



Introduction
DNA polymerases are an ancient family of enzymes responsible for replicating the genomes of organisms during cell division. Their movement on a template associates them to molecular motors that are powered by the free energy of nucleotide polymerization (Gelles and Landick, Reference Gelles and Landick1998). However, in contrast to most molecular motors, which are largely responsible for transporting cargo along protein tracks (Schliwa and Woehlke, Reference Schliwa and Woehlke2003), polymerases are a class of enzymes whose movements facilitate the transfer of information from parent to daughter strands using well-established Watson–Crick base pairing rules (Watson and Crick, Reference Watson and Crick1953). This impressive feat of chemical synthesis is accomplished through a complicated reaction pathway where each cycle of nucleotide addition involves a set of carefully orchestrated conformational changes that allow the enzyme to form a covalent bond between the growing primer strand and the correct incoming nucleotide (Steitz, Reference Steitz1999). The superiority of these motors is further demonstrated through the use of accessory domains that enable the enzyme to recognize and correct mistakes that arise due to misincorporation events. Thus, polymerases can be thought of as biological scribes capable of forward and reverse motions that allow for the writing and editing of genetic messages with unparalleled speed and accuracy.
The process of nucleotide selection, insertion, and extension is regulated by a series of checkpoints that control the efficiency and fidelity of nucleotide synthesis. Elegant biochemical, kinetic, and structural studies reveal the importance of induced fit in distinguishing correct nucleotides from incorrect nucleotides (Bryant et al., Reference Bryant, Johnson and Benkovic1983; Johnson, Reference Johnson2008; Ludmann and Marx, Reference Ludmann and Marx2016), noting that Watson–Crick hydrogen bonds are not always necessary for the replication of a DNA base pair (Moran et al., Reference Moran, Ren, Rumney and Kool1997b). Other factors that affect nucleotide recognition include hydrogen bonding to minor groove heteroatoms, base stacking, solvent exclusion, and shape (Kool, Reference Kool2002). Once present in the active site, chemical bond formation requires the substrate to adopt a productive geometry that leads to phosphodiester bond formation. In all cases, this involves a combination of side chain and divalent metal ion interactions that orient the substrate in a position that is suitable for in-line nucleophilic attack by the terminal 3′ hydroxyl group on the primer strand (Steitz et al., Reference Steitz, Smerdon, Jager and Joyce1994; Genna et al., Reference Genna, Vidossich, Ippoliti, Carloni and De Vivo2016). In cases where a polymerase is able to incorporate a modified nucleotide, additional molecular recognition events are available to detect changes in the duplex geometry, which often leads to polymerase stalling (Miller and Grollman, Reference Miller and Grollman1997). Although these parameters can vary between individual polymerases, the checkpoints of nucleotide selection, chemical bond formation, and primer extension place severe limitations on the synthesis of unnatural nucleic acid polymers by natural DNA polymerases.
One striking example of substrate specificity is the ability for polymerases to discriminate between DNA and RNA substrates inside the cell. The molecular difference between 2′-deoxyribonucleoside triphosphates (dNTPs) and ribonucleoside triphosphates (NTPs) is the presence of a 2′-hydroxyl group on the ribose sugar, which causes the furanose ring to adopt a different sugar pucker (C2′-endo versus C3′-endo for DNA and RNA, respectively) (Anosova et al., Reference Anosova, Kowal, Dunn, Chaput, Van Horn and Egli2016). Even though intracellular NTP levels are elevated relative to dNTP levels (>10-fold) (Traut, Reference Traut1994), DNA polymerases, such as Escherichia coli (E. coli) DNA polymerase I, are able to discriminate against NTPs by a factor of up to 105-fold (Astatke et al., Reference Astatke, Ng, Grindley and Joyce1998). This remarkable level of substrate specificity is achieved by a single bulky amino acid residue, referred to as the ‘steric gate’, which packs against the 2′ sugar position, preventing the insertion of NTPs into the enzyme active site. The steric gate is now recognized as a common feature of most DNA polymerases (Bonnin et al., Reference Bonnin, Lazaro, Blanco and Salas1999; Brown and Suo, Reference Brown and Suo2011).
In this review, we examine the impact of polymerase engineering on the field of synthetic biology. Special emphasis is placed on examples in which engineered polymerases have enabled the synthesis, replication, and evolution of synthetic genetic polymers with new physicochemical properties, such as enhanced ligand binding, catalysis, and biological stability. Such activities represent the forefront of polymerase engineering, as functional non-natural polymers are expected to drive future applications in synthetic biology, biotechnology, and healthcare. We begin with a review of polymerase function and structure, illustrating the latest techniques that have been used to answer fundamental questions about the mechanism of DNA synthesis. Next, we discuss examples where natural polymerases are able to recognize non-cognate and synthetic congeners as substrates either in the template or as nucleoside triphosphates. We then examine several techniques that have been applied to engineer polymerases with desired functional properties. Here we focus our attention on avant-garde strategies that are rapidly advancing the field of polymerase engineering. Finally, we conclude with examples of synthetic biology applications that have arisen due to the availability of engineered polymerases.
Natural polymerases
Fundamentals of DNA synthesis
DNA polymerases follow a primer extension mechanism in which a single strand of parental DNA is used as a template to synthesize the complementary daughter strand. In this reaction, the growing daughter strand is recognized as a primer that is extended in the 5′-3′ direction by sequentially adding the corresponding dNTP to the terminal 3′-hydroxyl group. As illustrated in Fig. 1, the template dictates the sequence of nucleotide addition following the classic Watson–Crick base pairing rules of adenine (A) pairing with thymine (T) and guanine (G) pairing with cytosine (C). Because the polymerase moves down the template in the 3′-5′ direction and the new DNA strand is generated in the 5′-3′ direction, the resulting product is an antiparallel DNA duplex.
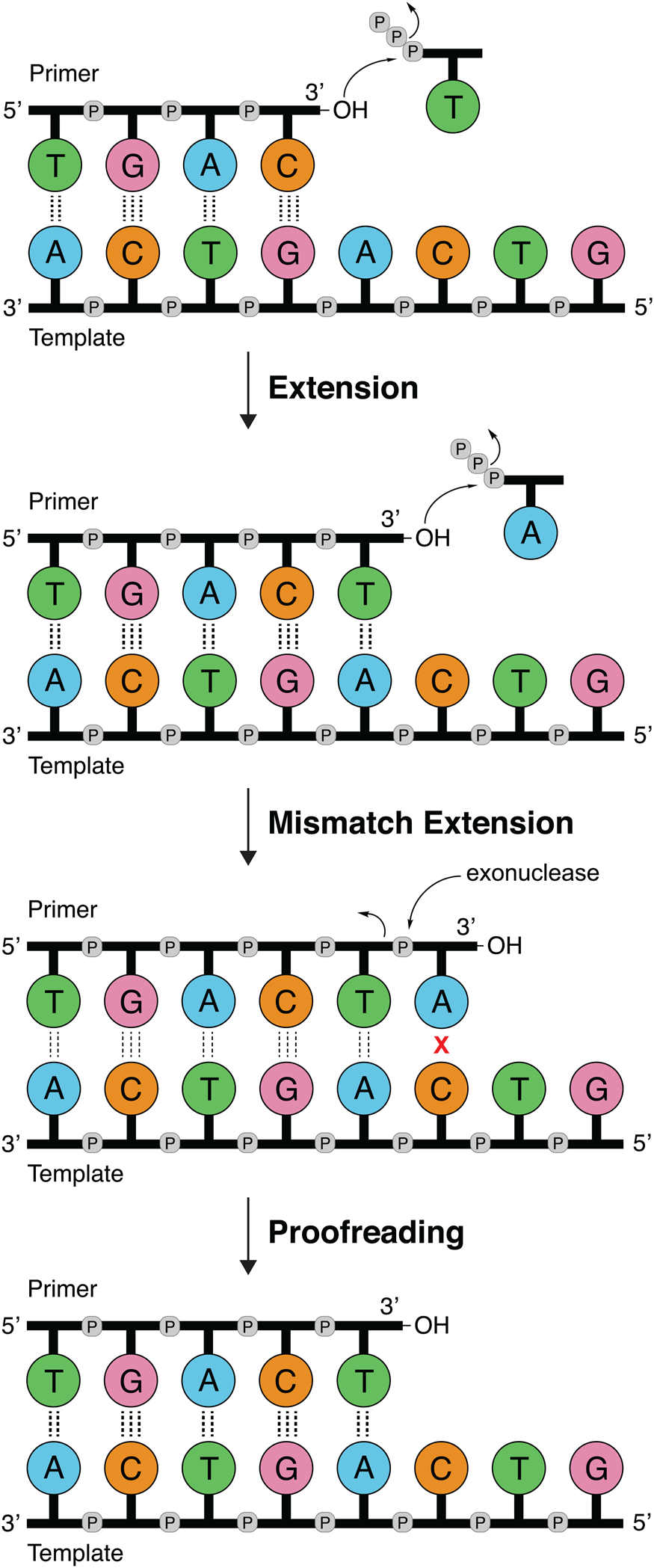
Fig. 1. DNA synthesis and mismatch repair. Natural DNA polymerases extend a DNA primer in the 5′-3′ direction using the template to determine the sequence of the growing strand. Polymerases with 3′-5′ exonuclease activity have the ability to correct mistakes by removing terminal nucleotides that are incorrectly paired with the template.
