jinek and Mccammon2017a, Reference Palermo, Ricci, Fernando, Rajshekhar, Jinek, Rivalta, Batista and Mccammon2017b; Zuo and Liu, Reference Zuo and Liu2017). However, the detailed conformational rearrangements leading up to the catalytically active Cas9 protein have not been clarified. Specifically, it is unclear how HNH would dock at the cleavage site and, importantly, how the REC domains would facilitate this process mechanistically. Understanding the role of the REC lobe in the activation of Cas9 is of key importance for improving the system toward controlled functionality. Indeed, mutations within the REC lobe have been shown to reduce off-target cleavage events in newly evolved CRISPR–Cas9 systems (Casini et al., Reference Casini, Olivieri, Petris, Montagna, Reginato, Maule, Lorenzin, Prandi, Romanel, Demichelis, Inga and Cereseto2018; Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Kleinstiver et al., Reference Kleinstiver, Pattanayak, Prew, Tsai, Nguyen, Zheng and Joung2016).
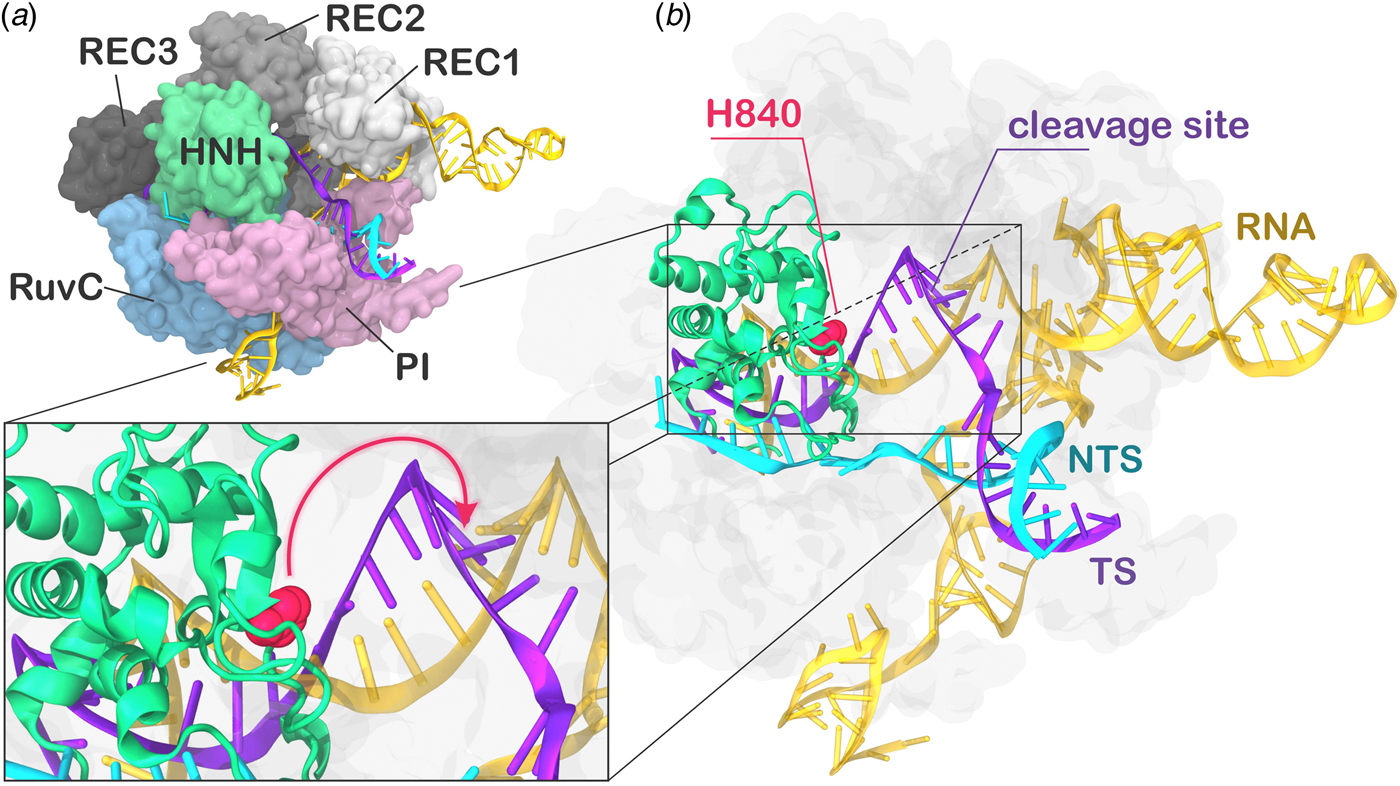
Fig. 1. (a) X-ray structure of the Streptococcus pyogenes CRISPR-Cas9 system (5F9R.pdb) in the pre-activated state (Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016). Cas9 is shown in molecular surface, highlighting protein domains in different colors. The RNA (orange), the target DNA (TS, magenta), and non-target DNA (NTS, cyan) strands are shown as ribbons. (b) The catalytic residue H840 (magenta) of the HNH domain is located ~19 Å from the cleavage site on the TS. A close-up view of the active site shows the additional conformational change needed to attain the formation of an activated state (shown using an arrow).
Here, multi-microsecond length molecular dynamics (MD) simulations reveal the activation process in ~16 µs of continuous simulation. Remarkably, the active conformation reached via MD simulations matches the structural and conformational transitions indicated by smFRET, while also being in accord with the newly available cryo-EM data. We show that the transition of the HNH domain depends on the structural remodeling of the REC domains, and is driven by favorable interactions with the REC lobe that form ‘on the fly’ during MD, eventually leading to the stable docking of HNH at the cleavage site. The observed conformational changes of the REC components pinpoint on an atomic scale how the recognition domains REC1–3 ‘sense’ nucleic acids, ‘regulate’ the HNH conformational change, and ultimately ‘lock’ HNH at the cleavage site, contributing to its catalytic activation. Finally, tight coupling between the REC lobe and the HNH domain is observed upon activation, ultimately ensuring catalytic competence (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017).
