


Introduction
The ribosome translates a messenger RNA (mRNA) into a chain of amino acid residues to produce proteins. In order to proceed at a speed that is required to sustain life translation factors are needed to catalyze the process. Several of these factors bind in complex with GTP molecules and dissociate after GTP hydrolysis. Non-cleavable GTP analogues inhibit the participation of these factors in protein synthesis. These GTP-depending factors can be called translational GTPases (referred as trGTPases). The induction of GTP hydrolysis has for a long time been an important question.
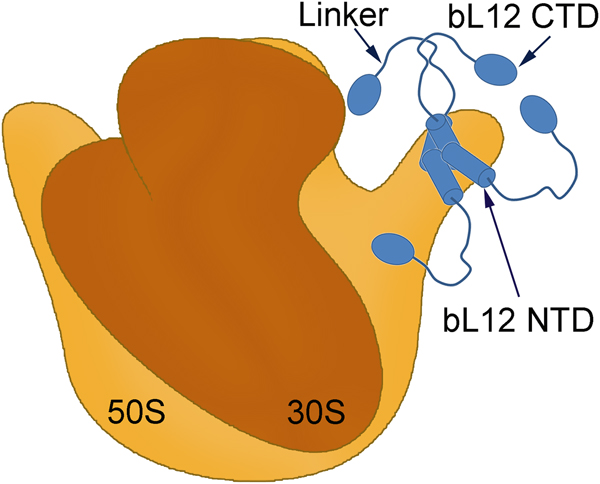
The two subunits of the ribosome in bacteria are usually composed of two large and one smaller RNA molecule and a large number of ribosomal proteins (See the book by Liljas & Ehrenberg, Reference Liljas and Ehrenberg2013). The early electron microscopic studies of the ribosomal large subunit identified a prominent stalk-like extension composed of the protein bL12 (Fig. 1) (Boublik et al., Reference Boublik, Hellmann and Roth1976; Lake, Reference Lake1976; Strycharz et al., Reference Strycharz, Nomura and Lake1978). Removing protein bL12 has detrimental effects on the functionality of the trGTPases (Kischa et al., Reference Kischa, Möller and Stöffler1971; Hamel et al., Reference Hamel, Koka and Nakamoto1972; Koteliansky et al., Reference Koteliansky, Domogatsky and Gudkov1978; Pettersson and Kurland, Reference Pettersson and Kurland1980; Mohr et al., Reference Mohr, Wintermeyer and Rodnina2002; Huang et al., Reference Huang, Mandava and Sanyal2010).
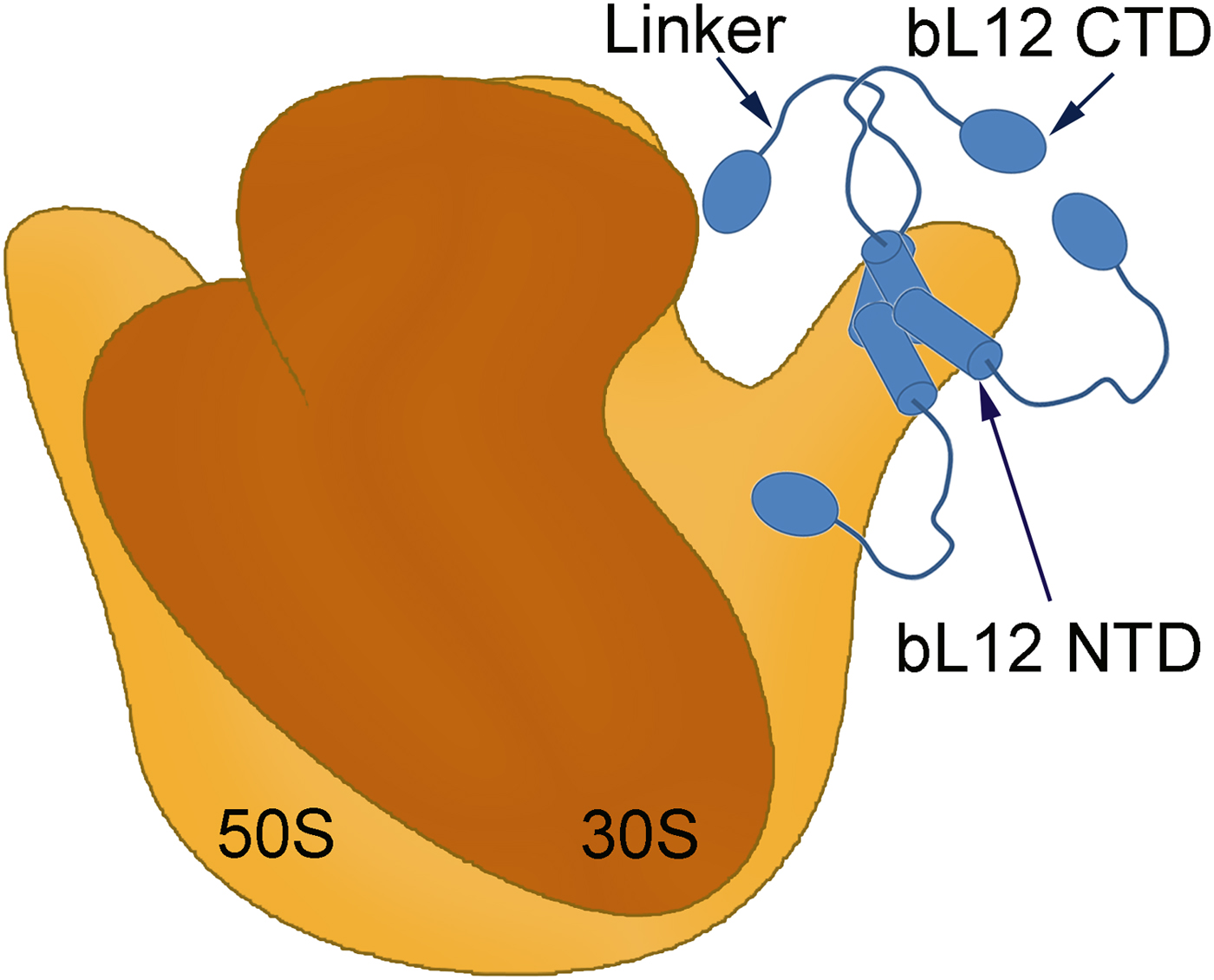
Fig. 1. A symbolic representation of the bacterial ribosome with its small (brown) and large (orange) subunits labelled as the 30S and 50S, respectively. Four copies of the protein bL12 are seen on the right-hand side (blue). Each monomer is composed of two domains (bL12 NTD and bL12 CTD) joined by a flexible linker. The bL12 NTD bind to the protein uL10 of the 50S subunit while the bL12 CTDs are free to move held only by the linker.
The organization of the ribosomal stalk proteins in different kinds of ribosomes and their role in relation to the GTP hydrolysis has remained enigmatic for decades but is gradually approaching an understanding.
The L7/L12 stalk
The protein bL12
The protein L7/L12 is an acidic protein that was first characterized in Escherichia coli, where its N-terminus can be acetylated (Terhorst et al., Reference Terhorst, Wittmann-Liebold and Möller1972). The acetylated form of L12 is referred to as L7; this is rarely seen in other species. A new naming system has been introduced to accentuate whether ribosomal proteins are universal (and get the prefix u) or occur only in bacteria (prefix b), archaea (a) or (e) for uniquely eukaryotic proteins (Ban et al., Reference Ban, Beckmann, Cate, Dinman, Dragon, Ellis, Lafontaine, Lindahl, Liljas, Lipton, McAlear, Moore, Noller, Ortega, Panse, Ramakrishnan, Spahn, Steitz, Tchorzewski, Tollervey, Warren, Williamson, Wilson, Yonath and Yusupov2014). According to this naming system, L7/L12 is now called bL12. For reviews on bL12 see Liljas, Reference Liljas1982, Reference Liljas1991; Gudkov, Reference Gudkov1997; Sanyal and Liljas, Reference Sanyal and Liljas2000; Wahl and Möller, Reference Wahl and Möller2002; Gonzalo and Reboud, Reference Gonzalo and Reboud2003.
The stoichiometry of bL12
The primary analysis of the stoichiometry of ribosomal proteins demonstrated that all proteins are present in one copy per ribosome except bL12, which was found in more than two copies per ribosome (Hardy, Reference Hardy1975; Subramanian, Reference Subramanian1975). Later chemical crosslinking and small angle X-ray scattering studies showed that bL12 forms dimers (Österberg et al., Reference Österberg, Sjöberg, Liljas and Pettersson1976). Thus the stoichiometry of the protein on the ribosome should always be in multiples of two. In E. coli there are two bL12 dimers (Österberg et al., Reference Österberg, Sjöberg, Pettersson, Liljas and Kurland1977), but some species (e.g. Deinococcus radiodurans, Thermus thermophilus) can have three dimers of bL12 (Ilag et al., Reference Ilag, Videler, McKay, Sobott, Fucini, Nierhaus and Robinson2005). The dimers of bL12 bind to the ribosome through protein uL10 (Diaconu et al., Reference Diaconu, Kothe, Schlunzen, Fischer, Harms, Tonevitsky, Stark, Rodnina and Wahl2005). The tight pentameric complex (bL12)4.uL10 was initially identified as a unique protein (called L8) not only in E. coli (Kaltschmidt and Wittmann, Reference Kaltschmidt and Wittmann1969, Reference Kaltschmidt and Wittmann1970; Stöffler, Reference Stöffler, Hasenbank, Bodley and Highland1974) but also in Bacillus stearothermophilus (Cohlberg and Nomura, Reference Cohlberg and Nomura1976). It was shown that by mixing bL12 with uL10 in vitro, the ‘unique protein’, L8, could be formed (Pettersson et al., Reference Pettersson, Hardy and Liljas1976).