Phylogenetic analysis reveals that DNA polymerases organize into seven different highly homologous sequence families (A, B, C, D, X, Y, and RT) (Table 1) (Ito and Braithwaite, Reference Ito and Braithwaite1991) that allow the activity of one member to predict the activity of another member. For example, the mutations required to imbue a natural DNA polymerase with RNA synthesis activity have similar activity when transferred to homologous enzymes (Dunn et al., Reference Dunn, Otto, Fenton and Chaput2016). As expected, some polymerase families have been more widely studied than others. Thermostable DNA polymerases belonging to the A- and B-family categories have been extensively studied due to their importance in DNA synthesis and sequencing applications. For example, A-family DNA polymerase I isolated from the thermophilic bacterial species Thermus aquaticus (Taq) is widely used in quantitative polymerase chain reaction (qPCR) applications due to its 5′-3′ exonuclease activity, which allows for the digestion of a downstream donor–quencher fluorescent probe that quantitatively measures DNA synthesis during polymerase extension (Holland et al., Reference Holland, Abramson, Watson and Gelfand1991). Taq DNA polymerase is also routinely used for T–A ligation and cloning strategies due to its proclivity for adding a single-untemplated adenosine residue to the 3′ end of the daughter strand (Clark, Reference Clark1988). Hyperthermophilic archaeal B-family DNA polymerases, which include such members as Tgo (Thermococcus gorgonarius), Kod (Thermococcus kodakarensis), Pfu (Pyrococcus furiosus), and 9°N (Thermococcus 9°N-7), are the basis of several DNA-sequencing applications (Zhang et al., Reference Zhang, Kang, Xu and Huang2015a). These enzymes are known to function with enhanced fidelity due to the presence of a strong 3′-5′ exonuclease proofreading domain. They are also reported to be more resistant than standard Taq polymerase to the inhibitory effects of blood components and detergents (Miura et al., Reference Miura, Tanigawa, Fujii and Kaneko2013). Interestingly, B-family polymerases have the ability to recognize and stall DNA replication when they encounter uracil residues in the template (Greagg et al., Reference Greagg, Fogg, Panayotou, Evans, Connolly and Pearl1999). Structural studies indicate that uracil discrimination is caused by a binding pocket in the amino-terminal domain of the polymerase that accommodates uracil but prevents binding to the four natural DNA bases (Fogg et al., Reference Fogg, Pearl and Connolly2002).
Table 1. Properties of natural DNA polymerases

Despite extensive sequence diversity, X-ray crystal structures reveal that nearly all polymerases adopt a catalytic domain that closely resembles a human right hand (Steitz, Reference Steitz1999). The one exception is X-family polymerases, which adopt a left-handed polymerase domain (Beard and Wilson, Reference Beard and Wilson2000). The catalytic domain is further divided into three subdomains that are commonly referred to as the palm, fingers, and thumb (Fig. 2a). The palm subdomain is composed of a β-sheet that forms the base of a deep cleft containing the catalytic residues responsible for promoting phosphodiester bond formation. The fingers subdomain is an α-helical structure lining one side of the cleft, while the thumb subdomain is another α-helical structure lining the opposite side of the cleft. The fingers are responsible for recognizing the incoming nucleoside triphosphate, while the thumb positions the DNA primer–template duplex in the cleft and plays a role in translocation and processivity (Brautigam and Steitz, Reference Brautigam and Steitz1998).
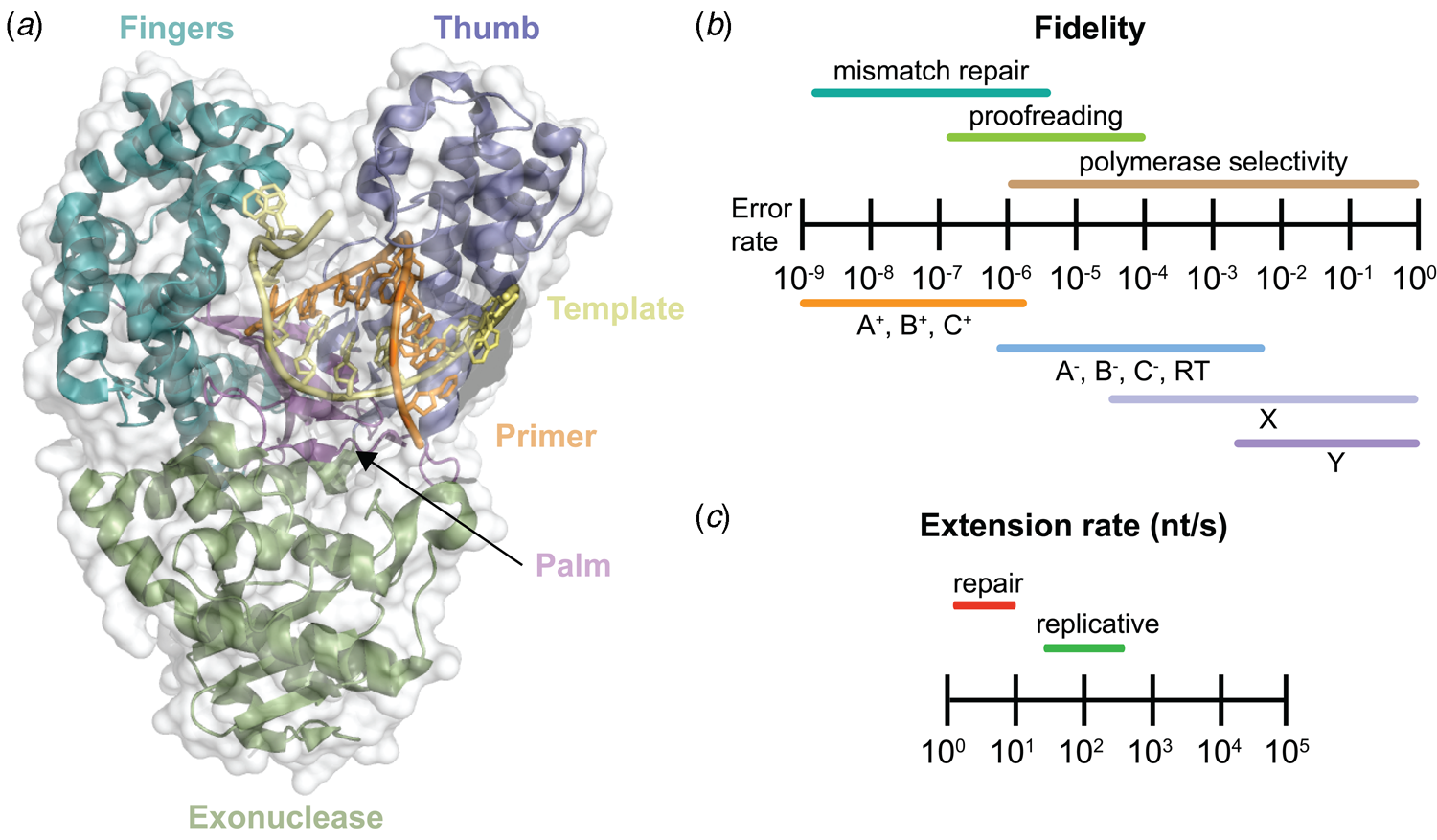
Fig. 2. DNA polymerase structure and catalysis. (a) Structure of the binary complex of Bst DNA polymerase bound to the DNA duplex (PDB: 6DSY). (b) Fidelity of DNA replication summarized according to different steps that enhance the fidelity of DNA synthesis and polymerase family. +/− Indicates the presence or absence of exonuclease activity. (c) Differences in the extension rate between repair and replicative DNA polymerases under in vitro conditions independent of accessory proteins that lead to faster rates in the cellular environment.
Speed and fidelity are critical parameters for DNA synthesis in rapidly dividing cells. For each nucleotide incorporation, a polymerase must distinguish the correct nucleoside triphosphate from an excess of incorrect and non-cognate (NTP) substrates. Due to their functional roles, the rate and fidelity of DNA synthesis can vary widely between different DNA polymerases (Fig. 2b and c). Replicative DNA polymerases found in A- and B-families have rates that can exceed 100 nt s−1 and intrinsic fidelities in the range of one error in 105–106 incorporation events (Kunkel, Reference Kunkel2004). For example, Kod polymerase functions with a rate of ~200 nt s−1, making it one of the fastest B-family DNA polymerases (Griep et al., Reference Griep, Kotera, Nelson and Viljoen2006). In addition, many polymerases have 3′-5′ exonuclease proofreading activity that exists as a separate domain or a tightly bound subunit, which can remove non-complementary nucleotides after phosphodiester bond formation (Fig. 1) (Kunkel and Bebenek, Reference Kunkel and Bebenek2000). These domains increase the fidelity of DNA synthesis by 10-fold (106–107 nt s−1) relative to polymerases lacking a proofreading domain (Loeb and Monnat, Reference Loeb and Monnat2008). By comparison, repair polymerases, such as pol β (X-family), are much slower at DNA synthesis and less faithful than replicative DNA polymerases, often functioning with rates in the range of ~10 nt s−1 and fidelities on the order of 1 error in 102–104 incorporation events (Fig. 2b and c) (Wu et al., Reference Wu, Yang and Tsai2017). However, the reduced activity of repair polymerases is expected given their functional role in repairing damaged sites in genomic DNA by various cellular repair mechanisms.