Results
Conformational transition of the HNH domain
Several studies have been conducted to understand the kinetics of the HNH conformational change, revealing a slow overall conformational transition (i.e. from the inactivated to activated state over milliseconds to seconds) (Raper et al., Reference Raper, Stephenson and Suo2018; Shibata et al., Reference Shibata, Nishimasu, Kodera, Hirano, Ando, Uchihashi and Nureki2017; Singh et al., Reference Singh, Sternberg, Fei, Doudna and Ha2016). However, the final conformational adjustment of the pre-activated crystal structure (Fig. 1) could rapidly occur (i.e. within micro-to-milliseconds), leading to an activated Cas9 (Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016). Due to the high flexibility of the HNH domain (Sternberg et al., Reference Sternberg, Lafrance, Kaplan and Doudna2015), the precise determination of its conformational change and the associated kinetics has been limited. In order to test the hypothesis of a fast sub-millisecond conformational change, in our previous study, we have carried out MD simulations of the pre-activated CRISPR–Cas9 (5F9R.pdb) (Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016) in an enhanced sampling regime (Palermo et al., Reference Palermo, Miao, Walker, Jinek and Mccammon2017a). We employed a Gaussian-accelerated MD (GaMD) approach, which can access conformational states of proteins and nucleic acids over milliseconds (and in some cases beyond) by running much shorter simulations (i.e. of hundreds of nanoseconds) (Miao and McCammon, Reference Miao and Mccammon2016a, Reference Miao and Mccammon2018; Miao et al., Reference Miao, Feher and Mccammon2015). During ~400 ns of GaMD, the catalytic H840 approached the scissile phosphate on the TS (i.e. phosphate −3) at a distance of ~15.0 Å, from its original location at ~19.4 Å (Fig. S1). Although the catalytic domain remained beyond the range required for catalysis, these simulations have highlighted the tendency for a fast conformational change of HNH in the late step of activation, in agreement with experiments (Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017; Shibata et al., Reference Shibata, Nishimasu, Kodera, Hirano, Ando, Uchihashi and Nureki2017; Sternberg et al., Reference Sternberg, Lafrance, Kaplan and Doudna2015). Encouraged by these outcomes, here we performed continuous simulations with the aim of capturing the activation process of the HNH domain in real time and on an atomic scale. Noteworthy, recent studies have employed enhanced sampling methods to access kinetic information (Stelzl and Hummer, Reference Stelzl and Hummer2017), while the efficiency of accelerated MD methodologies in exploring the conformational space has been used in conjunction with Markov models to construct solid kinetic models (Paul et al., Reference Paul, Wehmeyer, Abualrous, Wu, Crabtree, Schoneberg, Clarke, Freund, Weikl and Noé2017). However, while enhanced sampling MD describes well thermodynamic properties and conformational ensembles, it does not directly provide kinetic information, the latter is however preserved via unbiased simulations (Abrams and Bussi, Reference Abrams and Bussi2014; Miao and McCammon, Reference Miao and Mccammon2016b).
In order to obtain a continuous MD trajectory encompassing the microsecond time scale, we carried out MD on a specialized supercomputer – Anton-2 – that enables for micro-to-millisecond length simulations (Shaw et al., Reference Shaw, Grossman, Bank, Batson, Butts, Chao and Deneroff2014). CRISPR–Cas9 was simulated for ~16 µs revealing that the HNH domain approaches the cleavage site on the NTS after ~7 µs of MD and stably reaches a catalytically competent state after ~10 µs of MD (Fig. 2a–b). During the conformational transition, the distance between the Cα atom of the HNH catalytic residue (H840) and the scissile phosphate on the target DNA strand (i.e. the phosphate atom at position −3) shows a gradual decrease starting at ~7 µs, stabilizing at a distance of ~8 Å from the cleavage site after ~10 µs of MD (Fig. 2b, Fig. S2B). Remarkably, upon ~7 µs of unbiased MD, H840 approached the scissile phosphate at ~15.0 Å, reaching the configuration previously observed via GaMD simulations (Fig. S1). This result further establishes the ability of GaMD to capture long time scale events. However, longer GaMD trajectories might be required to access the complete conformational transition. In the final conformation, the catalytic H840 Cα is located ~8 Å from the cleavage site, while the imidazole side chain (and the reactive nitrogen) locates at a distance of ~5–6 Å from the cleavage site, priming the HNH domain for the hydrolysis of the TS (Fig. S3) (Jinek et al., Reference Jinek, Jiang, Taylor, Sternberg, Kaya, Ma, Anders, Hauer, Zhou, Lin, Kaplan, Iavarone, Charpentier, Nogales and Doudna2014). This configuration of the HNH domain agrees well with smFRET experiments (Raper et al., Reference Raper, Stephenson and Suo2018; Sternberg et al., Reference Sternberg, Lafrance, Kaplan and Doudna2015). Indeed, the S867–S355 distance, previously used to characterize the activated state of the HNH domain, reaches the experimental value of ~21 Å (Fig. S4). In the course of the simulation, we detect significant conformational plasticity of the REC lobe, in agreement with previous characterizations, performed using smFRET experiments (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017). Figure 2c–d reports the time evolution during MD of the E60–D273 and S960–S701 distances, which have been used in smFRET to distinguish the conformational states adopted by the REC2 and REC3 domains, respectively (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017). Here, we observe a conformational change of REC2 (Fig. 2a–c). After ~7 µs, simultaneously with the initiation of the conformational rearrangement of HNH, REC2 starts an outward transition that, upon ~11 µs of MD, results in an overall translation by ~8 Å relative to the starting position (i.e. the E60–D273 distance reaches ~40 Å from the initial 32.6 of the X-ray structure 5F9R.pdb). REC3 also shows significant conformational transitions (Fig. 2d). Indeed, the S960–S701 distance broadly fluctuates but overall increases by ~3–3.5 Å (from the initial value of 40.3 of the X-ray structure 5F9R.pdb), resulting in the opening of the groove hosting the RNA:DNA hybrid. Remarkably, in the final configuration the E60–D273 and S960–S701 distances reach values that well agree with the smFRET ranges (Figure 2c–d) (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017). Interestingly, the transitions of HNH and REC2 appear to be concerted, as it starts for both domains upon ~7 µs of simulation (Fig. 2b–c). However, HNH reaches an active conformation by 10 µs, while REC2 fully adopts the conformational transition at ~11 µs. Together, these observations suggest that the conformational changes of the recognition lobe assist the conformational activation of the HNH domain. Specifically, we observe an opening of REC3 and the mutual conformational adaptation of REC2 and HNH, whereby HNH approaches the cleavage site on the TS and REC2 moves apart with an outward translation of ~8 Å, enabling the active site to access the cleavage site.