Ribosomal location and basic structure
Electron microscopy showed that bL12 (formerly L7/L12) forms a stalk-like extension, the so-called L7/L12 stalk on the right-hand side of the large subunit when viewed from the subunit interface (Fig. 1; Boublik et al., Reference Boublik, Hellmann and Roth1976; Strycharz et al., Reference Strycharz, Nomura and Lake1978). This elongated protein has two structured domains: N-terminal domain (NTD) and C-terminal domain (CTD) interconnected by a highly flexible ‘hinge’ segment. The NTDs form the tight dimer and can be seen closely associated with the C-terminal long α8 helix of the protein uL10 (Diaconu et al., Reference Diaconu, Kothe, Schlunzen, Fischer, Harms, Tonevitsky, Stark, Rodnina and Wahl2005). Due to the flexible nature of the hinge, the CTDs could be crosslinked to various locations on the ribosome (Traut et al., Reference Traut, Dey, Bochkariov, Oleinikov, Jokhadse, Hamman and Jameson1995; Dey et al., Reference Dey, Bochkariov, Jokhadze and Traut1998). The flexibility explains why the bL12 stalk is poorly, if at all, visible in cryo-EM or crystallographic structures (Agrawal et al., Reference Agrawal, Heagle, Penczek, Grassucci and Frank1999; Ban et al., Reference Ban, Nissen, Hansen, Moore and Steitz2000; Yusupov et al., Reference Yusupov, Yusupova, Baucom, Lieberman, Earnest, Cate and Noller2001; Valle et al., Reference Valle, Zavialov, Li, Stagg, Sengupta, Nielsen, Nissen, Harvey, Ehrenberg and Frank2003; Harms et al., Reference Harms, Wilson, Schluenzen, Connel, Stachelhaus, Zaborowska, Spahn and Fucini2008; Gao et al., Reference Gao, Selmer, Dunham, Weixlbaumer, Kelley and Ramakrishnan2009; Schmeing et al., Reference Schmeing, Voorhees, Kelley, Gao, Murphy, Weir and Ramakrishnan2009; Voorhees et al., Reference Voorhees, Schmeing, Kelley and Ramakrishnan2010; Chen et al., Reference Chen, Feng, Kumar, Ero and Gao2013; Lin et al., Reference Lin, Gagnon, Bukley and Steitz2015).
The structure of the bL12 dimer
The bL12 dimer structures have been solved using X-ray crystallography and NMR (Fig. 2) (Wahl et al., Reference Wahl, Bourenkov, Bartunik and Huber2000; Bocharov et al., Reference Bocharov, Sobol, Pavlov, Korzhnev, Jaravine, Gudkov and Arseniev2004; Christodoulou et al., Reference Christodoulou, Larsson, Fucini, Connell, Pertinhez, Hanson, Redfield, Nierhaus, Robinson, Schleucher and Dobson2004). These structures clearly depict the structural features of different domains of bL12 and the basis of their dimerization. Additionally, they also demonstrate how the flexible hinge leads to different extended and compact conformations of the bL12 dimer.
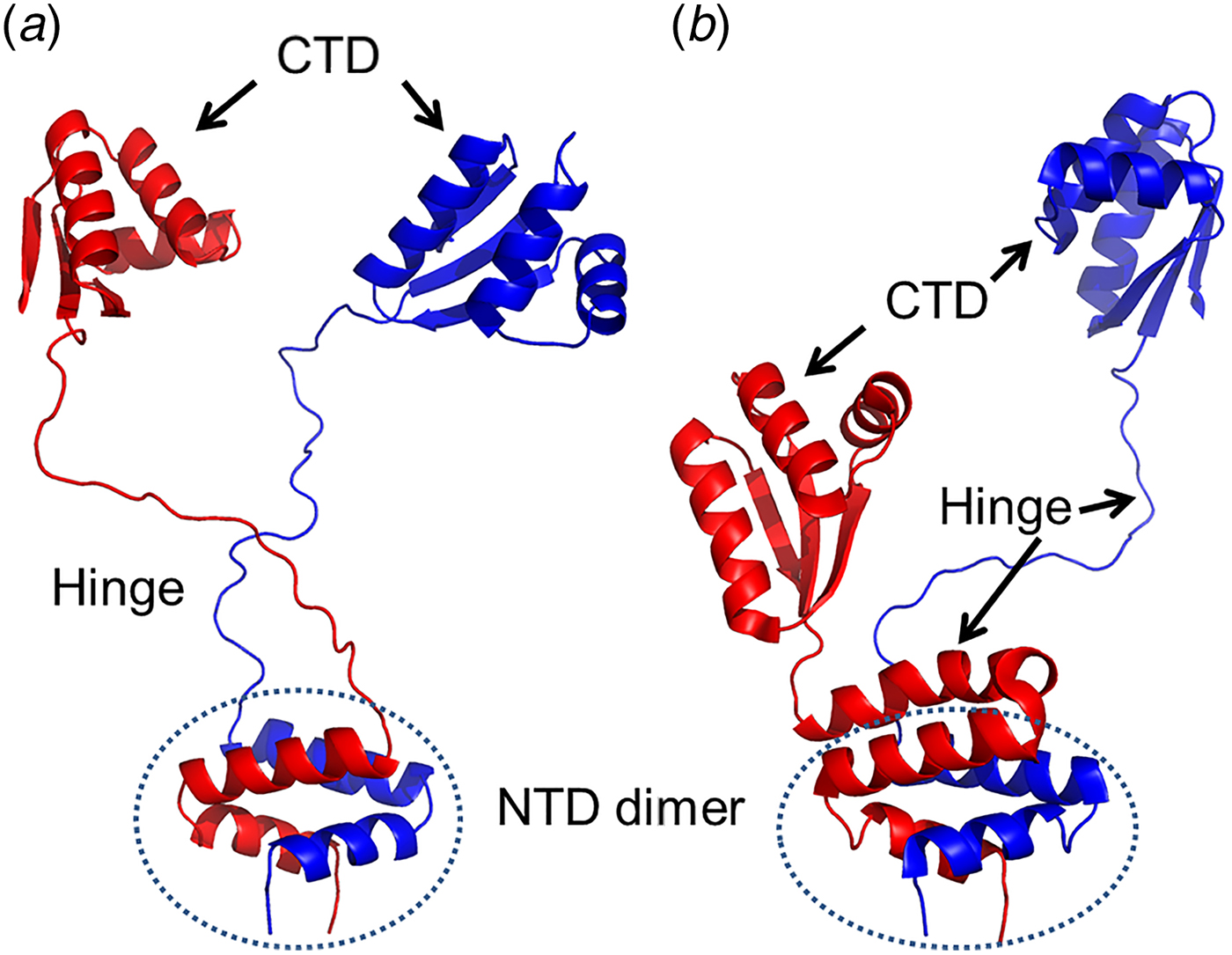
Fig. 2. Ribbon diagrams illustrating different conformations of the bL12 dimer. a, b – The conformations of the bL12 dimer from NMR (Bocharov et al., Reference Bocharov, Sobol, Pavlov, Korzhnev, Jaravine, Gudkov and Arseniev2004; PDB ID: 1RQT and 1RQS) showing both hinges extended (a) and one in extended and one in a compact state (b). The different domains are marked as CTD (C-terminal domain), NTD dimer (N-terminal domain dimer) and hinge.
The NTD of the bL12 monomer is 35 amino acid long in Escherichia coli and composed of two α-helices (α1 and α2; Bocharov et al., Reference Bocharov, Gudkov and Arseniev1996; 1998). The helices of the NTDs form a hook around its corresponding partner to form the bL12 dimer. In a bL12 dimer, the monomers are arranged in an anti-parallel fashion with respect to each other (Fig. 2; Sanyal and Liljas, Reference Sanyal and Liljas2000; Wahl et al., Reference Wahl, Bourenkov, Bartunik and Huber2000; Bocharov et al., Reference Bocharov, Sobol, Pavlov, Korzhnev, Jaravine, Gudkov and Arseniev2004).
The CTD of bL12 (residue number 52–120 in E. coli) was the first component of the ribosome for which the crystal structure was determined at atomic resolution (Leijonmarck and Liljas, Reference Leijonmarck and Liljas1987; Leijonmarck et al., Reference Leijonmarck, Eriksson and Liljas1980). It has a globular structure with a conserved, positively charged surface, which interacts with the trGTPases (Leijonmarck and Liljas, Reference Leijonmarck and Liljas1987; Wahl et al., Reference Wahl, Bourenkov, Bartunik and Huber2000; Bocharov et al., Reference Bocharov, Sobol, Pavlov, Korzhnev, Jaravine, Gudkov and Arseniev2004). Two α-helices (α4 and α5) of the bL12 CTDs constitute the universal translation factor interaction sites (Savelsbergh et al., Reference Savelsbergh, Mohr, Kothe, Wintermeyer and Rodnina2005; Helgstrand et al., Reference Helgstrand, Mandava, Mulder, Liljas, Sanyal and Akke2007).
NTD and CTD of bL12 are joined by a flexible link or hinge involving residues 36–51 (Bushuev et al., Reference Bushuev, Gudkov, Liljas and Sepetov1989; Bocharov et al., Reference Bocharov, Sobol, Pavlov, Korzhnev, Jaravine, Gudkov and Arseniev2004; Mulder et al., Reference Mulder, Bauakaz, Lundell, Liljas, Akke and Sanyal2004). The length and composition of the bL12 hinge, rich in alanyl and glycyl resides, varies considerably between different organisms (Liljas et al., Reference Liljas, Kirsebom, Leijonmarck, Hardesty and Kramer1986; Bushuev et al., Reference Bushuev, Gudkov, Liljas and Sepetov1989). The hinge is the source of the flexibility of bL12 proteins. From NMR studies it has been suggested that this hinge may be fully extended. Alternatively, it may also transiently form an α-helix (Fig. 2a, b) (Bocharov et al., Reference Bocharov, Sobol, Pavlov, Korzhnev, Jaravine, Gudkov and Arseniev2004). In the x-ray structure of bL12 from T. maritima (Sanyal and Liljas, Reference Sanyal and Liljas2000; Wahl et al., Reference Wahl, Bourenkov, Bartunik and Huber2000), where the crystallographic asymmetric unit contains two complete bL12 and two NTDs, both the hinges (α3) were seen to adopt the compact helical conformations.