Visualizing DNA synthesis through snapshots of trapped intermediates
Since its discovery in 1958, DNA polymerase I has been viewed as a model system for DNA synthesis in cells (Lehman et al., Reference Lehman, Bessman, Simms and Kornberg1958). Structural insights into the mechanism of DNA synthesis have been obtained from crystal structures that trap the enzyme at different stages of the catalytic cycle (Fig. 3) (Chim et al., Reference Chim, Jackson, Trinh and Chaput2018). Some of the most insightful data have been obtained from high-resolution structures of a thermostable bacterial DNA polymerase I member isolated from Geobacillus stearothermophilus (Bst, Fig. 2a) and its bacteriophage homolog T7 RNA polymerase (Kiefer et al., Reference Kiefer, Mao, Braman and Beese1998; Yin and Steitz, Reference Yin and Steitz2002, Reference Yin and Steitz2004). Starting from the Bst binary complex produced from one round of dNTP addition, Tyr714 on the O-helix occupies the insertion site, stacking above the newly added nucleotide on the growing primer strand (Chim et al., Reference Chim, Jackson, Trinh and Chaput2018). In this same structure, Tyr719 on the O1-helix forms a second stacking interaction with the n + 1 templating base, thereby preventing the next templating base from entering the active site. In step 2, the polymerase undergoes a conformational change to adopt a pre-insertion complex with the incoming nucleotide paired opposite Tyr714 in the enzyme active site (Chim et al., Reference Chim, Jackson, Trinh and Chaput2018). This intermediate, commonly referred to as the open ternary complex, is achieved by releasing the n + 1 templating base from its stacking interaction with Tyr719 and retracting Tyr714 to a position above the n templating base in the post-insertion site. In step 3, the enzyme undergoes a more significant conformational change to adopt a closed ternary complex, which defines the pre-catalytic state of the enzyme (Johnson et al., Reference Johnson, Taylor and Beese2003). Here, the n + 1 templating base finally enters the insertion site and forms a Watson–Crick base pair with the incoming nucleotide. In this structure, the fingers have rotated ~40° to allow several lysine and arginine residues on the O-helix to contact the triphosphate moiety of the dNTP substrate. In step 4, the enzyme adopts a post-catalytic complex in which chemical bond formation has occurred and the primer has been extended by one nucleotide (Yin and Steitz, Reference Yin and Steitz2004). Close examination of the enzyme active site reveals the presence of the pyrophosphate leaving group, suggesting that pyrophosphate departure coincides with opening of the fingers. To complete the cycle, the polymerase must translocate to the next position on the template to reform the binary complex. Together, crystal structures of the binary complex, pre-insertion site, closed ternary complex, and post-catalytic complex provide a structural view of DNA synthesis by a replicative DNA polymerase.
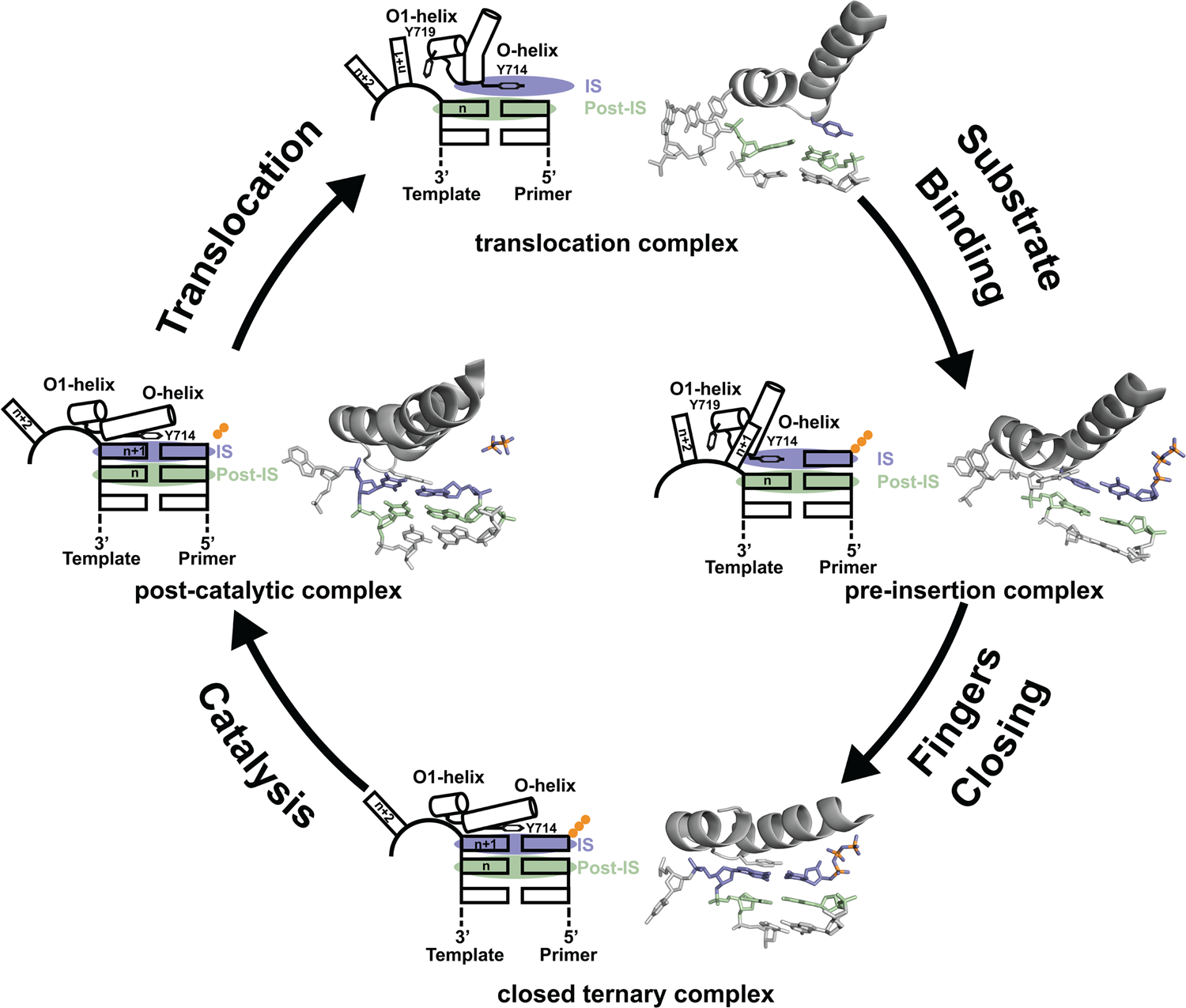
Fig. 3. Mechanism of DNA synthesis. The four key mechanistic steps depict a replication cycle for DNA synthesis. The translocation complex (top) is stabilized by π-stacking interactions between Tyr719 and the n + 1 templating base and between Tyr714 and the primer strand. Tyr714 occupies the insertion site (IS, purple) while a newly formed base pair is located in the post insertion site (post-IS, green). In the pre-insertion complex (right), the O-helix adjusts to accommodate the incoming dNTP substrate, which binds opposite Tyr714 in the IS. In the closed ternary complex (bottom), the polymerase undergoes a major conformational change to allow the n + 1 templating base to form a nascent base pair with the dNTP substrate in pre-catalytic state. Following catalysis, the finger subdomain remains closed with a trapped pyrophosphate moiety observed in the active site of the post-catalytic complex (left). To complete the cycle, the finger subdomain opens, pyrophosphate is released, and the enzyme translocates to the next position on the template. The translocation (6DSY), pre-insertion (6DSU), and closed ternary complexes (1LV5) are based on crystal structures of Bst DNA polymerase. The post-catalytic complex is T7 RNA polymerase (1S77), a homolog of Bst DNA polymerase. Adapted from Chim et al. (Reference Chim, Jackson, Trinh and Chaput2018).
Structural and kinetic data reveal that DNA polymerase fidelity is governed by subtle local rearrangements that are distinct from the major conformational domain movements observed in the binding and catalysis of cognate Watson–Crick base pairs. In particular, researchers have identified a distinct conformation in A-family polymerases that was suggested to be a fidelity checkpoint for correct nucleotide selection. X-ray crystal structures of Bst DNA polymerase containing mismatched substrates reveal a kink in the O-helix of the finger subdomain that results in a partially closed ternary complex, termed the ‘ajar’ conformation (Fig. 4a) (Wu and Beese, Reference Wu and Beese2011). The wobble base pair between the templating G nucleotide and the incoming TTP substrate places the α-phosphate at a distance that is too far from the 3′-OH group of the primer to facilitate efficient in-line attack on the dNTP substrate. This observation is supported by a reduction of at least 100-fold in the rate of nucleotide addition compared to the complementary dCTP substrate (Wu and Beese, Reference Wu and Beese2011).
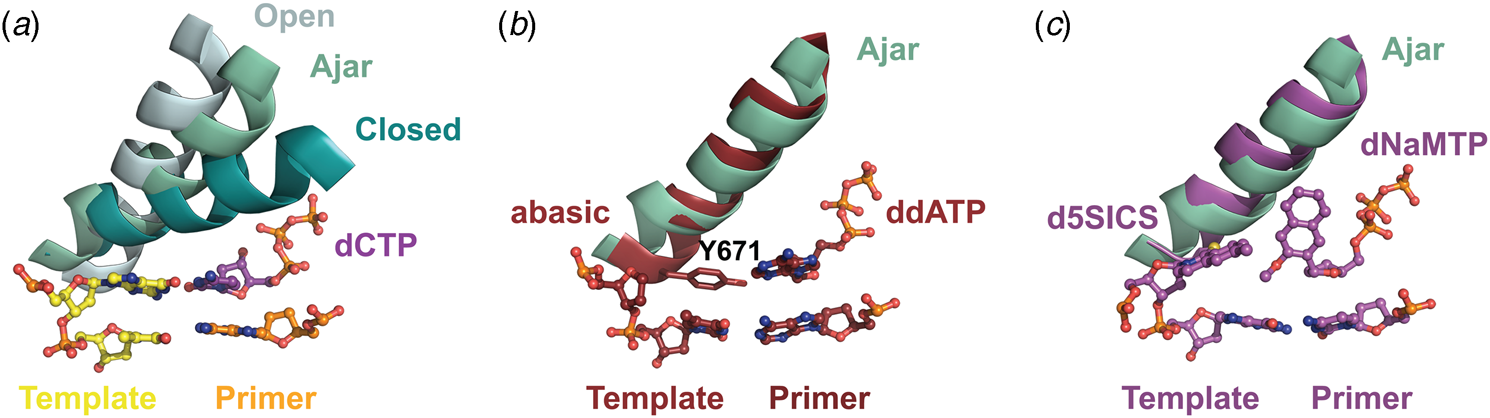
Fig. 4. X-ray crystal structures capturing the ajar conformation. (a) Structural comparison of the open (1L3S), ajar (3HP6), and closed (1LV5) ternary conformations of Bst DNA polymerase. The Bst ajar conformation was obtained using a mismatch template-dNTP combination. Structural comparisons showing KlenTaq with an abasic template (3LWL, red, panel b) and unnatural 5SICS-NaMTP base pair (4C8K, purple, panel c), both structures superimpose on the ajar conformation of Bst DNA polymerases (green).