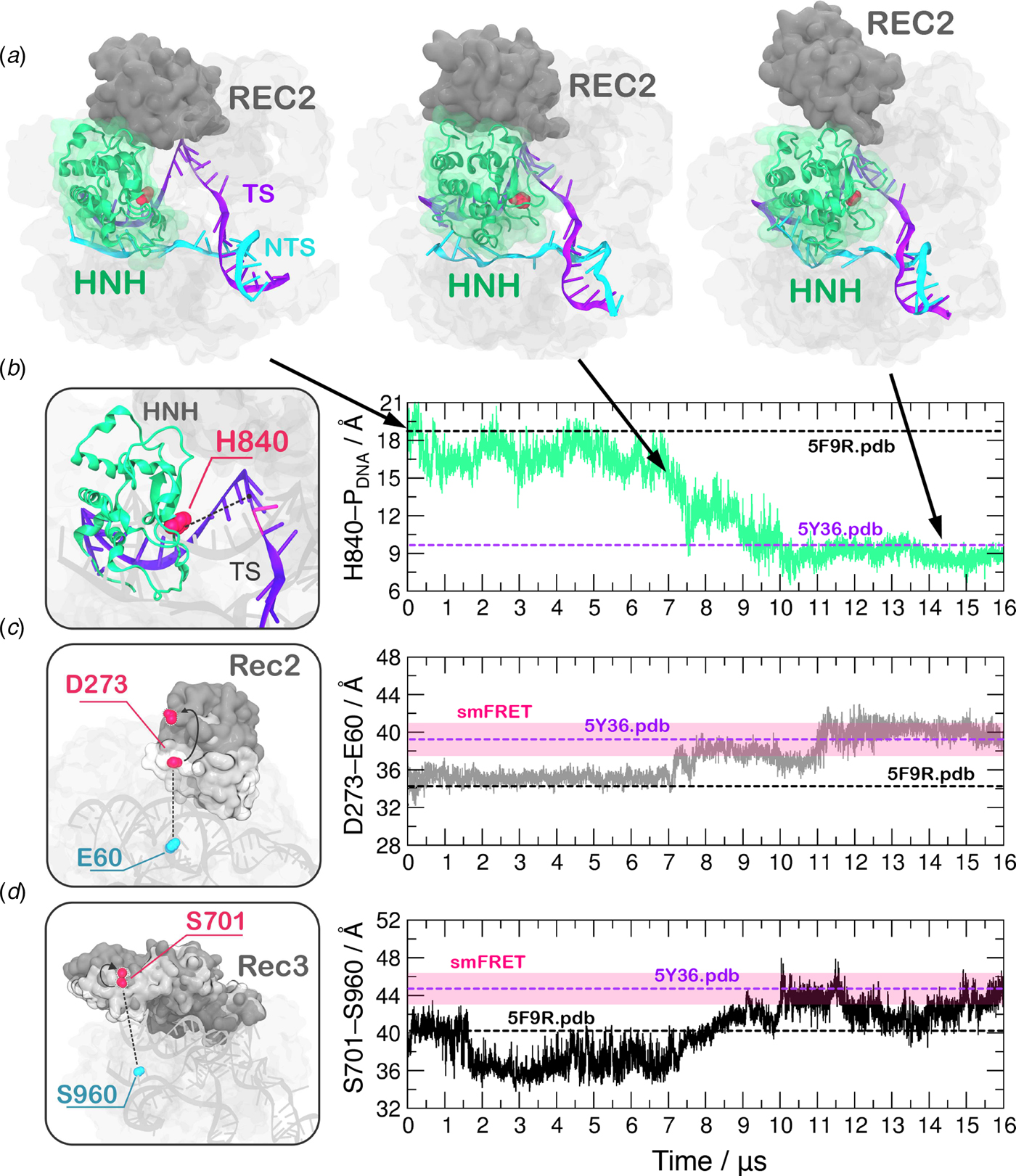
Fig. 2. (a) Conformational change of the HNH domain and structural adaptation of REC2 during ~16 µs of continuous MD simulations. (b–d) Time evolution along MD of: (b) the distance between H840 Cα and the scissile phosphate on the target DNA strand (phosphate at position −3), indicating the approach of the HNH domain at the cleavage site; (c) the distance between Cα atoms of E60 and D273 indicating the outward translation of REC2; (d) the distance between Cα atoms of S960 and S701 indicating the conformational change of REC3. Horizontal bars are used to indicate the value of the three distances in the X-ray structure of the pre-activated state (5F9R.pdb, starting configuration for MD) and in the model structure obtained via cryo-EM fitting of the EMD-8236 map (5Y36.pdb) (Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017; Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016). Transparent boxes are used to indicate the range assumed by the distances in single molecule Förster Resonance Energy Transfer (smFRET) experiments (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017).
A recent cryo-EM structure, solved at a 5.2 Å resolution (EMD-8236), shows that the HNH domain is closer to the recognition lobe than in the pre-cleavage state crystal structure (Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017). The final configuration obtained from MD is in good agreement with the all-atom model fitted in the EMD-8236 EM map (5Y36.pdb, Fig. 2b–d). In detail, in the 5Y36.pdb, the E60–D273 and S960–S701 distances reach values of 38.4 and 42.6 Å, respectively. MD simulations access these values by ~11 µs (for the E60–D273 distance, Fig. 2c) and ~10 µs (for the S960–S701 distance, Fig. 2d). In the fitted structure, the H840 Cα atom remains at a distance of 10.1 Å from the scissile phosphate (Fig. 2b), which is beyond the range required for catalysis (Fig. S3). In summary, the continuous MD simulations overall access the conformation obtained via cryo-EM, and further explore the conformational space. In this respect, it is worth noting given its limited resolution (5.2 Å), the available EMD-8236 structure can in fact represent multiple conformational states that could not be resolved (Nogales, Reference Nogales2016). As such, the fluctuations captured by the extensive MD simulations can be considered representative of the conformational landscape surrounding the EMD-8236 structure.
In order to track the large-scale collective motions of the REC lobe during the long time scale dynamics, we performed Principal Component Analysis (PCA) (Amadei et al., Reference Amadei, Linssen and Berendsen1993). This analysis captures how the protein domains move with respect to each other, highlighting conformational changes that are difficult to observe by visual inspection of the MD trajectory. As a result, PCA confirms large amplitude motions for HNH and the REC2–3 domains and the concerted nature of the conformational changes, while also revealing a conformational change for REC1 (Movie S1, Fig. S5). The latter moves in an opposite direction with respect to REC2 and REC3, toward the HNH domain. This result agrees well with the experimental vector map of global Cas9 conformational changes, from the RNA-bound state (4ZT0.pdb) to the DNA-bound state (5F9R.pdb, Fig. S4D) (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017). During the transition, the HNH domain forms a series of salt-bridge interactions with the REC lobe (Fig. 3). These ionic interactions mainly involve the REC1–2 regions, while HNH and REC3 remain separated from each other. This result well agrees with the novel cryo-EM structure (EMD-8236) (Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017). In this structure, HNH approaches the REC1 domain, but moves away from REC3 (Fig. S6). At ~7 µs of MD, charged residues of HNH and REC1 start engaging in ionic interactions, which increase in strength and number along the dynamics, stabilizing after ~10 µs, locking HNH at the TS (Fig. 3a). These interactions are key for the stable docking of HNH at the cleavage site. REC2 is also involved in ionic interactions, which are maintained along the simulation, indicative of the concerted conformational change of the two domains (Fig. 3b). Remarkably, interactions between HNH–REC1 are maintained in the activated state (i.e. during the last ~4 µs of MD), ensuring the positioning of HNH at the cleavage site. REC1 therefore cooperates with REC2 and REC3 in favoring HNH activation.