NMR spectra of large subunits or whole ribosomes show that bL12 CTD behaves, as if it would be essentially free in solution (Tritton, Reference Tritton1980; Gudkov et al., Reference Gudkov, Gongadze, Bushuev and Okon1982; Cowgill et al., Reference Cowgill, Nichols, Kenny, Butler, Bradbury and Traut1984; Bushuev et al., Reference Bushuev, Gudkov, Liljas and Sepetov1989; Mulder et al., Reference Mulder, Bauakaz, Lundell, Liljas, Akke and Sanyal2004; Bernadó et al., Reference Bernadó, Modig, Grela, Svergun, Tchorzewski, Pons and Akke2010). Furthermore, NMR spectra of bL12 in 50S subunits or 70S ribosomes indicate that only two of the four hinge regions are mobile (Christodoulou et al., Reference Christodoulou, Larsson, Fucini, Connell, Pertinhez, Hanson, Redfield, Nierhaus, Robinson, Schleucher and Dobson2004; Mulder et al., Reference Mulder, Bauakaz, Lundell, Liljas, Akke and Sanyal2004). It is not known whether the flexible bL12 hinges belong to the same or separate dimers (Sanyal and Liljas, Reference Sanyal and Liljas2000).
The interactions of bL12 with uL10
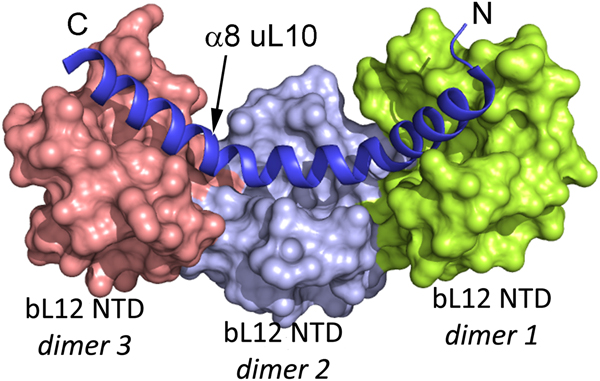
The two or three NTD dimers of bL12 (Ilag et al., Reference Ilag, Videler, McKay, Sobott, Fucini, Nierhaus and Robinson2005) all bind to the C-terminal helix (α8) of protein uL10 (Fig. 3; Diaconu et al., Reference Diaconu, Kothe, Schlunzen, Fischer, Harms, Tonevitsky, Stark, Rodnina and Wahl2005). This helix has a short sequence motif that repeats twice or three times. Each such motif can bind a dimer of bL12. From investigations of the bL12 dimer (Bocharov et al., Reference Bocharov, Gudkov and Arseniev1996; Mulder et al., Reference Mulder, Bauakaz, Lundell, Liljas, Akke and Sanyal2004) or the pentameric L8 complex (Bocharov et al., Reference Bocharov, Gudkov, Budovskaya and Arseniev1998) it is clear that the bL12 CTDs neither interact with each other nor with their NTDs.
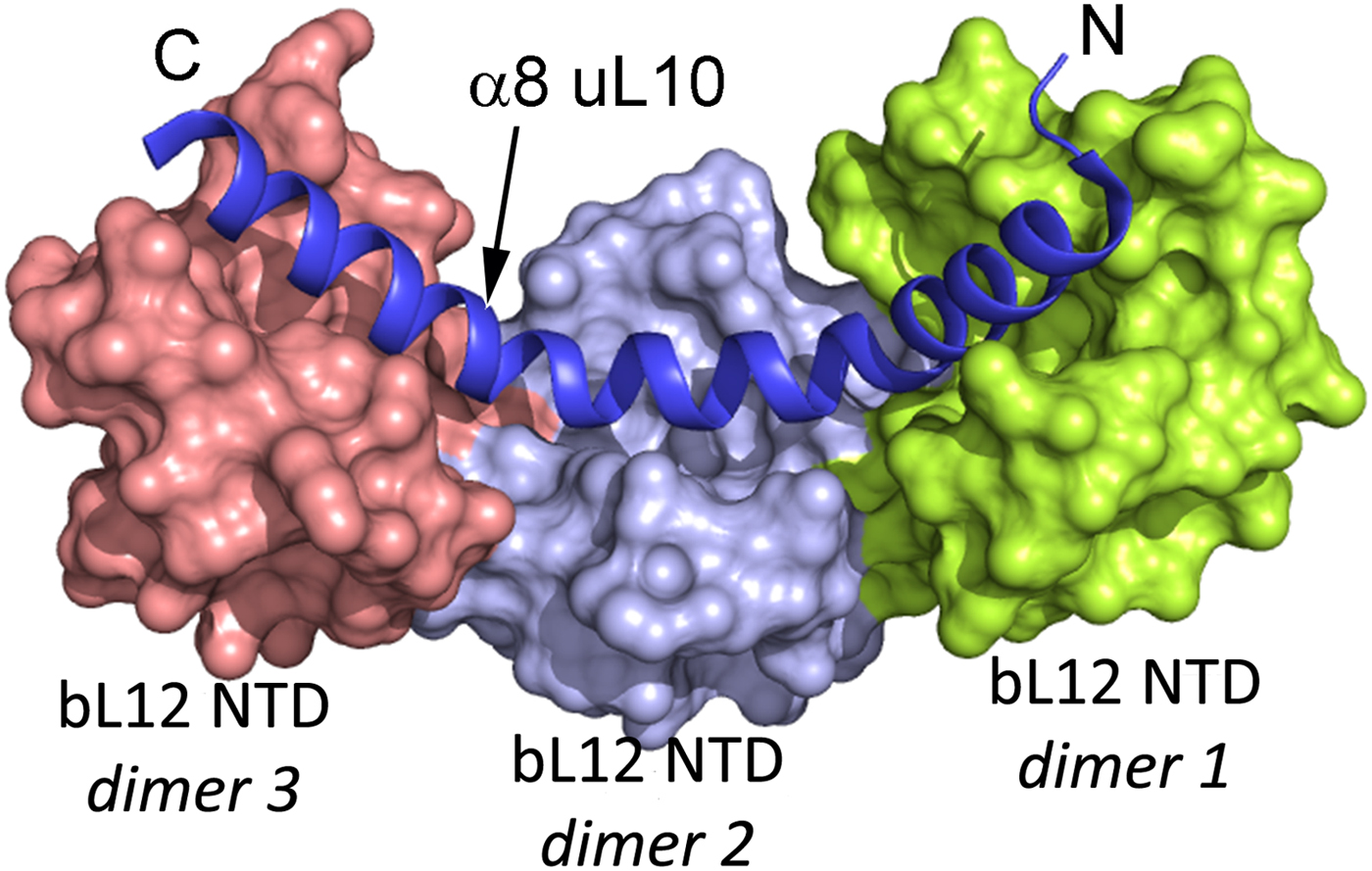
Fig. 3. The structure of three dimers of bL12 NTDs bound to α8 of uL10 (blue) extracted from PDB: 1ZAX.
The ‘stalk’ on archaeal or eukaryotic ribosomes
The P proteins
The proteins corresponding to bacterial bL12 and uL10 in eukaryotes and archaea can be phosphorylated and are therefore called P-proteins (Zinker and Warner, Reference Zinker and Warner1976). The protein P0 corresponds to uL10 (Wool et al., Reference Wool, Chan, Gluck and Suzuki1991). Its N-terminal part is an orthologue of the bacterial form of uL10 (Shimmin et al., Reference Shimmin, Ramirez, Matheson and Dennis1989). In eukarya there are two related proteins, eP1 and eP2, which functionally correspond to bL12 in that they bind to the long C-terminal helix of uL10 as two heterodimers (Tchorzewski et al., Reference Tchorzewski, Boguszewska, Dukowski and Grankowski2000a; Maki et al., Reference Maki, Hashimoto, Zhou, Naganuma, Ohta, Nomura, Robinson and Uchiumi2007) to form the bL12 corresponding stalk of the large subunit (Uchiumi et al., Reference Uchiumi, Wabha and Traut1987; for a review see Liljas, Reference Liljas1991). eP1/eP2 have no sequence similarity with bL12 but share the acidic nature. In archaea, there is only one protein related to eP1 and eP2. It is called aP1. Like in eukaryotes, the archaeal uL10 can bind three dimers of aP1. In addition, the C-terminal region of P0 in both archaea and eukaryotes contains an extension of about 30 residues identical with the C-terminal residues of P1 and P2 (Santos and Ballesta, Reference Santos and Ballesta1995).
The eP1 and eP2 have NTDs of about 70 residues followed by a flexible region of about 30 amino acid residues (Wool et al., Reference Wool, Chan, Gluck and Suzuki1991; Bailey-Serres et al., Reference Bailey-Serres, Vangala, Szick and Lee1997; Tchorzewski, Reference Tchorzewski2002). This hinge-region is acidic and highly variable in length and amino acid sequence but is rich in alanyl and glycyl residues. The C-termini of all P proteins has a conserved stretch of 10–13 residues. Several protein kinases can phosphorylate a conserved seryl residue in this region (Ballesta et al., Reference Ballesta, Rodriguez-Gabriel, Bou, Briones, Zambrano and Remacha1999). Like for bL12, the C-terminal regions of the eP1/eP2-proteins interact with trGTPases (Bargis-Surgey et al., Reference Bargis-Surgey, Lavergne, Gonzalo, Vard, Filhol-Cochet and Reboud1999). Similar to bL12, the NTDs of aP1 in archaea and eP1 and eP2 in eukaryotes are involved in the dimerization (Fig. 4) as well as binding to uL10 (Ballesta and Remacha, Reference Ballesta and Remacha1996). The mode of dimerization involving helical NTDs is more complex in archaea and eukaryotes than in bacteria and involves four α-helices from each molecule than two α-helices in bacteria (Fig. 4).