Förster resonance energy transfer (FRET) studies performed on the large (Klenow) fragment of E. coli DNA polymerase I provide additional evidence for the existence of the ajar conformation (Berezhna et al., Reference Berezhna, Gill, Lamichhane and Millar2012; Hohlbein et al., Reference Hohlbein, Aigrain, Craggs, Bermek, Potapova, Shoolizadeh, Grindley, Joyce and Kapanidis2013). Here, an intermediate FRET species, which appears to be a distinct conformation between the open and closed structures, was found to persist in the presence of mismatched substrates but only transiently exists when the complementary dNTP is present. Interestingly, structures of KlenTaq (Klenow-fragment analog of Taq DNA polymerase) containing an abasic site in the template reveal that the conserved gating tyrosine residue (Fig. 4b) can pair opposite an incoming substrate to allow for primer extension, albeit at significantly reduced rates due to the formation of a sub-optimal enzyme active site (Obeid et al., Reference Obeid, Blatter, Kranaster, Schnur, Diederichs, Welte and Marx2010, Reference Obeid, Welte, Diederichs and Marx2012). More recently, the ajar conformation was witnessed in a ternary structure of KlenTaq bound to the unnatural d5SICS:dNaMTP base pair (Fig. 4c) (Betz et al., Reference Betz, Malyshev, Lavergne, Welte, Diederichs, Romesberg and Marx2013). Collectively, these data suggest that the ajar conformation plays a functional role in nucleotide discrimination in which base pair mismatches stabilize an intermediate conformation that is not catalytically active.
Capturing phosphodiester bond formation by time-resolved crystallography
Data acquired from structural studies into the mechanism of DNA synthesis confirm the prediction that all polymerases catalyze the same nucleotide-transfer reaction, which involves the formation of a phosphodiester bond through nucleophilic attack of the 3′-OH group of the primer on the α-phosphate of the incoming nucleoside triphosphate with concomitant displacement of the pyrophosphate leaving group (Steitz et al., Reference Steitz, Smerdon, Jager and Joyce1994). The reaction is pH-dependent and analogous to acid-base catalysis, where the nucleophile (3′-OH) needs to be deprotonated and the leaving group (pyrophosphate) needs to be protonated. It requires two-metal ions that stabilize a pentacoordinate transition state in a bimolecular substitution (SN2) reaction mechanism (Fig. 5). Metal ion A activates the 3′-OH for nucleophilic attack, while metal ion B stabilizes the buildup of negative charge on the pyrophosphate leaving group via coordination to the β- and γ-phosphates. The reaction may be further activated through the formation of an intramolecular hydrogen bond between the 3′-hydroxyl and β-phosphate groups of the incoming dNTP substrate (Genna et al., Reference Genna, Vidossich, Ippoliti, Carloni and De Vivo2016).

Fig. 5. Conventional two-metal mechanism for DNA synthesis. MgA2+ assists in deprotonation of the 3′ OH and MgB2+ stabilizes the transition state and protonation of the pyrophosphate leaving group (2FMS).
Despite the accumulation of significant structural data showing polymerases from all domains of life trapped in various stages of DNA synthesis, the actual step of chemical bond formation has long remained elusive. This problem was elegantly solved when Yang and colleagues applied the technique of time-resolved X-ray crystallography to follow the course of phosphodiester bond formation by human polymerase η (pol η) (Nakamura et al., Reference Nakamura, Zhao, Yamagata, Hua and Yang2012). In this study, inactive pol η crystals were obtained by crystallizing a ternary complex of the polymerase bound to DNA and dATP in the presence of Ca2+ ions, a catalytically inactive divalent metal ion. The nucleotide-transfer reaction was then initiated in crystallo by transferring individual crystals first to a wash buffer and then to a reaction buffer containing Mg2+ ions, which displace the Ca2+ ions and allow phosphodiester bond formation to proceed. The reaction was stopped at defined times by freezing the crystals in liquid nitrogen for structural analysis. Electron density maps reveal that Mg2+ ions displace the Ca2+ ion within the first 40 s, forming the two-metal ion complex required for nucleotide transfer. From 40 to 230 s, the structures show a steady increase in the nucleotide addition product, thus capturing the chemical bond forming step (Fig. 6). Transient densities identified the rate limiting step of the reaction as deprotonation of the 3′-OH group, which is accompanied by a change in the sugar pucker conformation of the terminal nucleotide from C2′ endo to a C3′ endo. Interestingly, a third Mg2+ ion was found to be essential for DNA synthesis (Gao and Yang, Reference Gao and Yang2016). Similar results have also been observed for time-resolved reactions performed on DNA polymerase β (pol β) (Freudenthal et al., Reference Freudenthal, Beard, Shock and Wilson2013).

Fig. 6. Phosphodiester bond formation visualized by time-resolved X-ray crystallography. Two views of 2Fo–Fc maps (1.5σ) of 40 s (4ECR) and 230 s (4ECV) structures reveal progression of catalysis by pol η.
Promiscuous activities of natural polymerases
Although DNA polymerases are generally thought of as remarkably specific catalysts, many examples now exist where natural DNA polymerases are able to incorporate limited numbers of non-cognate or unnatural substrates into an otherwise natural DNA strand. The catalytic activity and fidelity of these reactions varies significantly depending on the type of chemical modification and the number of chemically modified nucleotides incorporated into the growing strand. In general, natural polymerases are more accepting of base modifications made to the 5-position of pyrimidines and the 7-position of purines than modifications made to the sugar moiety. Reactions of this type are typically performed using polymerases that are either naturally or intentionally deficient in exonuclease activity (exo-), which prevents removal of the modified residue after nucleotide incorporation. Most of the examples cataloged to date involve the incorporation of one or a small number of modified nucleotides into an otherwise natural DNA strand. However, a few cases are known where the template or extension product is composed entirely of non-natural nucleotides.
The varying degrees of tolerance exhibited by natural polymerases for unnatural substrates have played an important role in elucidating the mechanistic underpinnings behind how polymerases recognize their substrates. These details are not easily discerned from crystal structures obtained for polymerases caught at a specific step in the DNA synthesis cycle. Instead, they require chemical analogs that probe the enzyme active site in ways that are not possible purely with natural substrates. As illustrated in the section below, such experiments demonstrate that: (1) Watson–Crick hydrogen bonding groups can be rearranged or removed altogether, and (2) substrate tolerance varies considerably depending on the type of polymerase and chemical modification. Such information has provided insights into the limits of substrate specificity and identified starting points for evolving new variants with improved activity.
Natural DNA polymerases that function with reverse transcription activity
In 1973, Loeb and colleagues were the first to discover the promiscuous activities of natural polymerases by demonstrating that natural RNA templates can be copied into DNA using E. coli DNA polymerase I (Loeb et al., Reference Loeb, Tartof and Travaglini1973). This activity, commonly known as reverse transcription (RT), makes it possible to synthesize the cDNA products of RNA sequences. In nature, reverse transcription is mediated by reverse transcriptases, a class of polymerases that are responsible for replicating the genomes of RNA viruses (Coffin and Fan, Reference Coffin and Fan2016). Nearly two decades later, other laboratories recognized that Taq and Thermus thermophilus (Tth) DNA polymerases have measurable RT activity with Tth exhibiting 100-fold greater activity than Taq (Jones and Foulkes, Reference Jones and Foulkes1989; Tse and Forget, Reference Tse and Forget1990; Myers and Gelfand, Reference Myers and Gelfand1991). Although this activity helped establish the first examples of a coupled RT-PCR process for detecting and quantifying cellular RNAs, Tth's requirement for manganese ions results in higher error rates during cDNA synthesis. More recently, Bergquist and coworkers identified polymerases from other thermophilic organisms that exhibit RT-PCR activity under standard magnesium conditions (Shandilya et al., Reference Shandilya, Griffiths, Flynn, Astatke, Shih, Lee, Gerard, Gibbs and Bergquist2004).
Expanding the genetic alphabet with new hydrogen-bonding base pairs
In 1987, Benner and coworkers suggested that the functional activity of nucleic acid catalysts could be improved by incorporating additional chemical diversity into DNA and RNA (Benner et al., Reference Benner, Allemann, Ellington, Ge, Glasfeld, Leanz, Krauch, MacPherson, Moroney, Piccirilli and Weinhold1987). Toward this goal of augmenting nature's genetic alphabet, several non-natural base pairs were envisioned that would allow for novel hydrogen-bonding schemes between the various hydrogen-bond donor and acceptor groups found on the Watson–Crick face of designer nucleobases (Fig. 7). In 1989, Switzer and Benner demonstrated that this concept was physically possible by enzymatically synthesizing natural genetic polymers containing an unnatural iso-guanine:iso-cytosine (iso-G:iso-C) base pair (Switzer et al., Reference Switzer, Moroney and Benner1989). These experiments were performed using Klenow DNA polymerase and T7 RNA polymerase to synthesize DNA and RNA, respectively. Although iso-G was found to suffer from a minor enol tautomer that leads to mispairing opposite T and iso-C was susceptible to deamination, this foundational study paved the way for what would eventually become an artificially expanded genetic information system that includes the four canonical bases found in DNA plus four additional genetic letters that make up the S:B and Z:P base pairs (Piccirilli et al., Reference Piccirilli, Krauch, Moroney and Benner1990; Benner, Reference Benner2004; Hoshika et al., Reference Hoshika, Leal, Kim, Kim, Karalkar, Kim, Bates, Watkins, Santalucia, Meyer, Dasgupta, Piccirilli, Ellington, Santalucia, Georgiadis and Benner2019).