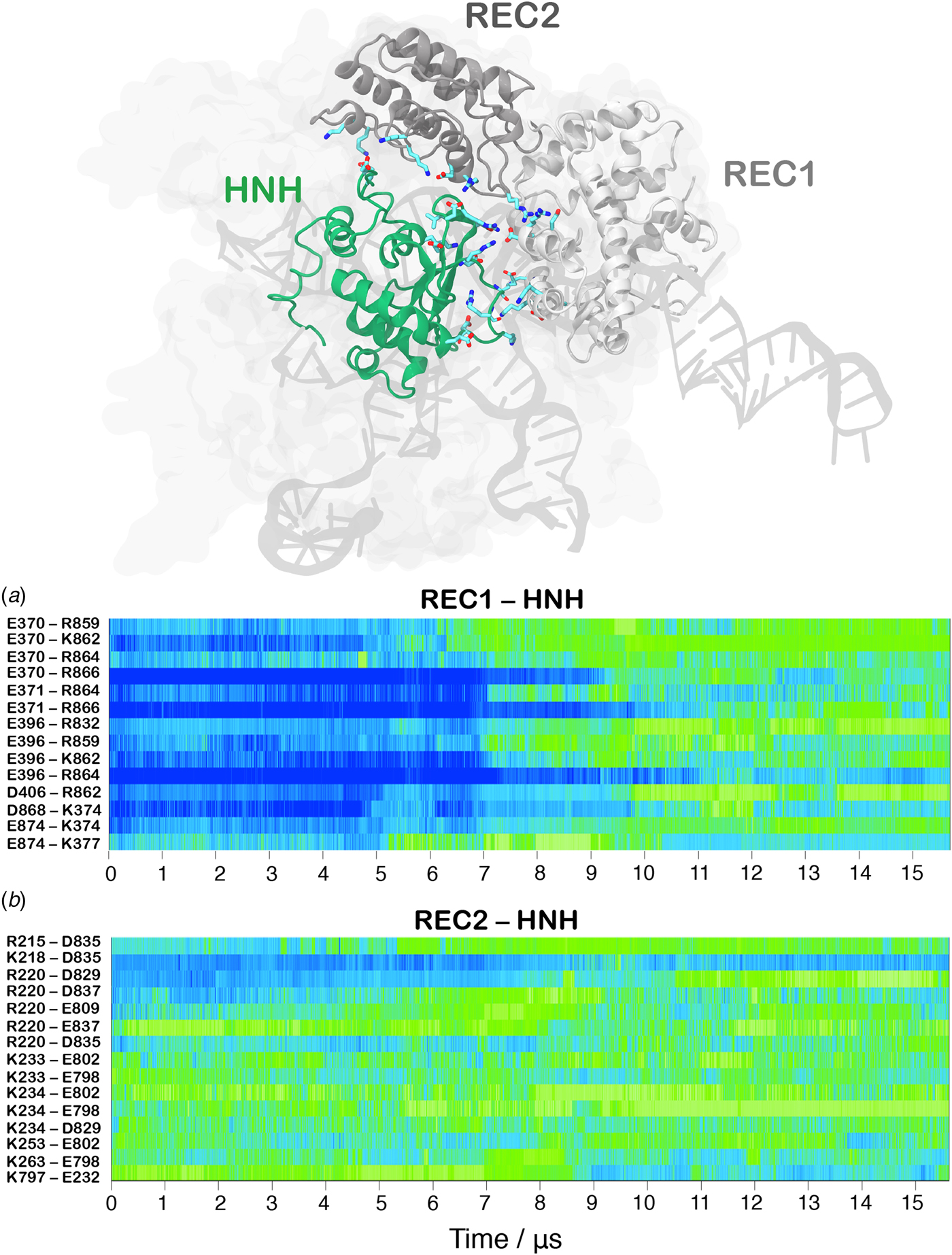
Fig. 3. Time evolution along ~16 µs of continuous MD simulations of the CRISPR-Cas9 system of the salt-bridge interactions established between the HNH and REC1 domains (a) and between the HNH and REC2 domains (b). Salt bridges are computed as distance between the center of mass of the oxygen atoms in the acidic side chain and center of mass of the nitrogen atoms in the basic side chain. The scale on the right shows the change in strength of salt-bridge interactions: from weak (blue) to strong (green). The most important ionic interactions established by HNH and the REC1/REC2 regions are also shown on the three-dimensional structure of the activated CRISPR–Cas9.
Insights on the on-target specificity
Here, we monitored the dynamic interactions of key positively charged residues, which belong to the HNH domain and intervene in the specificity of CRISPR–Cas9 (Fig. 4). Among them, K810 and K848 have been shown to reduce off-target cleavage events when mutated to alanine (Slaymaker et al., Reference Slaymaker, Gao, Zetsche, Scott, Yan and Zhang2016). K913 has been shown to bind the NTS in shorter time scales (i.e. ~0.8 µs), suggesting a possible role in facilitating the approach of HNH toward the scissile phosphate (Palermo et al., Reference Palermo, Miao, Walker, Jinek and Mccammon2016). In the pre-activated state (Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016), these residues do not interact with the nucleic acids. With the approach of HNH to the cleavage site on the TS (i.e. phosphate −3), K810 binds the phosphate −4 (Fig. 4a). Besides, K848 establishes multiple interactions with the nucleic acids and with the protein residues (Fig. 4b), finally contacting the DNA:RNA hybrid via the RNA phosphate backbone. Remarkably, in the 5Y36.pdb (all-atom model of the EMD-8236 map), K810 approaches the TS, while K848 binds the RNA backbone similarly to the configuration obtained upon ~15 µs of MD (at position −8) (Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017). K913 engages the NTS at several positions throughout the simulations (Fig. 4c). This finding confirms previous evidences from shorter MD simulations, suggesting that the interactions between the NTS and the loop formed by 906–918 residues would favor the approach of HNH toward the TS (Palermo et al., Reference Palermo, Miao, Walker, Jinek and Mccammon2016), and also clarifies smFRET experiments showing that the docking of HNH at the TS in its active configuration requires the presence of the NTS (Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017). Overall, these residues act as anchors of HNH at the DNA, favoring its docking at the TS cleavage site. As such, the disruption of these interactions with K810A/K848A substitutions may destabilize HNH at the cleavage site, thereby altering the dynamics of its conformational activation and consequently its cleavage activity (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Slaymaker et al., Reference Slaymaker, Gao, Zetsche, Scott, Yan and Zhang2016)
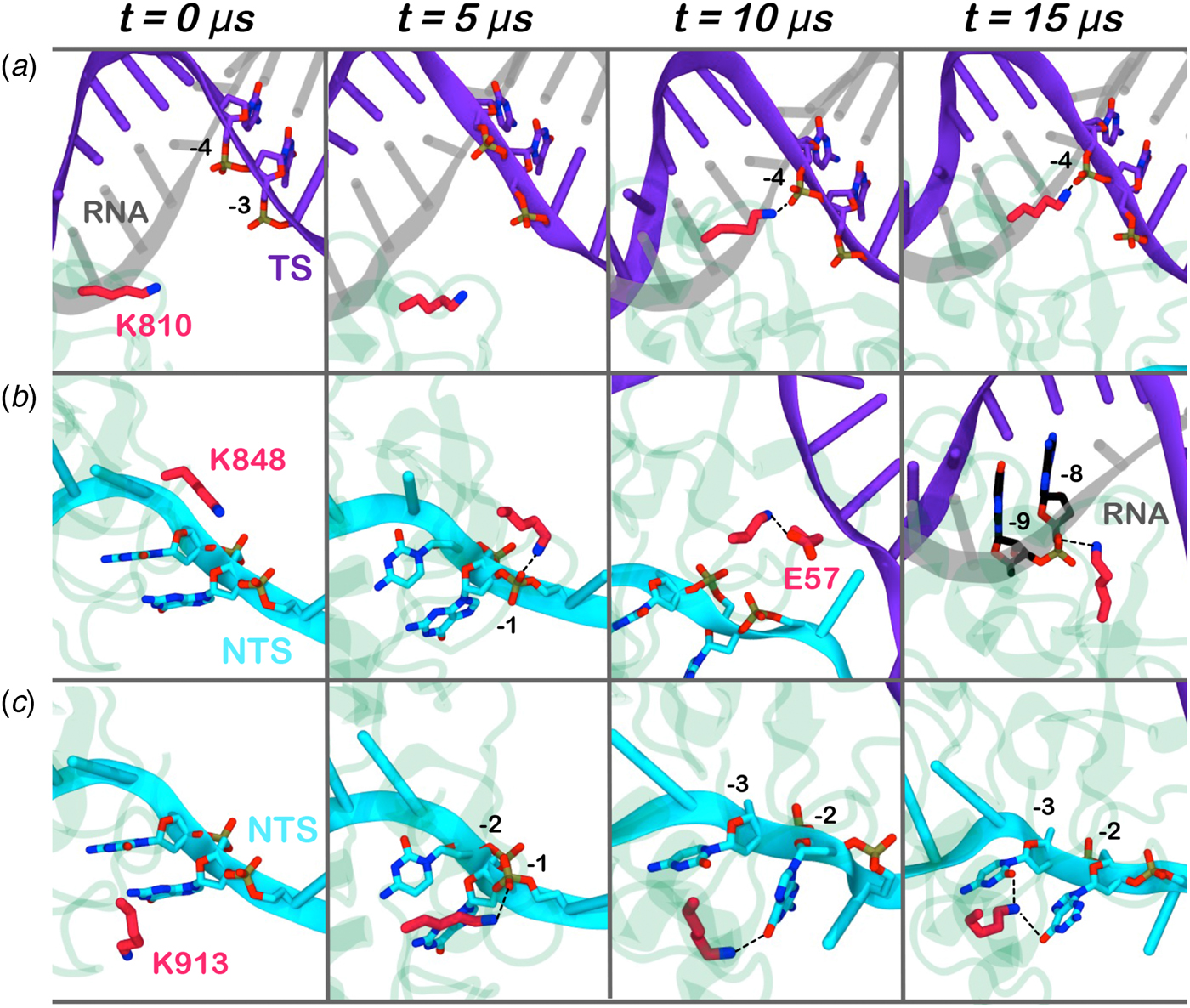
Fig. 4. Interaction established by the K810 (a), K848 (b), and K913 (c) residues of the HNH domain at ~0, 5, 10, 15 µs of MD. The RNA (gray), target DNA (TS, magenta), and non-target DNA (NTS, cyan) strands are shown as ribbons. The HNH domain (green) is shown as cartoon. Interacting residues are shown as sticks.
Multiple repeats along the conformational change