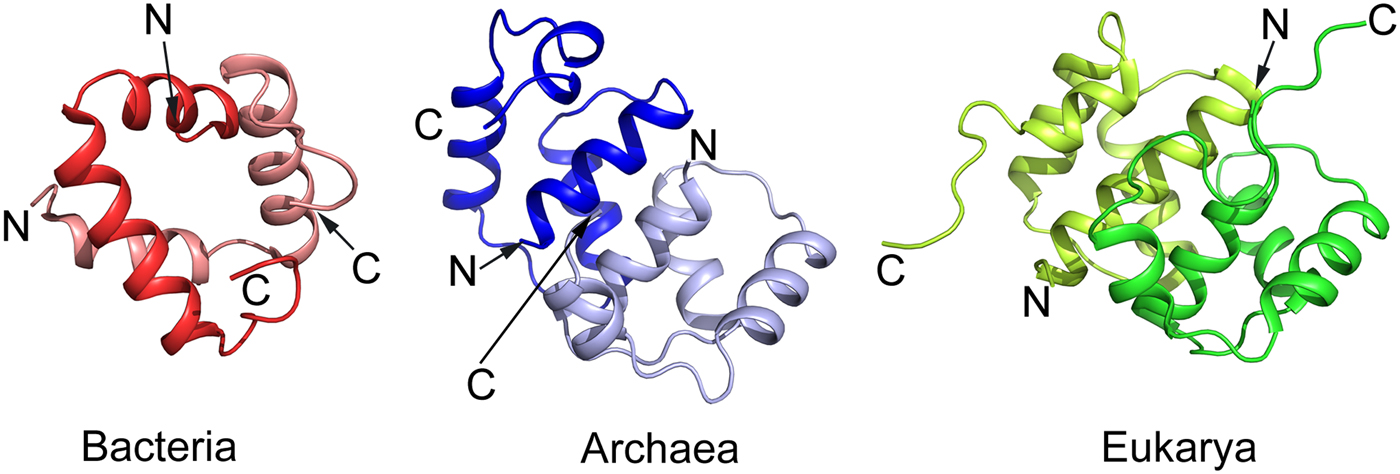
Fig. 4. Comparison of the structure of the NTD dimer of bacterial bL12 with archaeal protein aP1 and eukaryotic eP1/P2 proteins adopted from the PDBs 1RQT, 3A1Y, and 2LBF, respectively. While the archaeal and eukaryotic domains are similar, in comparison with the bacterial domain it is difficult to identify a structural similarity apart from the helical structure.
In yeast, there are four variants of the P proteins: P1A, P1B, P2A and P2B. P1A forms a heterodimer with P2B and P2A forms a heterodimer with P1B (Tchorzewski et al., Reference Tchorzewski, Boldyreff, Issinger and Grankowski2000b; Guarinos et al., Reference Guarinos, Remacha and Ballesta2001). In plants, there is one additional P protein, P3 (Bailey-Serres et al., Reference Bailey-Serres, Vangala, Szick and Lee1997). If all P-proteins are deleted, the ribosomes do not function. If only the P1 and P2 proteins are missing, the ribosome is able to support protein synthesis with the C-terminally extended uL10 alone (Santos & Ballesta, Reference Santos and Ballesta1994; Remacha et al., Reference Remacha, Jimenez-Diaz, Bermejo, Rodriguez-Gabriel, Guarinos and Ballesta1995), but a different subset of mRNAs is translated. However, if the ~30 amino acid long C-terminal residues of uL10 corresponding to the C-terminus of P1/P2 are also removed from uL10, the ribosome is no more functional.
The structure of the P-protein stalk
The structures of uL10 and the stalk complex from the archaeal species have been determined at high resolution (Kravchenko et al., Reference Kravchenko, Mitroshin, Nikonov, Piendl and Garber2010; Naganuma et al., Reference Naganuma, Nomura, Yao, Mochizuki, Uchiumi and Tanaka2010). The latter structure contains uL10 with three dimers of the N-termini of aP1. The structure of archaeal uL10 is closely similar to bacterial uL10, whereas the NTDs of aP1 are each composed of four α-helices in two layers. The dimer interface is formed by the two N-terminal helices (Fig. 4).
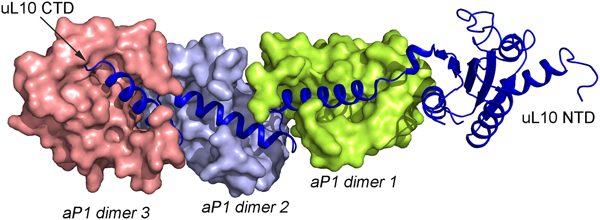
In all species, this stalk of the ribosome is long and flexible. The archaeal uL10 C-terminal helix binds over the 2-fold symmetry axis of the dimer of aP1, like in the uL10–bL12 interaction. The first and the fourth helix of aP1 interact almost orthogonally with the long helix of uL10 (Fig. 5). The possible arrangement of the archaeal P proteins in the ribosome will not be much different from the situation in the bacterial ribosome. One difference is that the C-terminal domain of the P proteins is smaller and that there is a C-terminal extension of uL10 of the same nature as the C-terminal domain of the P1 and P2 proteins.
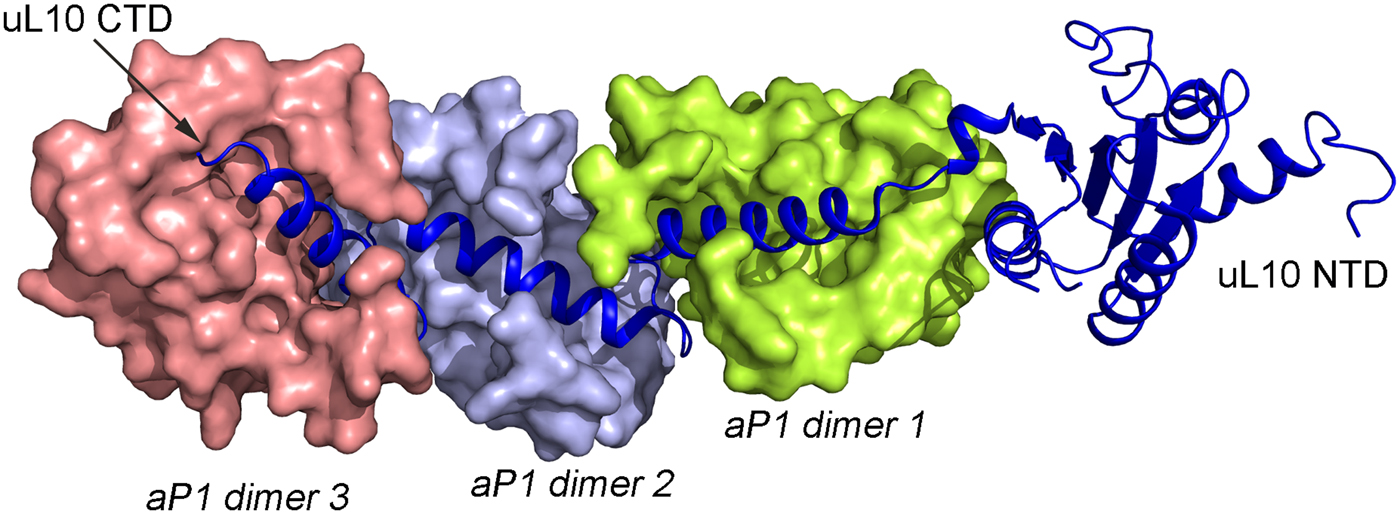
Fig. 5. Structure of the archaeal stalk complex (PDB: 3A1Y). uL10 is colored blue, while three aP1 dimers bound to the spine helices are colored in salmon, light blue and lemon. The aP1 dimers bind to the long C-terminal helix of uL10.
The trGTPases and their interactions with bL12 on the ribosome
The trGTPases
There are a number of trGTPases involved in protein synthesis (Table 1).
Table 1. Some trGTPases involved in protein synthesis on the ribosome

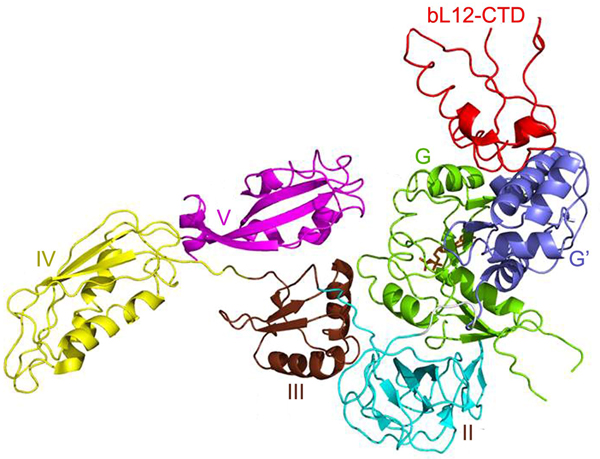
The trGTPases catalyze different steps of translation. These proteins, in their GTP-bound states catalyze different steps of translation on the ribosome. Further, they hydrolyze the bound GTP molecule and dissociate from the ribosome. The different roles of the trGTPases necessitate their different structures (Fig. 6), but they all have two domains in common, the GTP binding domain (G domain), normally at the N-terminus and the subsequent domain (domain II). Two of the trGTPases, EF-G and RF3, have an insert in the G-domain called G′ domain (Fig. 6). EF4 and BipA are paralogs of EF-G, but among other differences, they lack the G′ domain. For further structural and functional comparisons see Liljas and Ehrenberg (Reference Liljas and Ehrenberg2013); Heller et al. (Reference Heller, Kamalampeta and Wieden2017); Gibbs and Fredrick (Reference Gibbs and Fredrick2018).