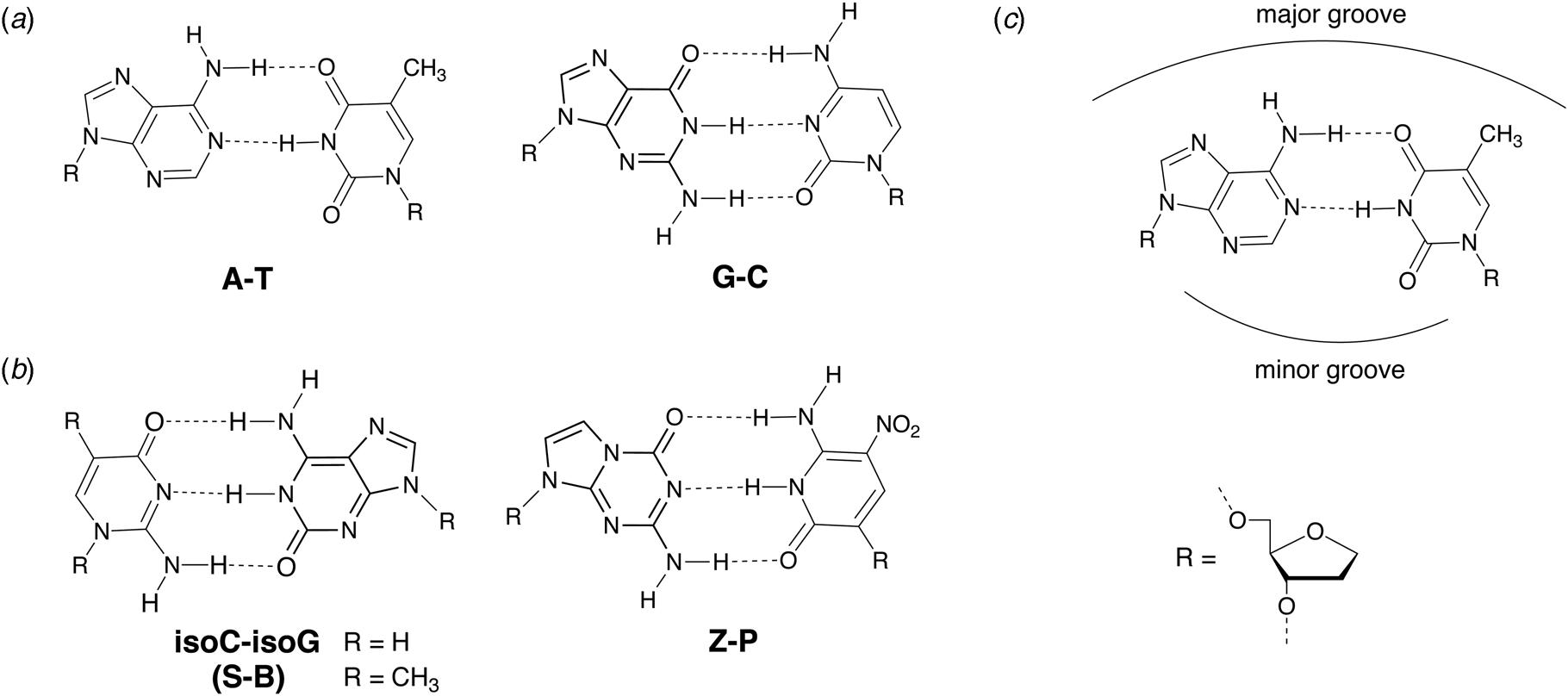
Fig. 7. Chemical structures of natural and alternative DNA base pairs with complementary hydrogen bonding groups. (a) The natural A:T and G:C base pairs. (b) Examples of alternative base pairs obtained by shuffling the hydrogen bond donor and acceptor groups. (c) Schematic view of the major and minor groove regions of a Watson–Crick base pair.
Being the first unnatural base pair, iso-G:iso-C was widely studied in a variety of different contexts. Tor and Dervan used N 6-(6-aminohexyl)isoguanine (6-AH-isoG) to establish a general protocol for site-specifically labeling RNA (Tor and Dervan, Reference Tor and Dervan1993). Accordingly, T7 RNA polymerase is used to transcribe RNA molecules that contain the 6-AH-isoG nucleotide at a defined position that is then post-transcriptionally modified by coupling biotin or a fluorescent dye to the primary amino group attached to the iso-G nucleobase. Whereas iso-C was originally reported to be prone to deamination, Horn and colleagues found that the 5-methyl iso-C (iso-CMe) analog ameliorates this problem (Horn et al., Reference Horn, Chang and Collins1995). The ability to chemically synthesize oligonucleotides containing iso-C and iso-G led to thermal and thermodynamic studies on duplexes arranged in both the antiparallel and parallel strand configurations (Roberts et al., Reference Roberts, Bandaru and Switzer1997a; Seela et al., Reference Seela, He and Wei1999) as well as the formation of iso-G tetraplex and pentaplex motifs that self-assemble around monovalent cations (Roberts et al., Reference Roberts, Chaput and Switzer1997b; Chaput and Switzer, Reference Chaput and Switzer1999; Kang et al., Reference Kang, Heuberger, Chaput, Switzer and Feigon2012). Moreover, iso-G:iso-C base pairing has been visualized inside the duplex of DNA crystals (Robinson et al., Reference Robinson, Gao, Bauer, Roberts, Switzer and Wang1998), evaluated in the context of the hammerhead ribozyme (Ng et al., Reference Ng, Benseler, Tuschl and Eckstein1994), shown to replicate nonenzymatically (Chaput and Switzer, Reference Chaput and Switzer2000), and found to be a viable substrate for RecA-mediated DNA recombination (Rice et al., Reference Rice, Chaput, Cox and Switzer2000). More recently, the iso-G:iso-CMe base pair has been renamed the S:B pair in honor of its inventors, Switzer and Benner (Hoshika et al., Reference Hoshika, Leal, Kim, Kim, Karalkar, Kim, Bates, Watkins, Santalucia, Meyer, Dasgupta, Piccirilli, Ellington, Santalucia, Georgiadis and Benner2019).
Expanding the genetic alphabet with hydrophobic base pairs
An alternative approach to generating unnatural base pairs began in 1997 when Kool and colleagues made the surprising discovery that hydrogen bonding is not an absolute requirement for DNA synthesis (Moran et al., Reference Moran, Ren, Rumney and Kool1997b). Steady-state kinetic measurements showed that Klenow DNA polymerase recognizes difluorotoluene (F), a non-hydrogen bonding isostere of thymine (Fig. 8a), only ~4-fold less efficiently than natural TTP (Moran et al., Reference Moran, Ren and Kool1997a). Additional polymerase studies revealed that selectivity for the insertion of A opposite F rather than C, T, or G was strikingly similar to that of T, making F a strong shape mimic of T (Moran et al., Reference Moran, Ren and Kool1997a). Subsequent study on 4-methylbenzimidazole (Z), a nonpolar analog of adenine (Fig. 8a), led to the first demonstration in which a hydrophobic base pair was replicated by a DNA polymerase (Morales and Kool, Reference Morales and Kool1998). This study showed that the unnatural Z:F base pair exhibits strong selectivity against natural nucleotides, with the noted exception of dATP mispairing opposite F in the template. Nevertheless, the ability to replicate hydrophobic base pairs in vitro cultivated the notion that hydrophobicity and shape complementarity contribute to the recognition of DNA substrates (Kool, Reference Kool2002).

Fig. 8. Chemical structures of hydrophobic base pairs developed for DNA replication. (a) First-generation base pairs designed as hydrophobic shape mimics (isosteres) of natural Watson–Crick base pairs. (b) Second-generation analogs established for higher replication efficiency and fidelity using an iterative chemical optimization approach of design, synthesize, and test.
Inspired by the success of the Z:F base pair, Schultz and Romesberg applied a more traditional medicinal chemistry approach to identify an array of nonpolar molecules that are recognized as base pairs by natural DNA polymerases (Ogawa et al., Reference Ogawa, Wu, Mcminn, Liu, Schultz and Romesberg2000). One of the more successful early examples was 7AI, an indole ring system that is capable of self-pairing (7AI:7AI) in duplex DNA (Fig. 8a) (Tae et al., Reference Tae, Wu, Xia, Schultz and Romesberg2001). Using Klenow DNA polymerase, 7AI showed modest incorporation efficiency (~200-fold less efficient than natural bases), but high selectivity against natural nucleotides (Tae et al., Reference Tae, Wu, Xia, Schultz and Romesberg2001). However, 7AI is poorly extended after nucleotide incorporation, which limits its utility as an orthogonal third base pair. This problem was partially solved using a second DNA polymerase, mammalian polymerase β (pol β), which allows DNA synthesis to continue from a primer that has been extended with 7AI (Tae et al., Reference Tae, Wu, Xia, Schultz and Romesberg2001).