Continuous MD simulations of huge macromolecular systems reaching the microsecond time scale are challenging to be achieved via conventional supercomputers and the data obtained using Anton-2 are at the limit of the state-of-the-art technology. As such, the MD performed using Anton-2 in the present study, as well as in the previous studies by our and other research groups (Lindorff-Larsen et al., Reference Lindorff-Larsen, Maragakis, Piana and Shaw2016; Mouchlis et al., Reference Mouchlis, Bucher, Mccammon and Dennis2015), enabled to capture conformational changes and biophysical processes in a single trajectory. While this approach certainly preserves the kinetic features of the system, considering the stochastic nature of the biomolecular processes, multiple simulations are required to precisely estimate the time scale of the events. However, due to the high computational cost of each single trajectory, it is challenging to produce additional and/or longer simulation runs, recovering the observed events. In order to cope with this issue and to understand the statistical relevance of the observed conformational changes, we have extracted eight equally distributed snapshots (at times 1, 3, 5, 7, 9, 11, 13, and 15 µs of the Anton-2 trajectory) to perform independent MD simulations of ~300 ns in two replicas (reaching additional ~4.8 µs of aggregate statistics). Figures S6–S8 report the time evolution along the simulated runs of the H840–PDNA, E60–D273, and S960–S701 distances, enabling comparison with the continuous simulation obtained with Anton-2 (Fig. 2). As a result, in the systems extracted at 1, 3, and 5 µs, in which Cas9 assumes a pre-activated configuration, the computed distances stably oscillate around the initial values. The three distances also remain stable in the activated configurations (i.e. extracted at 11, 13, and 15 µs). In the case of the systems extracted at 7 and 9 µs, at which the system undergoes the conformational transition, we observe higher fluctuations in the time evolution of the computed distances. Moreover, in the hundreds-of-nanoseconds, these latter follow the trend observed in the microsecond length dynamics, which shows the transition toward the activated state (Figs S6–S8). This highlights the tendency for the conformational transition toward activation, supporting the results obtained with single trajectory obtained with Anton-2 and previous enhanced sampling simulations (Fig. S1) (Palermo et al., Reference Palermo, Miao, Walker, Jinek and Mccammon2017a). In order to understand the factors underlying this consistent behavior, we examined the structures extracted at times 1, 3, 5, 7, 9, 11, 13, and 15 µs of the Anton-2 trajectory and used as a starting point of the independent MD runs (Fig. S9). As a main difference between the pre-activated systems (i.e. extracted at times 1, 3, and 5 µs) and the system initiating the conformational transition (i.e. extracted at time 7 µs), in this latter we observe the approach of the side chains of charged residues belonging to HNH and REC1 (Fig. S9D), preluding their engagement in salt-bridge interactions, which will be fully formed at time 9 µs. At this point (9 µs, Fig. S9E), the outward transition of REC2 with respect to HNH is observed. Considering that these newly formed salt-bridge interactions are stably maintained in the activated state (i.e. for snapshots extracted at 11, 13, and 15 µs, as well as throughout the last ~6 µs of the continuous MD simulation, Fig. 3), these results suggest that the approach of the side chains of the charged residues of HNH and REC1 at ~7 µs is the first triggering event making the system prone to the larger conformational change leading to its final activation.