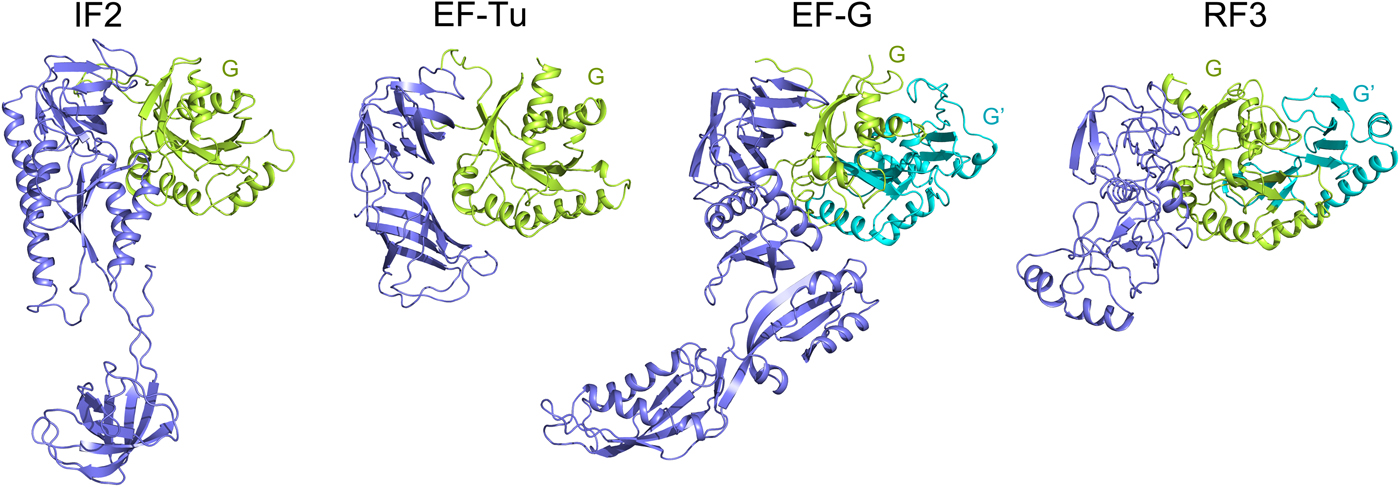
Fig. 6. The structure of the major trGTPases showing G and G′ domains in green and cyan. The structures are adopted from the following PDBs: IF2 (PDB: 3JCN), EF-Tu (PDB: 4PC7), EF-G (PDB: 4V9O) and RF3 (PDB: 4V89).
The binding of trGTPases to the ribosome
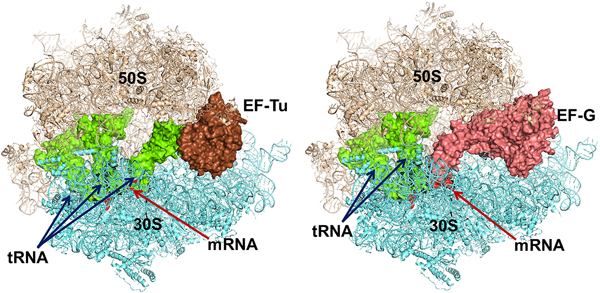
The trGTPases in complex with GTP bind to overlapping sites on the ribosome (Fig. 7) (Heimark et al., Reference Heimark, Hershey and Traut1976). The two common domains of the factors, G and II, interact with the ribosome in similar ways. These interactions, identified by cryo-EM and crystallography, show that the G-domains interact primarily with the sarcin-ricin loop (SRL) and the GTPase activating region (GAR) of the large subunit at the base of the bL12 stalk. The binding sites include RNA-helices H43 and H44 as well as proteins uL11 and uL6 (Gao et al., Reference Gao, Selmer, Dunham, Weixlbaumer, Kelley and Ramakrishnan2009; Schmeing et al., Reference Schmeing, Voorhees, Kelley, Gao, Murphy, Weir and Ramakrishnan2009). The anticodon-stem-loop of the tRNA in the ternary complex, EF-Tu·GTP·aa-tRNA interacts with the decoding region of the small subunit and so does the domain IV of EF-G. IF2, on the contrary, interacts initially only with the 30S subunit. When the subunits associate IF2 makes substantial contacts with the GAR of the 50S subunit. RF3 interacts mostly with the 50S subunit in a similar fashion as EF-Tu (Gao et al., Reference Gao, Zhou, Rawat, Huang, Bouakaz, Wang, Cheng, Liu, Zavialov, Gursky, Sanyal, Ehrenberg, Frank and Song2007; Pallesen et al., Reference Pallesen, Hashem, Korkmaz, Koripella, Huang, Ehrenberg, Sanyal and Frank2013).
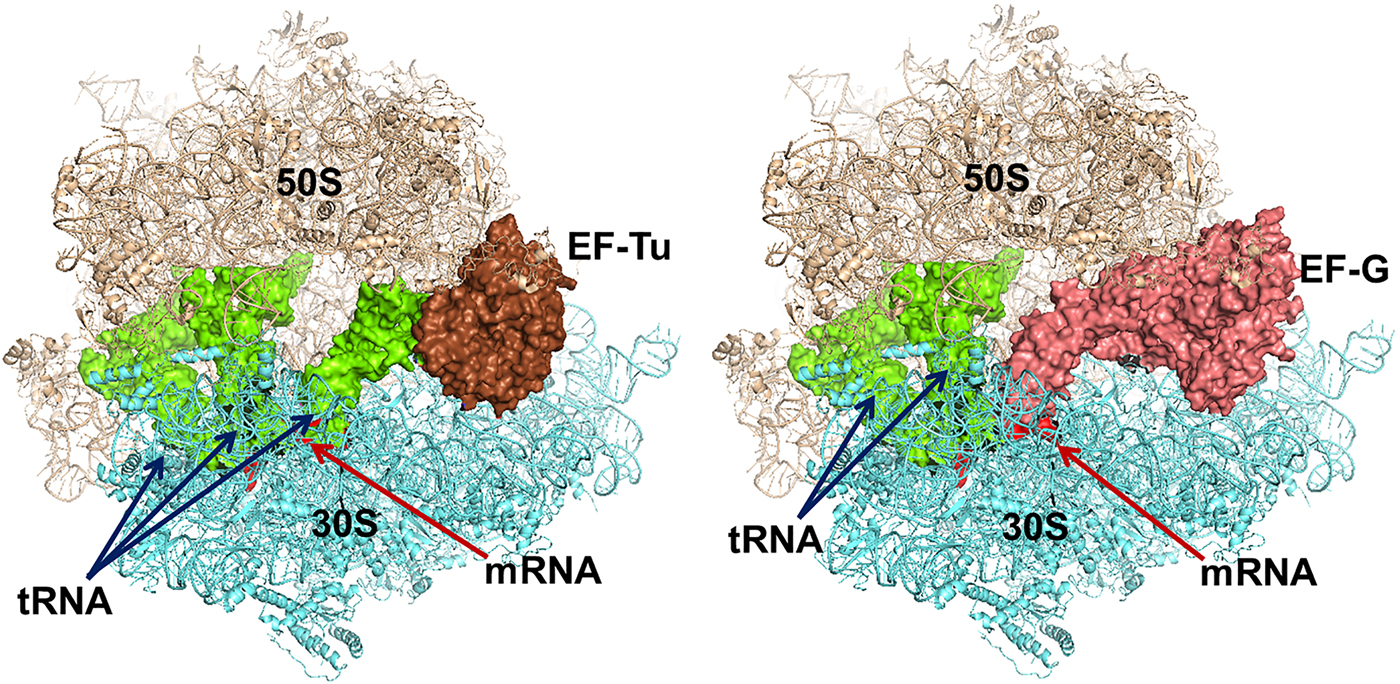
Fig. 7. Left - Elongation factor Tu (EF-Tu) in complex with an aminoacyl tRNA and a GTP analogue bound to the ribosome. Right – Elongation factor G with a GTP analogue bound to the ribosome. The similarity in binding is striking. The structures are adopted from the following PDBs: EF-Tu (PDB: 4V5R), EF-G (PDB: 4V5F).
What induces GTPase activity in the trGTPases?
The induction of GTPase activity in the trGTPases by the ribosome has been a challenging question. The trGTPases are part of a large family of GTPases where generally a GTPase activating protein (GAP) participates at the right stage and induces GTP hydrolysis. Furthermore, a residue in a loop of the GTPase, the so-called Switch II, has been thoroughly discussed. In most GTPases, this residue is a glutamine. Its ability to function as a general base is severely limited (Langen et al., Reference Langen, Schweins and Warshel1992). The corresponding residue in the trGTPases is a histidine, which would normally be able to function as a general base. However, since the GTPases have a common evolution it is most likely that the mechanism for GTP hydrolysis is the same, and that the histidine has a different role.
Several investigations have focused on the question of whether bL12 could be the ribosomal GAP and whether the histidine in Switch II of the trGTPases could function as a general base. Previously, it was proposed that isolated bL12 could independently stimulate GTP hydrolysis of EF-G on the ribosome unlike EF-Tu (Savelsbergh et al., Reference Savelsbergh, Mohr, Wilden, Wintermeyer and Rodnina2000). Subsequent kinetic studies revealed that depletion of bL12 from the 70S ribosome selectively affects stimulation of GTP hydrolysis in both EF-Tu and EF-G under multiple turnovers (Mohr et al., Reference Mohr, Wintermeyer and Rodnina2002; Savelsbergh et al., Reference Savelsbergh, Mohr, Kothe, Wintermeyer and Rodnina2005) or single turnover condition (Diaconu et al., Reference Diaconu, Kothe, Schlunzen, Fischer, Harms, Tonevitsky, Stark, Rodnina and Wahl2005). However, it was demonstrated that for GTPase stimulation on IF2, bL12 does not play any role (Huang et al., Reference Huang, Mandava and Sanyal2010). Also, high-resolution structures of EF-Tu and EF-G on the ribosome demonstrated that bL12 is not in contact with the G-domains of the trGTPases (Voorhees et al., Reference Voorhees, Schmeing, Kelley and Ramakrishnan2010; Carlson et al., Reference Carlson, Bassam, Weis, Colby, Shelton, Wuerth, Walter and Spiegel2017). Therefore, the precise role of bL12 in the context of GTP hydrolysis by trGTPases remains elusive and awaits further investigation.