In 2003, Hirao and colleagues extended the number of non-polar base pairs that are recognized by DNA polymerases by demonstrating strong shape complementarity between the adenosine analog Q and a new pyrimidine analog pyrrole-2-carbaldehyde (Pa) (Mitsui et al., Reference Mitsui, Kitamura, Kimoto, To, Sato, Hirao and Yokoyama2003). The Q:Pa base pair (Fig. 8a) was designed to be more selective than the original Q:F base pair, which permits modest to high levels of misincorporation opposite A and T nucleotides (Morales and Kool, Reference Morales and Kool1999). Unlike the 7AI:7AI base pair, Klenow DNA polymerase is able to efficiently incorporate and extend the Q:Pa base pair in both sequence contexts with Q or Pa present in the template strand. Mispairing experiments reveal that dATP inserts opposite either Q or Pa, but that the resulting mispair leads to chain termination in the subsequent extension step. However, PaTP is inserted and extended with low efficiency opposite A, indicating that the geometry of the terminal Pa nucleotide is not a complete impediment to further extension. These data indicate that the Q:Pa pair is an improvement over the original F:Q pair in terms of selectivity and extension efficiency but that further engineering would be required to achieve true orthogonality.
Expanding the genetic alphabet with metal-mediated base pairs
Metal-mediated base pairs represent a third approach for expanding the genetic alphabet beyond the four bases found in nature. Metal-mediated base pairs consist of two artificial bases that coordinate a suitable metal ion in the Watson–Crick base pairing region of a natural base pair (Jash and Muller, Reference Jash and Muller2017). Dozens of examples have been described that coordinate metal ions, such as Cu2+, Ag1+, Hg2+, Pd2+, and Cd2+, in synthetic DNA produced by solid-phase synthesis. Although metal-mediated base pairs have been described in the architectures of several DNA nanostructures (Jash and Muller, Reference Jash and Muller2017), significantly less is known about their recognition properties in the context of DNA replication. One of the more successful examples is the dS–Cu–dS base pair (Fig. 9), which is fully orthogonal and can be PCR amplified in the presence of the canonical A:T and G:C base pairs (Kaul et al., Reference Kaul, Muller, Wagner, Schneider and Carell2011). However, the requirement for an organic co-factor (ethylene diamine) in addition to the inorganic co-factor (Cu2+) may limit the application of the dS–Cu–dS base pair relative to other pairs that rely on an inorganic co-factor alone (Kim and Switzer, Reference Kim and Switzer2013; Kobayashi et al., Reference Kobayashi, Takezawa, Sakamoto and Shionoya2016; Rothlisberger et al., Reference Rothlisberger, Levi-Acobas, Sarac, Marliere, Herdewijn and Hollenstein2017). Despite this minor weakness, the ability to design unnatural base pairs based on metal ion coordination chemistry provides ample room for further development. For example, Shionoya and colleagues recently found that Cu2+-mediated artificial base pairing offers a novel approach for controlling the allosteric regulation of catalytic DNA molecules (Nakama et al., Reference Nakama, Takezawa, Sasaki and Shionoya2020). One could imagine applying similar design principles toward the development of metal-responsive materials and logic circuits.

Fig. 9. Chemical structure of a metal-mediated DNA base pair. Metal-mediated base pairs consist of two ligands in the DNA nucleobase position that coordinate a metal ion.
Replicating six-letter genetic alphabets with increased efficiency and fidelity
Early efforts toward the development of orthogonal base pairs led to the realization that many first-generation base pairs suffer from problems that limit their use in practical applications. In some cases, the efficiency of nucleotide incorporation was low when compared to natural bases, while other cases witnessed poor extension kinetics with the polymerase pausing after nucleotide insertion (Hamashima et al., Reference Hamashima, Kimoto and Hirao2018). Another common problem was nucleotide selectivity in the enzyme active site with unnatural bases mispairing to varying degrees with natural bases (Hamashima et al., Reference Hamashima, Kimoto and Hirao2018). To overcome these problems, organic chemistry was used to design new versions of unnatural base pairs that replicate with higher catalytic efficiency and fidelity. Benner and colleagues, for example, developed the Z:P base pair (Fig. 7), which is more stable than a conventional G:C base pair (Wang et al., Reference Wang, Hoshika, Peterson, Kim, Benner and Kahn2017). In the context of a six-letter genetic alphabet, the Z:P base pair is sufficiently robust that it can be enzymatically synthesized (Yang et al., Reference Yang, Sismour, Sheng, Puskar and Benner2007), amplified by PCR and sequenced (Yang et al., Reference Yang, Chen, Alvarado and Benner2011), transcribed into RNA and reverse transcribed back into DNA (Leal et al., Reference Leal, Kim, Hoshika, Kim, Carrigan and Benner2015), subjected to iterative rounds of in vitro selection, and used to evolve aptamers, a type of synthetic antibody (Dunn et al., Reference Dunn, Jimenez and Chaput2017), that bind to breast and liver cancer cell lines (Sefah et al., Reference Sefah, Yang, Bradley, Hoshika, Jimenez, Zhang, Zhu, Shanker, Yu, Turek, Tan and Benner2014; Zhang et al., Reference Zhang, Yang, Sefah, Bradley, Hoshika, Kim, Kim, Zhu, Jimenez, Cansiz, Teng, Champanhac, Mclendon, Liu, Zhang, Gerloff, Huang, Tan and Benner2015b). In subsequent study, DNA aptamers containing Z and P were generated with high specificity to mammalian cells overexpressing glypican 3, a known biomarker for liver cancer (Zhang et al., Reference Zhang, Yang, Trinh, Teng, Wang, Bradley, Hoshika, Wu, Cansiz, Rowold, Mclendon, Kim, Wu, Cui, Liu, Hou, Stewart, Wan, Liu, Benner and Tan2016).
Similarly, Romesberg and Hirao also developed second generation unnatural base pairs that faithfully replicate using natural DNA polymerases (Malyshev and Romesberg, Reference Malyshev and Romesberg2015; Hamashima et al., Reference Hamashima, Kimoto and Hirao2018). For example, the hydrophobic TPT3:NAM base pair (Fig. 8b) generated by Romesberg and coworkers achieves 99.98% selectivity per doubling by PCR using OneTaq DNA polymerase (Li et al., Reference Li, Degardin, Lavergne, Malyshev, Dhami, Ordoukhanian and Romesberg2014), and the hydrophobic Ds:Px base pair (Fig. 8b) produced by Hirao and colleagues achieves 99.97% selectivity per doubling by PCR using Deep Vent DNA polymerase (Okamoto et al., Reference Okamoto, Miyatake, Kimoto and Hirao2016). The Ds:Px base pair was used to evolve high affinity DNA aptamers containing five genetic letters (A,C,G,T,Ds) to the protein targets vascular endothelial growth factor 165 (VEGF165), interferon-γ (INFγ), and von Willebrand factor A1 domain (vWF) (Kimoto et al., Reference Kimoto, Yamashige, Matsunaga, Yokoyama and Hirao2013; Matsunaga et al., Reference Matsunaga, Kimoto and Hirao2017). The increased chemical diversity of these libraries led to the production of aptamers with significantly higher affinity for their targets than comparable libraries using only natural bases. In subsequent study, the Ds-containing DNA aptamers were shown to inhibit VEGF165 and INFγ binding to their cognate cellular receptors (Matsunaga et al., Reference Matsunaga, Kimoto, Hanson, Sanford, Young and Hirao2015; Kimoto et al., Reference Kimoto, Nakamura and Hirao2016), which advances the use of aptamers as synthetic affinity reagents.
Given the propensity for natural polymerases to replicate unnatural base pairs, structural studies were undertaken to compare the geometry of unnatural base pairs to those found in nature. Three different ternary structures have now been solved with an unnatural base pair occupying the insertion site of a KlenTaq DNA polymerase. The examples (Fig. 10) feature the unnatural base pairs of NaM–5SICS, Ds–Px, and P–Z in which the nucleotides NaM, Ds, and P occupy the templating position and 5SICS, Px, and Z are the incoming substrates, respectively (Betz et al., Reference Betz, Malyshev, Lavergne, Welte, Diederichs, Dwyer, Ordoukhanian, Romesberg and Marx2012, Reference Betz, Kimoto, Diederichs, Hirao and Marx2017; Singh et al., Reference Singh, Laos, Hoshika, Benner and Georgiadis2018). The collection of structures shows the artificial base pairs adopting planar geometries that are structurally similar to natural base pairs. Interestingly, a solution structure of duplex DNA containing a NaM–5SICS base pair unconstrained by a DNA polymerase reveals an intercalated structure rather than the more normal coplanar structure with edge-on-edge packing (Malyshev et al., Reference Malyshev, Pfaff, Ippoliti, Hwang, Dwyer and Romesberg2010). Similar structures have also been witnessed for other hydrophobic base pairs (Brotschi et al., Reference Brotschi, Haberli and Leumann2001; Matsuda et al., Reference Matsuda, Fillo, Henry, Rai, Wilkens, Dwyer, Geierstanger, Wemmer, Schultz, Spraggon and Romesberg2007; Wojciechowski and Leumann, Reference Wojciechowski and Leumann2011), indicating that the polymerase induces a Watson–Crick geometry required for DNA replication.

Fig. 10. Crystal structures of KlenTaq ternary complexes with unnatural Watson–Crick base pairs. Closed ternary structures of KlenTaq complexed with (a) NaM:5SICSTP (3SV3), (b) Ds:PxTP (5NKL), and (c) P:ZTP (5W6K).