Cooperative domain dynamics in the activated state
The activated state reached via MD simulations remains stable over the last ~6 µs (Fig. 2), allowing its conformational dynamics to be analyzed. To characterize the inter-dependent motions of the protein residues and understand the cooperative dynamics of the Cas9 domains in the activated state, we employed the Lange and Grubmüller method, which captures the overall correlations (i.e. both linear and non-linear) among protein residues (Lange and Grubmuller, Reference Lange and Grubmuller2006). The generalized correlation (GC) matrix is a sensitive method for detecting the interdependence in the motions of two spatially distant residues, and provides a measure of how much the motion of one residue is dependent on that of another residue. GC matrices have been computed for the activated configurations, over the last ~4 µs of MD. Visual inspection of the GC matrix shows that the HNH domain strongly correlates with the REC lobe in the active state (Fig. 5a). A quantitative evaluation of the inter-dependent couplings established by HNH with the REC lobe has been obtained by computing the inter-domain GC scores (GCs), which accumulate the GC coefficients of the single residues over each protein domain of interest (Fig. 5b, full details in the Methods section). The GCs are a measure of the overall inter-domain correlations, indicating the most relevant coupled motions occurring across the system during the simulations. The GCs have been useful in the characterization of the allosteric effects in CRISPR–Cas9 (Palermo et al., Reference Palermo, Ricci, Fernando, Rajshekhar, Jinek, Rivalta, Batista and Mccammon2017b) and other protein/nucleic acid complexes (Ricci et al., Reference Ricci, Silveira, Rivalta, Batista and Skaf2016), as well as in describing the inter-dependent conformational dynamics between the protein and RNA components of the human spliceosome (Casalino et al., Reference Casalino, Palermo, Spinello, Rothlisberger and Magistrato2018). As a result, the HNH and REC3 domains display the highest overall GCs of 0.64 (Fig. 5b), which indicates a highly cooperative conformational dynamics in the activated state. Notably, both MD simulations and recent cryo-EM data (EMD-8236) (Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017) show that HNH and REC3 are not in direct contact with each other in the activated configuration (Figs 2d and 5c). Considering that the RNA:DNA hybrid locates within the groove formed by HNH and REC3 and directly interacts with both domains, the high inter-dependency of the domains motions suggests that the hybrid mediates the coupling between the two domains. This provides a plausible explanation for recent experimental observations, showing that REC3 allows for HNH nuclease activation upon recognition of the formation of the RNA:DNA hybrid (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017). High inter-domain correlations are also detected for the HNH and REC2 (GCs of 0.48). This inter-dependent conformational dynamics corroborate the experimental evidence for tight coupling between the HNH and REC2 domains to ensure catalytic competence (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017). Specifically, smFRET has shown reciprocal changes in the conformational states assumed by HNH and REC2 across multiple DNA substrates, indicating that the conformational dynamics of HNH and REC2 is tightly coupled to ensure catalysis. Notably, the HNH domain has high conformational plasticity, as shown by biochemical and biophysical experiments (Osuka et al., Reference Osuka, Isomura, Kajimoto, Komori, Nishimasu, Shima, Nureki and Uemura2018; Sternberg et al., Reference Sternberg, Lafrance, Kaplan and Doudna2015) and previous shorter MD simulations (Palermo et al., Reference Palermo, Miao, Walker, Jinek and Mccammon2016). Additionally, in the cryo-EM structure EMD-3277, the HNH and REC2 are observed at a lower resolution (8–10 Å) than the overall structure (6 Å), highlighting their mobility (Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016). In light of this evidence, the data reported here explain how these flexible protein components are coupled to enable function. Finally, high correlations are also observed between the REC2 and REC3 (GCs of 0.47), indicating that in the activated state, the REC lobe engages in a tight interplay to enable function.
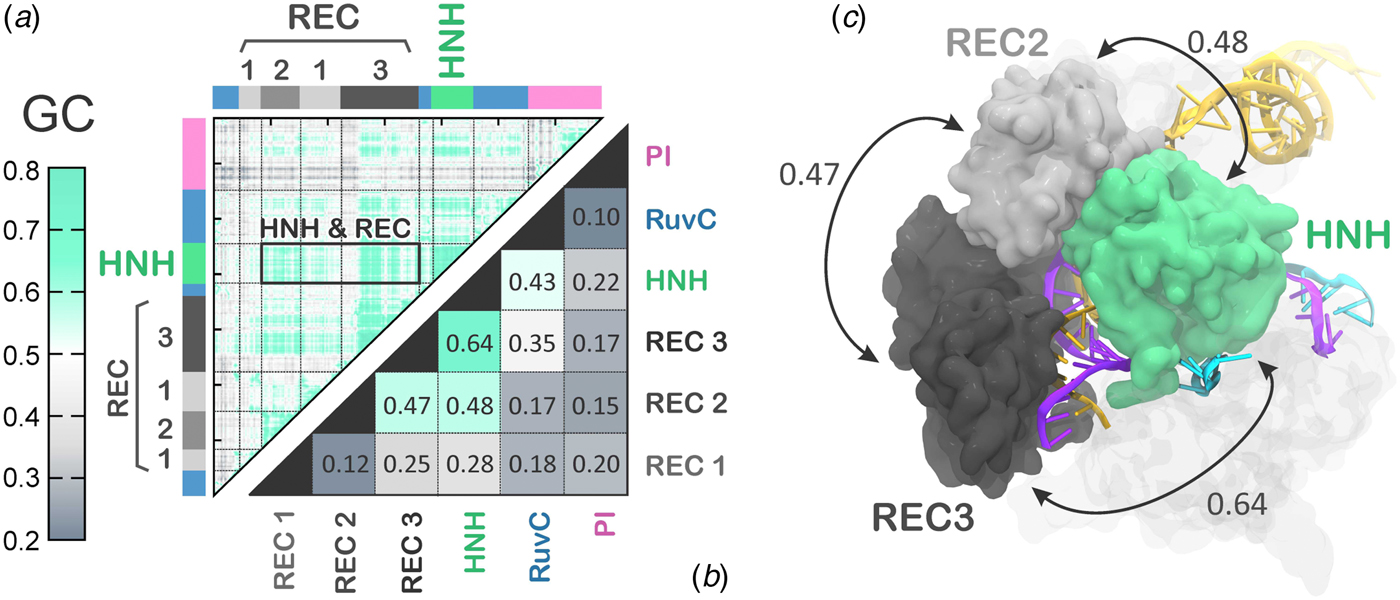
Fig. 5. (a) Matrix of the generalized correlation (GC), calculated over the last ~4 µs of MD of CRISPR–Cas9. The strength of the computed correlations is color-coded from green (highly correlated motions) to gray (poorly correlated motions). A box is used to highlight the high correlation between the HNH domain and the REC lobe. (b) Inter-domain GC scores (GCs) plotted for each Cas9 domain (REC1–3, HNH, RuvC, and PI domain) in a two-by-two matrix, measuring the strength of the overall correlation that each domain establishes with the others (details in the Method section). (c) The highest GCs are plotted on the three-dimensional structure of CRISPR–Cas9, revealing a tight coupling between HNH and the REC2–3 domains. Cas9 is shown in a molecular surface, highlighting the HNH and REC2–3 domains with different colors. The RNA (orange) and the target DNA (TS, magenta) forming a RNA:DNA hybrid, as well as the non-target DNA (NTS, cyan) are shown as ribbons.