For a considerable time, the SRL of the large subunit has also been considered to be an important region of the ribosome for factor interactions. Chemical footprinting showed that both EF-Tu and EF-G protect this loop (Moazed et al., Reference Moazed, Robertson and Noller1988). Crystallographic experiments on EF-Tu bound to the ribosome have made it clear that the phosphate of adenine 2662 in the SRL is critically involved in GTP hydrolysis (Voorhees et al., Reference Voorhees, Schmeing, Kelley and Ramakrishnan2010). It was shown that the phosphate of A2662 hydrogen bonds to His84 in EF-Tu and brings it up against the nucleophilic water molecule that is moved into close proximity of the γ-phosphate of the GTP molecule. His84 was suggested to act like a general base (Berchtold et al., Reference Berchtold, Reshetnikova, Reiser, Schirmer, Sprinzl and Hilgenfeldt1993; Voorhees et al., Reference Voorhees, Schmeing, Kelley and Ramakrishnan2010). However, insightful mechanistic analysis supported by in silico modeling and molecular dynamics (MD) simulations suggest that it is more likely that the water molecule, possibly through a second water molecule, donates one proton to the γ-phosphate and attacks the phosphate by an in-line mode, which leads to the hydrolysis of the γ-phosphate (Fig. 8; Liljas et al., Reference Liljas, Ehrenberg and Åqvist2011; Åqvist and Kamerlin, Reference Åqvist and Kamerlin2015; Mondal and Warshel, Reference Mondal and Warshel2018). This mechanism is probably universal for the trGTPases. Thus, the phosphate of an adenine in the SRL replaces the GTPase activating protein (GAP) for trGTPases in general.
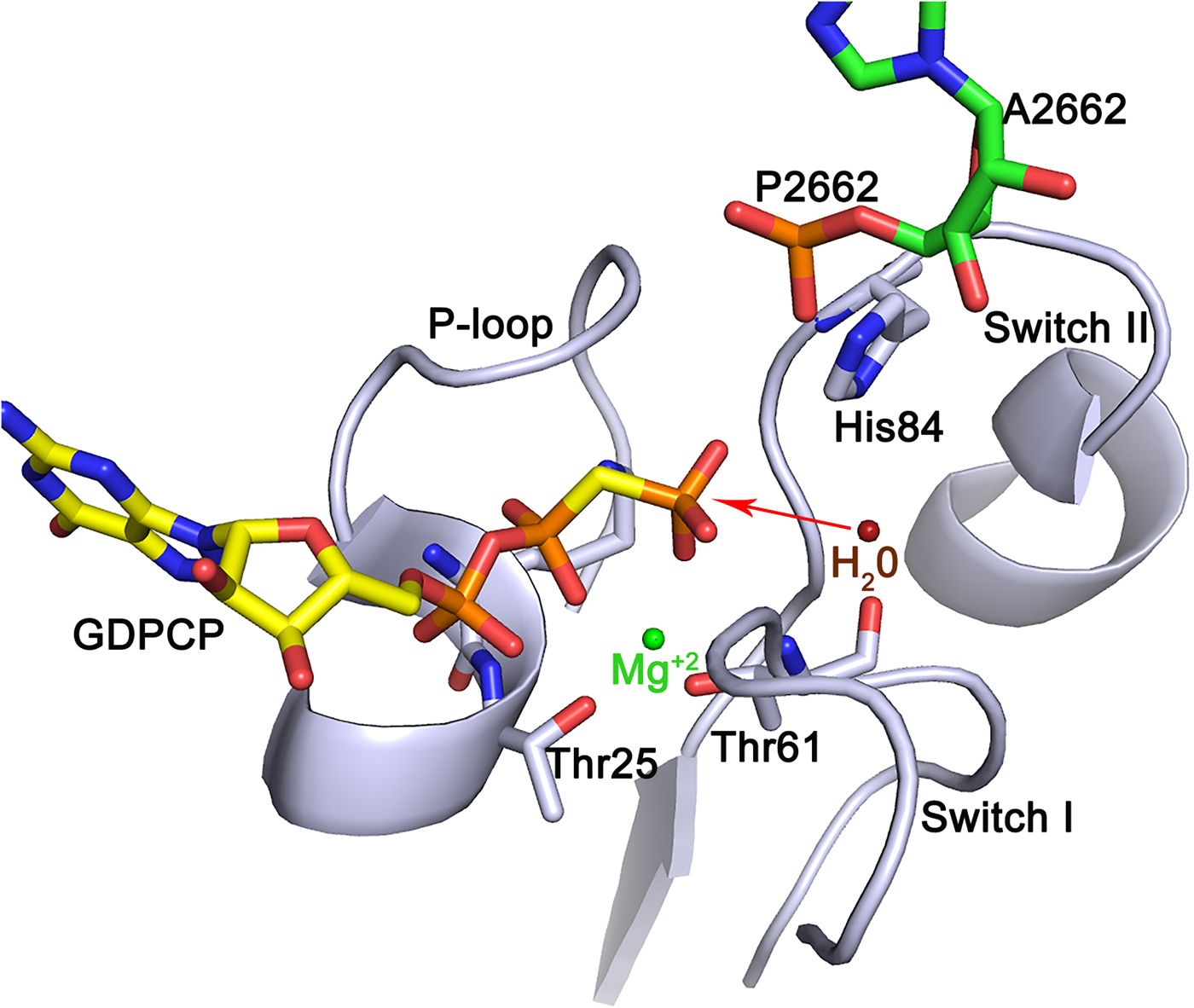
Fig. 8. A possible mechanism for GTP hydrolysis by the trGTPases. The phosphate of A2662 of SRL accepts a hydrogen bond from His 84 of the switch II of EF-Tu and positions it next to the water molecule close to the γ-phosphate. Both side chain nitrogens of the histidine are protonated making the histidine positively charged. The water molecule (red) donates a proton to the γ-phosphate leading to an in-line attack by the hydroxyl ion on the γ-phosphate (Liljas et al., Reference Liljas, Ehrenberg and Åqvist2011, Åqvist & Kamerlin, Reference Åqvist and Kamerlin2015). The structure was adopted from PDB: 4V5L (Voorhees et al., Reference Voorhees, Schmeing, Kelley and Ramakrishnan2010).
The role of bL12 in the functions of the trGTPases
The removal of the CTD of bL12 has the same effect as removing the whole protein on the activity of trGTPases (van Agthoven et al., Reference van Agthoven, Maassen, Schrier and Möller1975; Koteliansky et al., Reference Koteliansky, Domogatsky and Gudkov1978). Thus, it was concluded that bL12 interacts with the trGTPases through its CTD. Ribosomes with only one bL12 dimer are partially active (Griaznova and Traut, Reference Griaznova and Traut2000; Mandava et al., Reference Mandava, Peisker, Ederth, Kumar, Ge, Szaflarski and Sanyal2012). Ribosomes where one CTD of each bL12 dimer was removed also retain activity (Oleinikov et al., Reference Oleinikov, Jokhadze and Traut1998). The removal of all bL12 delays the release of inorganic phosphate from EF-G after GTP hydrolysis (Mohr et al., Reference Mohr, Wintermeyer and Rodnina2002). If the hinge of bL12 is significantly shortened, the ribosomes behave as if there was no CTD, but essentially doubling the length of the hinge has no negative effect (Gudkov et al., Reference Gudkov, Bubunenko and Gryaznova1991; Bubunenko et al., Reference Bubunenko, Chuikov and Gudkov1992).
We are faced with the dilemma that bL12 is important for GTP hydrolysis, but apparently not directly involved in the catalytic mechanism. What does it do? Which part of bL12CTD interacts with the factors and where on the factors does the interaction occur?
As mentioned before, bL12 was proposed to be responsible for GTPase activation of EF-G and EF-Tu (Savelsbergh et al., Reference Savelsbergh, Mohr, Wilden, Wintermeyer and Rodnina2000; Mohr et al., Reference Mohr, Wintermeyer and Rodnina2002; Savelsbergh et al., Reference Savelsbergh, Mohr, Kothe, Wintermeyer and Rodnina2005). However, with the advancement of the field with high-resolution structures and biochemistry, it became evident that bL12 is not a GAP. Rather it could act as a ‘propeller’ like instrument with multiple extended arms for continuous binding and recruitment of the trGTPases, especially EF-Tu to the ribosome (Diaconu et al., Reference Diaconu, Kothe, Schlunzen, Fischer, Harms, Tonevitsky, Stark, Rodnina and Wahl2005). However, so far there is no direct proof to this claim. Only a recent fluorescence-based localization study indicated simultaneous binding of several EF-Tu on each ribosome (Mustafi and Weisshaar, Reference Mustafi and Weisshaar2018). If true, this would mean that bL12 increases the local concentration of the translation factors, especially EF-Tu in a ternary complex with aminoacylated tRNAs to ensure their constant supply essential for a fast rate of translation.
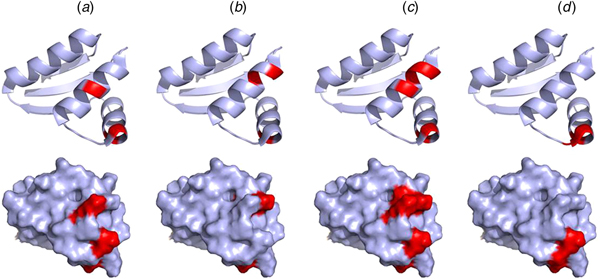
Mutations of V66, I69, K70, and R73 of the CTD of E. coli bL12 lead to an increased KM for the binding of EF-G but have little effect on GTP hydrolysis. Furthermore, these mutations inhibit the release of inorganic phosphate after GTP hydrolysis by EF-G (Savelsbergh et al., Reference Savelsbergh, Mohr, Kothe, Wintermeyer and Rodnina2005). Similarly, mutations of K65, V66, I69, K70, R73, and K84 have significant effects on ribosome binding of EF-Tu, and as a consequence of that, on GTP hydrolysis (Diaconu et al., Reference Diaconu, Kothe, Schlunzen, Fischer, Harms, Tonevitsky, Stark, Rodnina and Wahl2005). These mutational studies agree well with the NMR experiments suggesting that IF2, EF-Tu, EF-G, and RF3 all interact with helices 4 and 5 of bL12 CTD, in particular with residues V66, A67, V68, K70, G79, L80, E82 (Helgstrand et al., Reference Helgstrand, Mandava, Mulder, Liljas, Sanyal and Akke2007; Fig. 9). This surface corresponds with the conserved surface area of the CTD domain of bL12 (Leijonmarck and Liljas, Reference Leijonmarck and Liljas1987).