Testing hypotheses about polymerase recognition
Beyond the immediate implications of establishing new hydrophobic base pairs, the ability to construct synthetic analogs of natural bases provides a unique opportunity to test hypotheses about how polymerases recognize their substrates (Jung and Marx, Reference Jung and Marx2005). In the mid-1990s, some of the first crystal structures of polymerases bound to their substrates were solved to high resolution (Pelletier et al., Reference Pelletier, Sawaya, Kumar, Wilson and Kraut1994; Doublie et al., Reference Doublie, Tabor, Long, Richardson and Ellenberger1998). These structures, which include Bst DNA polymerase (Kiefer et al., Reference Kiefer, Mao, Braman and Beese1998), a close structural analog of Klenow DNA polymerase, reveal the presence of hydrogen bonding interactions between polar side chains and hydrogen bond acceptor atoms (N3 of purines and O2 of pyrimidines) found on the minor groove side of A:T and G:C base pairs. The observation of these interactions in the enzyme active site suggested that minor groove hydrogen bonding is an important aspect of DNA substrate recognition. To test this hypothesis, DNA synthesis reactions were performed using hydrophobic bases that either contain or lack minor groove hydrogen-bonding acceptor atoms (Morales and Kool, Reference Morales and Kool1999). The resulting data clearly show that minor groove hydrogen bonding is critical for base pair recognition. Moreover, these interactions are more prevalent at the nucleotide extension step than the nucleotide insertion step and are stronger for the growing primer strand than the templating strand (Morales and Kool, Reference Morales and Kool1999). Interestingly, each of the second-generation unnatural base pairs described above (Z:P, 5SICS:NaM, and Ds:Px, see Fig. 8b) have hydrogen bond acceptor atoms on the minor groove side of the Watson–Crick base pair to facilitate polymerase recognition.
Recognizing chemical modifications made to nucleobase positions
Structure–activity studies indicate that thermophilic DNA polymerases exhibit broad tolerance for chemical modifications made to the C5 position of pyrimidines and the C7 position of 7-deazapurines (Fig. 11) (Jager and Famulok, Reference Jager and Famulok2004; Jager et al., Reference Jager, Rasched, Kornreich-Leshem, Engeser, Thum and Famulok2005; Hollenstein, Reference Hollenstein2012; Kielkowski et al., Reference Kielkowski, Fanfrlik and Hocek2014; Cahova et al., Reference Cahova, Panattoni, Kielkowski, Fanfrlik and Hocek2016). Notable examples include the use of Kod and Vent DNA polymerases to evolve slow off-rate modified aptamers (SOMAmers) from diversity-enhancing libraries containing C5-modified deoxyuridine residues (Vaught et al., Reference Vaught, Bock, Carter, Fitzwater, Otis, Schneider, Rolando, Waugh, Wilcox and Eaton2010; Gawande et al., Reference Gawande, Rohloff, Carter, Von Carlowitz, Zhang, Schneider and Janjic2017). This strategy led to the development of an array-based platform for monitoring protein levels in human serum (Ostroff et al., Reference Ostroff, Bigbee, Franklin, Gold, Mehan, Miller, Pass, Rom, Siegfried, Stewart, Walker, Weissfeld, Williams, Zichi and Brody2010; Williams et al., Reference Williams, Kivimaki, Langenberg, Hingorani, Casas, Bouchard, Jonasson, Sarzynski, Shipley, Alexander, Ash, Bauer, Chadwick, Datta, Delisle, Hagar, Hinterberg, Ostroff, Weiss, Ganz and Wareham2019). Interestingly, the ability to synthesize DNA strands with multiple consecutive modifications uncovered strong substrate preferences between thermophilic A- and B-family DNA polymerases. Famulok and coworkers, for example, found that archaeal B-family DNA polymerases are more accepting of base-modified nucleotides than thermophilic A-family DNA polymerases (Jager et al., Reference Jager, Rasched, Kornreich-Leshem, Engeser, Thum and Famulok2005). Sawai and colleagues made similar observations for C5-modified pyrimidines (Kuwahara et al., Reference Kuwahara, Nagashima, Hasegawa, Tamura, Kitagata, Hanawa, Hososhima, Kasamatsu, Ozaki and Sawai2006). Together, these observations suggest that A- and B-family polymerases have different structural constraints in the major groove region of the polymerase active site.

Fig. 11. Chemical structure of nucleobase-modified DNA. (a) Numbering of pyrimidine and purine ring aromatic systems. (b) Examples of common aliphatic and aromatic side chains.
Marx and coworkers investigated the substrate specificity of A- and B-family DNA polymerases by solving high resolution crystal structures of KlenTaq and Kod DNA polymerases bound to natural and base-modified substrates (Bergen et al., Reference Bergen, Steck, Strutt, Baccaro, Welte, Diederichs and Marx2012, Reference Bergen, Betz, Welte, Diederichs and Marx2013; Kropp et al., Reference Kropp, Durr, Peter, Diederichs and Marx2018; Kropp et al., Reference Kropp, Diederichs and Marx2019). The structures indicate that bulky modifications pass through a cavity that extends outside the enzyme active site. This cavity enables members of both polymerase families to incorporate C5-modified pyrimidines and C7-modified purines into the growing DNA strand and to continue DNA synthesis afterward. Consistent with polymerase activity observed by Famulok and Sawai (Jager et al., Reference Jager, Rasched, Kornreich-Leshem, Engeser, Thum and Famulok2005; Kuwahara et al., Reference Kuwahara, Nagashima, Hasegawa, Tamura, Kitagata, Hanawa, Hososhima, Kasamatsu, Ozaki and Sawai2006), the cavity is larger and more accessible for Kod DNA polymerase than KlenTaq DNA polymerase (Fig. 12). In addition, the structures also show that substrate specificity is impacted by the location of the thumb subdomain. In the case of KlenTaq, the tip of the thumb (residues 506–509) extends into the major groove region of the DNA duplex, whereas the analogous region in Kod (residues 668–675) interacts with the phosphodiester backbone.
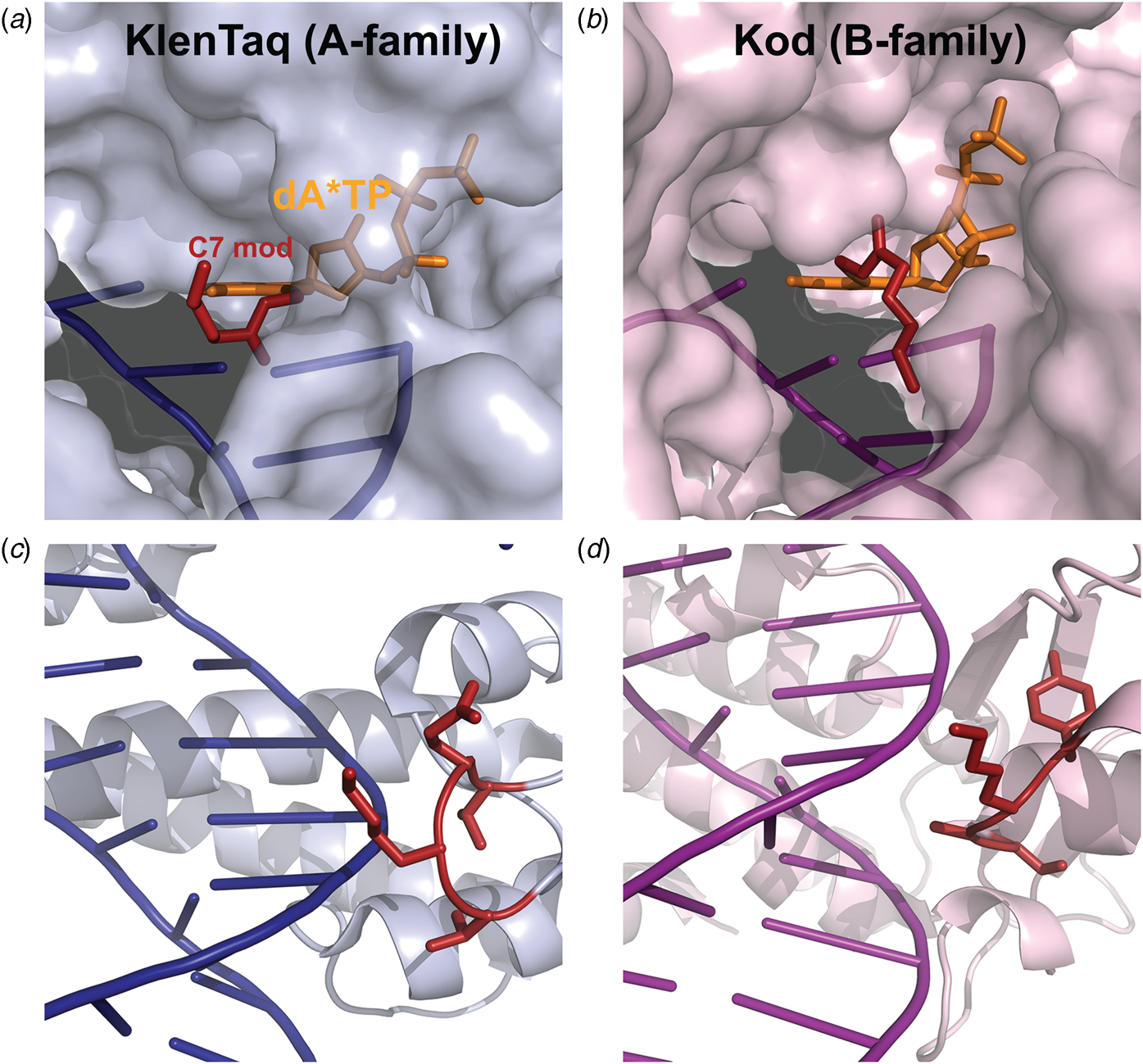
Fig. 12. Structural comparison of A- and B-family polymerases toward modified nucleotides. Surface representation of the closed ternary structures of (a) KlenTaq (6Q4U) and (b) Kod (6Q4T) in complex with a C7-modified dATP (denoted dA*TP) reveals a larger cavity in Kod. Consequently, the C7 modification (red) is well-resolved in KlenTaq but highly flexible in Kod. The thumb subdomains from (c) KlenTaq extends into the major groove, while the analogous region in (d) Kod interacts with the phosphodiester backbone.