Discussion
Activation mechanism
In the activation process of CRISPR–Cas9, a conformational change of the catalytic HNH domain is required to trigger catalysis (Fig. 1) (Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016). Here, MD simulations show that the docking of the HNH domain at the TS is facilitated by favorable interactions and by the conformational changes of the REC lobe. We show an opening of REC3 and an outward translation of REC2, which moves apart from its original position, resulting in the exposure of TS to the HNH active site for cleavage (Fig. 2). This result agrees well with the available experimental data (Chen and Doudna, Reference Chen and Doudna2017; Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017; Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017) and reveals that the activation of HNH is dependent on the conformational changes of the REC domains. Specifically, REC3 shows significant conformational transitions that result in the opening of the groove accommodating the RNA:DNA hybrid. This finding well agrees with recent cryo-EM data (Huai et al., Reference Huai, Li, Yao, Zhang, Cao, Kong, Jia, Yuan, Chen, Lu and Huang2017) and with the available smFRET experiments (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Dagdas et al., Reference Dagdas, Chen, Sternberg, Doudna and Yildiz2017), showing that the activation of the HNH domain for catalysis requires a change in the conformational state of REC3. In the activated configuration, the conformational dynamics of the HNH and REC3 domains is highly coupled (Fig. 5). Considering that both HNH and REC3 directly interact with the RNA:DNA hybrid, the high inter-dependency of the domains motions suggests that the hybrid mediates the coupling between the two domains. This finding provides further clarification on recent experiments, suggesting that REC3 would allow for HNH activation upon ‘sensing’ the formation of a complete RNA:DNA hybrid (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017). Accordingly, the simulations show that, while ‘sensing’ (i.e. interacting with) the RNA:DNA hybrid, REC3 affects the conformational dynamics of HNH and, in turn, its activation for cleavage. MD simulations also pinpoint a key role for the REC2 domain. During the conformational activation of HNH, REC2 rotates outward to enable the HNH to approach the cleavage site on the TS (Fig. 2). As well, the continuous simulation shows that during the activation process, the opening of REC2 and the approach of HNH to the TS start simultaneously (at ~7 µs, Fig. 2), indicating that the two domains cooperatively relocate to facilitate catalysis, likely in response to the ‘sensing’ of REC3 (Sung et al., Reference Sung, Park, Kim, Lee and Kim2018). In the activated state, HNH and REC2 show highly cooperative conformational dynamics (Fig. 5), corroborating the experimental evidence of their tight coupling to ensure catalytic competence (Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017). Taken together, these findings suggest that REC2 functions as a ‘regulator’ for HNH function (Chen and Doudna, Reference Chen and Doudna2017; Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017). MD simulations further reveal an unexpected role for REC1, which approaches HNH by establishing a series of ionic interactions, thereby enabling HNH to dock at the cleavage site on the TS. These observations suggest that REC1 acts as a ‘lock’ for HNH (Fig. 3). Overall, the tight interplay observed among the REC1–3 domains and the HNH domain reveals how the ‘sensor’ (REC3), the ‘regulator’ (REC2) of HNH activation (Chen and Doudna, Reference Chen and Doudna2017; Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017), together with the ‘lock’ (REC1), contribute to the formation of an activated CRISPR–Cas9 complex (Movie S2).
Conclusions
Here, we disclose the mechanism leading to the formation of a catalytically active CRISPR–Cas9 nuclease following substrate DNA binding. Unbiased all-atoms MD simulations reveal the approach of the HNH domain to the cleavage site on the TS in ~16 µs of continuous simulation. We characterize on an atomic scale the molecular determinants leading to the HNH conformational transition and the key role of the REC lobe in enabling the docking of HNH at the cleavage site. The remarkable conformational changes and the collective conformational dynamics of the REC1–3 domains enable ‘sensing’ of the nucleic acid binding, ‘regulation’ of the HNH conformational transition and ‘locking’ of the HNH at the cleavage site, eventually leading to the formation of a catalytically active CRISPR–Cas9 (Movie S2). Good agreement with biochemical experiments and structural data highlights the consistency of the activated conformation. As such, these simulations add detailed mechanistic information on the CRISPR–Cas9 activation process, clarifying the mechanistic role of the REC lobe components. Considering the key role of the REC lobe in the specificity of Cas9 (Casini et al., Reference Casini, Olivieri, Petris, Montagna, Reginato, Maule, Lorenzin, Prandi, Romanel, Demichelis, Inga and Cereseto2018; Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Kleinstiver et al., Reference Kleinstiver, Pattanayak, Prew, Tsai, Nguyen, Zheng and Joung2016), this study provides the foundation for understanding how the REC lobe domains control the cleavage of off-target sequences. Toward this aim, extensive characterization is currently ongoing in our laboratories. Overall, the knowledge on the HNH activation process and the role of the recognition lobe components, deciphered here, establishes a framework for future studies and novel structure-based engineering efforts for improved genome editing.
Material and method section
Structural model
MD simulations were based on the X-ray structure of the S. pyogenes Cas9 in complex with RNA and DNA (5F9R.pdb), solved at 3.40 Å resolution (Jiang et al., Reference Jiang, Taylor, Chen, Kornfeld, Zhou, Thompson, Nogales and Doudna2016). The model system was embedded in explicit waters, while Na+ ions were added to neutralize the total charge, leading to an orthorhombic periodic simulation cell of ~180 · 120 · 140 Å3, for a total of ~ 300 000 atoms.
MD simulations
A simulation protocol tailored for RNA/DNA endonucleases was adopted (Palermo et al., Reference Palermo, Cavalli, Klein, Alfonso-Prieto, Dal Peraro and De Vivo2015; Sponer et al., Reference Sponer, Bussi, Krepl, Banas, Bottaro, Cuhna, Gil-Ley, Pinamonti, Poblete, Jurecka, Walter and Otyepka2018), embracing the use of the Amber ff12SB force field, which includes the ff99bsc0 corrections for DNA (Perez et al., Reference Perez, Marchan, Svozil, Sponer, Cheatham, Laughton and Orozco2007) and the ff99bsc0 + χOL3 corrections for RNA (Banas et al., Reference Banas, Hollas, Zgarbova, Jurecka, Orozco, Cheatham, Sponer and Otyepka2010; Zgarbova et al., Reference Zgarbova, Otyepka, Sponer, Mladek, Banas, Cheatham and Jurecka2011). The TIP3P model was employed for explicit water molecules (Jorgensen et al., Reference Jorgensen, Chandrasekhar, Madura, Impey and Klein1983). The Åqvist (Aqvist, Reference Aqvist1990) force field was employed for Mg ions, as in previous studies on similar Mg-aided RNA/DNA nucleases (Casalino et al., Reference Casalino, Palermo, Rothlisberger and Magistrato2016; Reference Casalino, Palermo, Abdurakhmonova, Rothlisberger and Magistrato2017; Palermo et al., Reference Palermo, Stenta, Cavalli, Dal Peraro and De Vivo2013). An integration time step of 2 fs was used. All bond lengths involving hydrogen atoms were constrained using the SHAKE algorithm (Ryckaert et al., Reference Ryckaert, Ciccotti and Berendsen1977). Temperature control (300 K) was performed via Langevin dynamics (Turq et al., Reference Turq, Lantelme and Friedman1977), with a collision frequency γ = 1. Pressure control was accomplished by coupling the system to a Berendsen barostat (Berendsen et al., Reference Berendsen, Postma, Van Gunsteren, Dinola and Haak1984), at a reference pressure of 1 atm and with a relaxation time of 2 ps. The system was subjected to energy minimization to relax water molecules and counter ions, keeping the protein, the RNA, DNA, and Mg ions fixed with harmonic position restraints of 300 kcal/mol · Å2. Then, the system was heated up from 0 to 100 K in the canonical ensemble (NVT), by running two simulations of 5 ps each, imposing position restraints of 100 kcal/mol · Å2 on the above-mentioned elements of the system. The temperature was further increased up to 200 K in ~100 ps of MD in the isothermal–isobaric ensemble (NPT), reducing the restraint to 25 kcal/mol · Å2. Subsequently, all restraints were released and the temperature of the system was raised up to 300 K in a single NPT simulation of 500 ps. After ~1.1 ns of equilibration, ~10 ns of NPT runs were carried out allowing the density of the system to stabilize around 1.01 g/cm3. Finally, the production runs were carried out in the NVT ensemble. Simulations were performed using the GPU version of AMBER 16 (Case et al., Reference Case, Betz, Botello-Smith, Cerutti, Cheatham, Darden, Duke, Giese, Gohlke, Goetz, Homeyer, Izadi, Janowski, Kaus, Kovalenko, Lee, Legrand, Li, Lin, Luchko, Luo, Madej, Mermelstein, Merz, Monard, Nguyen, Nguyen, Omelyan, Onufriev, Roe, Roitberg, Sagui, Simmerling, Swails, Walker, Wang, Wolf, Wu, Xiao, York and Kollman2016; Salomon-Ferrer et al., Reference Salomon-Ferrer, Gotz, Poole, Le Grand and Walker2013) and the SPFP precision model (Le Grand et al., Reference Le Grand, Goetz and Walker2013). MD simulations were performed to equilibrate the system for ~120 ns prior to long time scale continuous MD. Indeed, the well-equilibrated system was used as a starting point for simulations on Anton-2 (described below). As well, eight equally distributed snapshots were extracted at times 1, 3, 5, 7, 9, 11, 13, and 15 µs of the Anton-2 trajectory and subjected to independent MD simulations in two replicas (i.e. ~300 ns for each snapshot in two replicas), reaching a total of additional 4.8 µs.