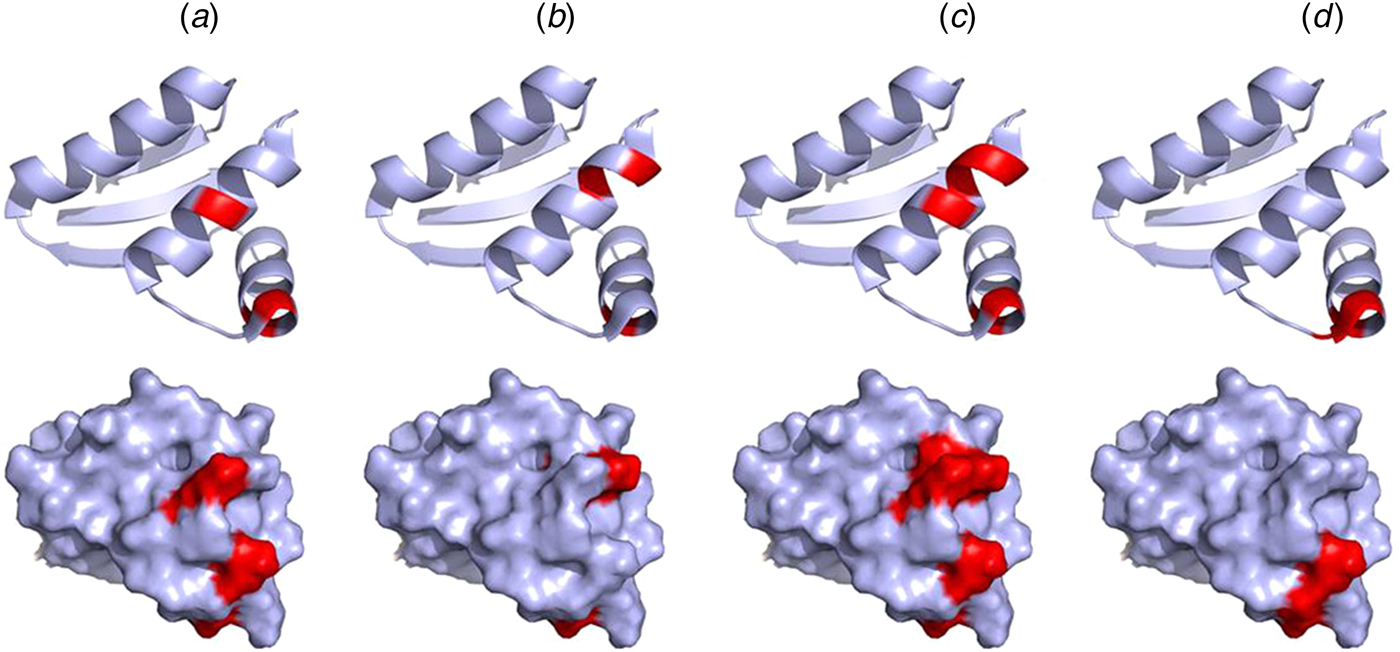
Fig. 9. Illustration of the interaction sites of the four major trGTPases IF2 (a), EF-Tu (b), EF-G (c) and RF3 (d) on the bL12 CTD as identified by NMR mapping (Helgstrand et al., Reference Helgstrand, Mandava, Mulder, Liljas, Sanyal and Akke2007). The top panel represents the ribbon diagram and the bottom panel represents the surface illustration of bL12-CTD. The interaction sites are highlighted in red.
EF–G interacts with the CTD of bL12 through its G′ domain as seen by crosslinking, cryo-EM and crystallography (Fig. 10; Datta et al., Reference Datta, Sharma, Qi, Frank and Agrawal2005; Nechifor & Wilson, Reference Nechifor and Wilson2007; Harms et al., Reference Harms, Wilson, Schluenzen, Connel, Stachelhaus, Zaborowska, Spahn and Fucini2008; Gao et al., Reference Gao, Selmer, Dunham, Weixlbaumer, Kelley and Ramakrishnan2009; Tourigny et al., Reference Tourigny, Fernández, Kelley and Ramakrishnan2013; Brilot et al., Reference Brilot, Korostelev, Ermolenko and Grigorieff2013; Chen et al., Reference Chen, Feng, Kumar, Ero and Gao2013). This is true also for RF3 (Pallesen et al., Reference Pallesen, Hashem, Korkmaz, Koripella, Huang, Ehrenberg, Sanyal and Frank2013). For the trGTPases lacking the G’-domain, EF-Tu (Stark et al., Reference Stark, Rodnina, Wieden, van Heel and Wintermeyer2000), IF2 (Allen et al., Reference Allen, Zavialov, Gursky, Ehrenberg and Frank2005; Simonetti et al., Reference Simonetti, Marzi, Billas, Tsai, Fabbretti, Myasnikov, Roblin, Vaiana, Hazemann, Eiler, Steitz, Puglisi, Gualerzi and Klaholz2013; Ge et al., Reference Ge, Mandava, Lind, Åqvist and Sanyal2018) and BipA (Kumar et al., Reference Kumar, Chen, Ero, Ahmed, Tan, Li, Wong, Bhushan and Gao2015), bL12 interacts with the G domain.

Fig. 10. The interaction of the G’ and G domains of EF-G with bL12CTD (red) on T. thermophilus 70S ribosomes (PDB: 4V5F) (Gao et al., Reference Gao, Selmer, Dunham, Weixlbaumer, Kelley and Ramakrishnan2009).
The main role for the bacterial trGTPase IF2 is to promote efficient ribosomal subunit association. bIF2 forms a preinitiation complex with 30S subunit programmed with mRNA and fMet-tRNAfMet. bL12 is needed for rapid factor–dependent subunit association with a 70S initiation complex (Huang et al., Reference Huang, Mandava and Sanyal2010; Mandava et al., Reference Mandava, Peisker, Ederth, Kumar, Ge, Szaflarski and Sanyal2012). Cryo-EM and crosslinking have shown that bL12 interacts with the G domain of IF2 (Heimark et al., Reference Heimark, Hershey and Traut1976; Allen et al., Reference Allen, Zavialov, Gursky, Ehrenberg and Frank2005). Recently, Ge et al. (Reference Ge, Mandava, Lind, Åqvist and Sanyal2018) analyzed a range of mutants in bL12 and IF2 to identify the sites involved in subunit association (Fig. 11). Mutations of the conserved K65, K70, R73, and K81 on the α4–α5 helices of bL12 caused a significant defect in subunit association. Similarly, mutations in D506, D508, E522, and E523 of G4-G5 helix of IF2 were found to be deleterious for fast subunit association. It can be easily noticed that the identified residues of bL12 and IF2 are basic and acidic in nature, respectively. Moreover, mutations to oppositely charged amino acids on both bL12 and IF2 caused largest defects, which could be rescued by using charge-swapped mutants on both the interacting partners. This mutational analysis pinpointed the amino acids on bL12 and IF2, which are the molecular determinants for their interaction. In addition, MD simulations identified the details of the interactions of these residues at the molecular level. It suggested several salt-bridge interactions between the bL12 CTD and the G domain of IF2 (Fig. 11). Based on the analysis, one can conclude that charge complementarity is the basis of bL12 and IF2 interaction (Ge et al., Reference Ge, Mandava, Lind, Åqvist and Sanyal2018). Moreover, it can be inferred that bL12 (CTD) on the 50S and IF2 on 30S-preinitiation complex act like recognition partners; their initial interaction leads to the rapid association of the ribosomal subunits. Based on the similarity of the G-domains of the trGTPases it can be speculated that the other trGTPases too interact with bL12 CTD through a similar mechanism based on electrostatic interaction. The available cryo-EM based pictures of bL12 interaction with RF3 (Pallesen et al., Reference Pallesen, Hashem, Korkmaz, Koripella, Huang, Ehrenberg, Sanyal and Frank2013) and EF-G (Gao et al., Reference Gao, Selmer, Dunham, Weixlbaumer, Kelley and Ramakrishnan2009) support that model.