Long time scale MD simulations of the CRISPR–Cas9 complex were performed using Anton-2 (Shaw et al., Reference Shaw, Grossman, Bank, Batson, Butts, Chao and Deneroff2014), a special-purpose supercomputer for micro-to-millisecond length MD, starting from a well-equilibrated configuration, obtained after ~120 ns of conventional MD (see above). Simulations on Anton-2 were performed using the same force-field parameters used for conventional MD simulations. A reversible multiple time step algorithm (Tuckerman et al., Reference Tuckerman, Berne and Martyna1992) was employed to integrate the equations of motion with a time step of 2 fs for short-range non-bonded and bonded forces and 6 fs for the long-range non-bonded forces, for a total of ~16 µs of simulations. Simulations were performed at constant temperature (300 K) and pressure (1 atm) using the multigrator integrator as implemented in Anton-2 (Lippert et al., Reference Lippert, Predescu, Ierardi, Mackenzie, Eastwood, Dror and Shaw2013). The k-Gaussian split Ewald method (Shan et al., Reference Shan, Klepeis, Eastwood, Dror and Shaw2005) was used for long-range electrostatic interactions. Hydrogen atoms were added assuming standard bond lengths and were constrained to their equilibrium position with the SHAKE algorithm (Ryckaert et al., Reference Ryckaert, Ciccotti and Berendsen1977). Overall, molecular simulations have been carried out for an overall sampling time of >20 µs.
Principal component analysis
In PCA, the covariance matrix of the protein Cα atoms is calculated and diagonalized to obtain a new set of generalized coordinates (eigenvectors) to describe the system motions. Each eigenvector, called Principal Component (PC), is associated to an eigenvalue corresponding to the mean square fluctuation contained in the system's trajectory projected along that eigenvector. The first PC1 corresponds to the system's largest amplitude motion, and the dynamics of the system along PC1 is usually referred to as ‘essential dynamics’ (Amadei et al., Reference Amadei, Linssen and Berendsen1993). In this work, each conformation sampled during MD was projected into the collective coordinate space defined by the first two eigenvectors (PC1 and PC2), characterizing the essential conformational sub-space sampled by Cas9. Full details are in the SI.
Correlation analysis
Cross-correlations of residues in the Cas9 protein were computed based on mutual information between all Cα atoms using the generalized correlation analysis approach developed by Lange and Grubmüller (Lange and Grubmuller, Reference Lange and Grubmuller2006), which is explained in detail in the SI. The g_correlation module in the Gromacs 3.3 (Lindahl et al., Reference Lindahl, Hess and Van Der Spoel2001) package has been employed. Based on the computed correlations, the Generalized Correlation score (GCs) has been employed to define quantitatively the inter-dependent couplings established by HNH with the REC lobe. The GCs has been originally introduced by Ricci et al. in the study of the conformational dynamics of the PPARγ–RXRα nuclear receptor complex (Ricci et al., Reference Ricci, Silveira, Rivalta, Batista and Skaf2016), and used to describe the allosteric effects in CRISPR–Cas9 (Palermo et al., Reference Palermo, Ricci, Fernando, Rajshekhar, Jinek, Rivalta, Batista and Mccammon2017b) and the inter-dependent dynamics of the human spliceosome (Casalino et al., Reference Casalino, Palermo, Spinello, Rothlisberger and Magistrato2018). The GCs are computed by processing the GC matrix as follows. For each amino acid residue a GCs can be defined:
 $${\rm GC}{\rm s}_i = \mathop \sum \limits_{\,j \ne i}^N {\rm G}{\rm C}_{ij}, $$
$${\rm GC}{\rm s}_i = \mathop \sum \limits_{\,j \ne i}^N {\rm G}{\rm C}_{ij}, $$representing a measure of both the number and the intensity of the GC coefficients displayed by each residue. To filter non-trivial correlations and eliminate the noise due to uncorrelated motions, per-residue GCs were computed considering only highly positive correlations (GC ⩾ 0.60). GCs were used to detail the overall inter-domain correlations as follows: GCs were calculated for each residue i belonging to a specific protein domain (e.g. HNH, Rec1–3), with the residues j belonging to another protein domain of interest. Then, GCs were accumulated over all residues j of each specific Cas9 domain and normalized by the number of coupling residues i and j, which display GC ⩾ 0.60. This resulted in a set of per-domain GCs, ranging from 0 (not-correlated) to 1 (correlated), measuring the strength of the overall correlation that each domain establishes with the others. The overall GCs are a measure of the most important correlated motions among protein domains taking place in the simulations, which help in identifying how specific protein regions mechanistically intervene in the overall correlation network (Palermo et al., Reference Palermo, Ricci, Fernando, Rajshekhar, Jinek, Rivalta, Batista and Mccammon2017b; Ricci et al., Reference Ricci, Silveira, Rivalta, Batista and Skaf2016).
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0033583518000070
Acknowledgements
G.P. is supported by the Swiss National Science Foundation with the postdoctoral award P300PA_164698. J.S.C. is supported by the NSF Graduate Research Fellowship. J.A.M. thanks NIH, NSF, HHMI, NBCR, and SDSC grants. Anton-2 computer time was provided by the Pittsburgh Supercomputing Center through grant PSCA16035P from the NIH. The Anton-2 machine at PSC was generously made available by D.E. Shaw Research. Computer time for additional MD has been awarded by XSEDE via the grant TG-MCB160059. G.P. thanks Dr. Lorenzo Casalino for useful discussions.
Author contribution
G.P., J.S.C., and M.J. designed research; G.P. performed the simulations; G.P., C.G.R., and I.R. analyzed the data. V.S.B, J.A.D., and J.A.M. supervised research. G.P. wrote the manuscript. All authors provided critical commentary and contributed to editing the manuscript.
Conflict of interest
None.








 Open access
Open access