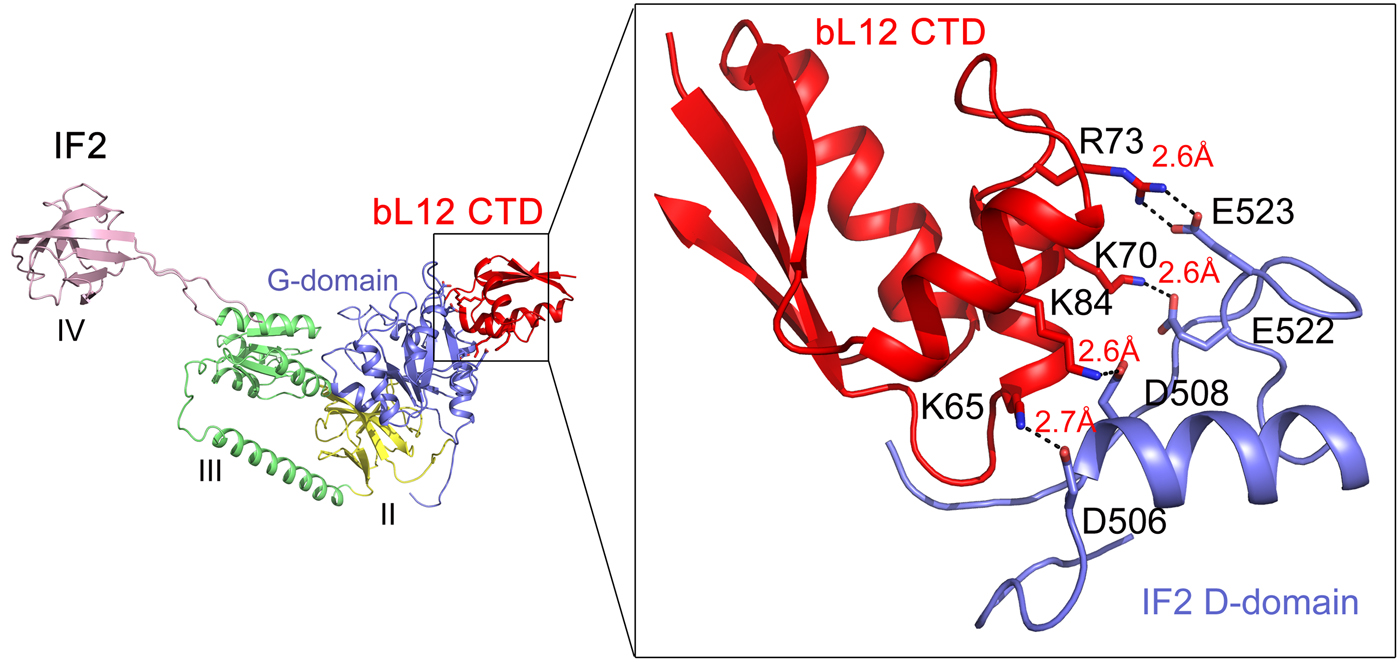
Fig. 11. The interaction of bL12 CTD (red) with IF2 on the 70S ribosome as suggested from the mutational studies combined with MD simulations (Ge et al., Reference Ge, Mandava, Lind, Åqvist and Sanyal2018).
Carlson et al., (Reference Carlson, Bassam, Weis, Colby, Shelton, Wuerth, Walter and Spiegel2017) have revisited some of the old experiments after significantly improved purification of the experimental components. First of all they used E. coli JE28 ribosomes in which all bL12 proteins carry chromosomally fused C-terminal His-tag (Ederth et al., Reference Ederth, Mandava, Dasgupta and Sanyal2009). With the aid of the His-tag, they ensured that bL12 was completely removed from the ribosomes. They also used an extensive denaturation and renaturation based method for purification of bL12 more extensively. In their study, bL12 did not induce GTPase activity in the isolated EF-G in contrast to what was reported earlier by Savelsbergh et al., Reference Savelsbergh, Mohr, Wilden, Wintermeyer and Rodnina2000. However, the GTPase activity by EF-G on the ribosome requires the presence of bL12. The binding of EF-G in complex with the non-hydrolysable GTP analogue GDPNP to the ribosome was dramatically reduced if bL12 was absent. If the G′ domain was removed from EF-G the GTPase activity was much reduced and the presence or absence of bL12 in the ribosome did not make any difference. Likewise, the binding of RF3 and IF2 to ribosomes also required the presence of bL12 (Carlson et al., Reference Carlson, Bassam, Weis, Colby, Shelton, Wuerth, Walter and Spiegel2017).
The interaction of P-proteins with trGTPases
In archaeal or eukaryotic translation the P-proteins are essential for the function of the trGTPases. However, all P1/P2 dimers can be removed despite maintaining the GTPase activity. This activity can be maintained by the C-terminus of uL10 alone. However, the lack of some or all of the P1/P2 dimers leads to losses in fidelity and a different subset of mRNAs is translated (Santos & Ballesta, Reference Santos and Ballesta1994; Remacha et al., Reference Remacha, Jimenez-Diaz, Bermejo, Rodriguez-Gabriel, Guarinos and Ballesta1995, Wawiórka et al., Reference Wawiórka, Molestak, Szahwai, Michalec-Wawiórka, Molon, Borkiewicz, Grela, Boguszewska and Tchórzewski2017). This may suggest a complex role of these proteins but primarily relates to the decoding process where different complexes of EF1A with GTP and aminoacylated tRNAs are tested against the current codon in the A-site. The rate needed for efficient discrimination of non-cognate and near-cognate tRNAs cannot be met by a single C-terminus.
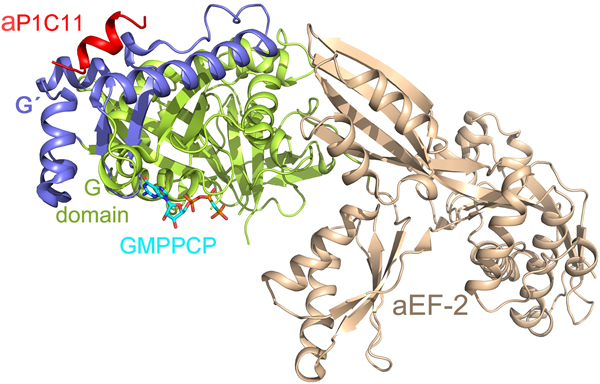
With regard to the CTD of the P-proteins, there has been a lack of information about its structure and interaction with the trGTPases. Recently Tanzawa et al. (Reference Tanzawa, Kato, Girodat, Ose, Kumakura, Wieden, Uchiumi, Tanaka and Yao2018) studied the binding of a C-terminal peptide of aP1 to aEF2. The peptide takes the conformation of a helix, which binds to the G4 and G5 elements of the factor similar to the observations in case of bacterial IF2 (Fig. 12) (Ge et al., Reference Ge, Mandava, Lind, Åqvist and Sanyal2018). However, in contrast to the charge complementarity-based interaction of bL12-CTD and IF2, the 11 amino acid long C-terminal helix of aP1 (aP1C11) binds to aEF2a in a hydrophobic groove.

Fig. 12. The C-terminal (C11) peptide of aP1 (red) binds to the archaeal EF2 in a hydrophobic groove between the G (green) and G’ (blue) domains (PDB ID: 5H7L) (Tanzawa et al., Reference Tanzawa, Kato, Girodat, Ose, Kumakura, Wieden, Uchiumi, Tanaka and Yao2018).
Ribosome inhibitory proteins high-jack the ribosomal stalk for binding
There is a range of ribosome inhibitory proteins (RIP) that generally modify the part of SRL, which is important for the GTPase activity. In some cases, like ricin, a central nucleotide is depurinated and in other cases, like alpha-sarcin, the large ribosomal RNA is cut. The RIPs are evolutionary related, but the sequence conservation is quite low. Interestingly, without any sequence or structural relationship to the trGTPases, the RIPs like the trGTPases benefit in binding to the ribosome from the presence of the P-proteins. The binding is through hydrophobic interactions of the very C-terminal 6–9 residues of the P-proteins (Shi et al., Reference Shi, Mak, Wong and Shaw2016).
Conclusions
The stalk proteins have very distinctive roles primarily in relation to the trGTPases. However, it remains enigmatic what their actual role is in relation to the activation of GTP hydrolysis in trGTPases. There are at least two options generally discussed:
(A) The ribosomal stalk, with multiple arms bearing the C-terminal structures of bL12 or the P-proteins, recruits trGTPases to their binding area on the ribosome by binding and thereby increasing the local concentration of trGTPases. This would, in turn, increase the rate of GTP hydrolysis.
(B) The ribosomal stalk binds the trGTPases on the ribosome and stabilizes their binding to the factor-binding site at the SRL loop on the ribosome thereby increasing the rate of GTP hydrolysis.
The first mechanism (A) is most widely accepted by the ribosome field at present. However, relevant experimental proof supporting this mechanism is still lacking. If the native-like high rate of GTP hydrolysis could be achieved by adding an increasing concentration of the trGTPases with a bL12-depleted ribosome, then this mechanism would be proven true. That would then justify the need for multiple bL12-CTDs for increasing the local concentration of the trGTPases for high GTPase rates. However, the recent study with super-resolution microscopy infers that the four bL12 proteins are essentially saturated with EF-Tu ternary complexes in vivo (Mustafi and Weisshaar, Reference Mustafi and Weisshaar2018). This observation again speaks in favor of the factor recruitment model.
Experimental evidence for the second mechanism (B) is also scarce. If a high GTPase rate can be achieved by titrating L12-CTDs with a bL12-depleted ribosome, it might support model (B). It would mean that the bL12-CTD interaction is important for stable binding of the trGTPases in the vicinity of the SRL and for attaining the correct conformation for GTPase activation. Carlson et al. (Reference Carlson, Bassam, Weis, Colby, Shelton, Wuerth, Walter and Spiegel2017) found reduced occupancy of EF-G-GDPNP on the ribosome when bL12 is deleted. This result clearly indicates the need of bL12, presumably the CTD, for stable binding of the trGTPases on the ribosome. The case of the initiation factors, bacterial IF2 or archaeal or eukaryotic eIF5B illustrates another aspect of the bL12 CTD (Ge et al., Reference Ge, Mandava, Lind, Åqvist and Sanyal2018). These factors initially bind to the small subunit and need the stalk proteins for association with the large subunit. These results also clearly support the B alternative. It is also possible that the ribosomal stalk proteins use different or specialized modes of action with respect to different translational GTPases. Its role in the function of other translation factors also remains elusive.
From our discussions of the available literature regarding bL12 it becomes obvious that despite about 50 years of research, the role of the ribosomal stalk remains somewhat enigmatic. On the contrary, the multifaceted research on bL12 and the P proteins has brought us much further into this intriguing story.
Acknowledgements
We are thankful to Dr. Chandra Sekhar Mandava and Dr. Xueliang Ge for helping with the artwork used in this article. SS acknowledges research funding support from Swedish Research Council (2014-4423 and Research Environment Grant 2016-06264), and Knut and Alice Wallenberg Foundation (KAW 2017·0055).







 Open access
Open access