Propagation and evolution of an artificial genetic system
In a striking example of enzyme promiscuity, we recently discovered two naturally occurring DNA polymerases that will faithfully replicate 2′-fluoroarabino nucleic acid (FANA) (Wang et al., Reference Wang, Ngor, Nikoomanzar and Chaput2018b), which is an unnatural genetic polymer that contains 2′-fluoroarabino residues in place of natural ribose or deoxyribose nucleotides (Damha et al., Reference Damha, Wilds, Noronha, Brukner, Borkow, Arion and Parniak1998). Kinetic measurements collected using polymerase kinetic profiling (PKPro), a technique that monitors nucleotide synthesis using high-resolution fluorescent dyes that intercalate into the growing duplex (Nikoomanzar et al., Reference Nikoomanzar, Dunn and Chaput2017), reveal that Tgo DNA polymerase catalyzes the synthesis of FANA polymers on DNA templates with a rate of ~15 nt min−1, while Bst DNA polymerase promotes DNA synthesis on FANA templates with a rate of ~1 nt min−1 (Wang et al., Reference Wang, Ngor, Nikoomanzar and Chaput2018b). The replication process occurs with a mutational rate of ~8 × 10−4 and an overall fidelity of 99.9% (Fig. 13a), making it the most faithful replication system for a xeno-nucleic acid (XNA) polymer (Chaput and Herdewijn, Reference Chaput and Herdewijn2019).

Fig. 13. A general RNA-cleaving FANA enzyme. (a) Molecular structures of DNA, RNA, and FANA. (b) Fidelity of FANA replication after a cycle of Tgo transcription and Bst reverse-transcription. (c) Predicted secondary structure showing the FANA enzyme (green) and RNA substrate (red) complex. (d) Time course of RNA cleavage by FANAzyme 12-7. S, RNA substrate; P, 5′-cleavage product. Adapted from Wang et al. (Reference Wang, Ngor, Nikoomanzar and Chaput2018b).
An obvious application of the FANA replication system is the evolution of XNA aptamers and catalysts with enhanced nuclease resistance for diagnostic and therapeutic applications (Houlihan et al., Reference Houlihan, Arangundy-Franklin and Holliger2017). Toward this goal, an efficient RNA-cleaving FANA enzyme (FANAzyme, Fig. 13b) was generated that functions at a rate of >106-fold over the uncatalyzed reaction and achieves substrate saturation with Michaelis–Menten kinetics (Fig. 13c) (Wang et al., Reference Wang, Ngor, Nikoomanzar and Chaput2018b). The enzyme comprises a small 25 nt catalytic domain that is flanked by substrate-binding arms that can be engineered to recognize diverse RNA targets. Divalent metal ion, pH profiles, and mass spectrometry analyses indicate that the reaction follows a metal and pH-dependent transesterification mechanism to produce an upstream cleavage product carrying a cyclic 2′,3′-monophosphate and a downstream strand with a 5′-OH group. In addition to expanding the chemical space of nucleic acid enzymes, this example provides a framework for evolving new types of FANA enzymes that can be generated using commercially available reagents, which is not the case for other XNA systems (Wang et al., Reference Wang, Ngor, Nikoomanzar and Chaput2018b).
Structural insights into Bst DNA polymerase as an XNA reverse transcriptase
Bst DNA polymerase is unusual among naturally occurring replicative DNA polymerases, as it exhibits innate reverse transcriptase activity on nucleic acid templates of diverse chemical composition. Primer-extension studies reveal that Bst will copy templates composed of non-cognate RNA (Shi et al., Reference Shi, Shen, Niu and Ma2015), and the synthetic congeners of glycerol nucleic acid (Tsai et al., Reference Tsai, Chen and Szostak2007), FANA (Wang et al., Reference Wang, Ngor, Nikoomanzar and Chaput2018b), and threose nucleic acid (TNA) (Dunn and Chaput, Reference Dunn and Chaput2016), into full-length DNA products. We obtained the first structural insights into an enzyme with XNA reverse transcriptase activity by solving crystal structures of Bst DNA polymerase that capture the post-translocated product of DNA synthesis on templates composed entirely of FANA and TNA (Fig. 14) (Jackson et al., Reference Jackson, Chim, Shi and Chaput2019). Comparison of these structures with Bst DNA polymerase bound to the natural DNA primer–template duplex (Chim et al., Reference Chim, Jackson, Trinh and Chaput2018) reveals differences, particularly at the enzyme active site as well as in protein interactions with the duplexes (Jackson et al., Reference Jackson, Chim, Shi and Chaput2019). The DNA/FANA and DNA/TNA duplexes within the active site adopt distinct conformations from the natural system (Fig. 14a), whereas the number of protein contacts to the phosphodiester backbone increase by 8 and 13, respectively, presumably to better position the template for DNA synthesis. Interestingly, despite strikingly different backbone conformations, both FANA and TNA adopt B-type helical structures when hybridized to DNA (Fig. 14b). Taken together, these data suggest the importance of structural plasticity as a possible mechanism for XNA-dependent DNA synthesis and offers preliminary rationale for designing variants with improved functional activity. However, it should be stressed that further structural studies are needed to fully understand how gain-of-function mutations are changing the active site conformation of engineering polymerases.

Fig. 14. X-ray crystal structures of natural Bst DNA polymerase exhibiting XNA reverse transcription activity. (a) Active site region with the polymerase bound to a DNA (gray), FANA (6MU4, green), and TNA (6MU5, orange) primer hybridized to a DNA template. Strands 7, 8, 12, and 13 comprise a portion of the antiparallel β-sheet of the palm subdomain. (b) Top-down view of bound the duplex structures. Adapted from Jackson et al. (Reference Jackson, Chim, Shi and Chaput2019).
Engineering polymerase functions by rational design
Rational design has been used to discover new polymerase activities without resorting to molecular evolution. Early strategies utilized natural sequence diversity and residue or domain swapping to change the substrate specificity or biological stability of a polymerase. Structure-guided approaches have also been used to predict specific amino acid changes that would lead to a desired activity. Together, these strategies provide insight into the mechanism of DNA synthesis, the functional role of accessory domains, and the potential for new or improved activities to arise from natural sequence variation. The following section illustrates a number of cases where the deletion or transfer of residues between DNA polymerases leads to enhancements in enzyme performance (Table 2). Other cases, however, show the limitations of rational design and the need for more advanced approaches to enzyme engineering.
Table 2. Engineered polymerases and applications
Structural permutations of natural DNA polymerases
In 1970, Klenow and Henningsen were the first to use limited proteolysis as a way to evaluate the mechanism of a DNA polymerase (Klenow and Henningsen, Reference Klenow and Henningsen1970). Using affinity chromatography to purify DNA polymerase I from crude cellular extracts of E. coli lysate, two distinct polymerase elution profiles emerged with different enzymatic properties and molecular weights. Although both enzymes retained their cognate polymerase and 3′-5′ exonuclease proofreading activities, only the larger enzyme (~150 kDa) exhibited 5′-3′ exonuclease activity. Speculating that the 5′-3′ exonuclease domain had been removed by proteolytic digestion, the larger DNA polymerase was treated with subtilisin to produce a smaller fragment (~70 kDa) with the same size and enzymatic properties observed for the smaller polymerase isolated by affinity chromatography. This version of DNA polymerase I, now commonly known as Klenow DNA polymerase, is routinely used to form blunt ended DNA by filling in 5′ overhangs and removing 3′ overhangs (Sambrook et al., Reference Sambrook, Fritsch and Maniatis T1989) and for second strand cDNA synthesis after reverse transcription of RNA back into DNA (Gubler, Reference Gubler1987). Klenow DNA polymerase holds a special place among synthetic biologists, as it was the first DNA polymerase used to replicate an unnatural base pair in DNA (Switzer et al., Reference Switzer, Moroney and Benner1989).
Following the invention of PCR (Saiki et al., Reference Saiki, Gelfand, Stoffel, Scharf, Higuchi, Horn, Mullis and Erlich1988), a significant effort was made to improve the isolation of Taq DNA polymerase by recombinant protein expression in E. coli so that the enzyme could be used as a tool for molecular biology research. In addition to optimizing the promoter sequence (Lawyer et al., Reference Lawyer, Stoffel, Saiki, Myambo, Drummond and Gelfand1989; Engelke et al., Reference Engelke, Krikos, Bruck and Ginsburg1990), researchers sought to increase protein expression levels by truncating the enzyme. In two separate cases, shorter versions of Taq DNA polymerase (94 kDa) were produced by removing segments of the gene encoding the 5′-3′ exonuclease domain (Fig. 15). The first example was a 705 bp 5′-truncation that yielded a 67 kDa variant called KlenTaq (67 kDa), which is the Klenow-fragment analog of Taq DNA polymerase (Barnes, Reference Barnes1992). The second example was a truncation that removed the first 867 bp region to yield a 61 kDa derivative known as Stoffel (Lawyer et al., Reference Lawyer, Stoffel, Saiki, Chang, Landre, Abramson and Gelfand1993). Although full-length Taq DNA polymerase is widely used in quantitative real-time PCR applications due to its 5′-3′ exonuclease activity, the smaller KlenTaq and Stoffel polymerases are often used to amplify DNA containing modified nucleotides and as starting points for directed evolution (Malyshev et al., Reference Malyshev, Seo, Ordoukhanian and Romesberg2009; Yamashige et al., Reference Yamashige, Kimoto, Takezawa, Sato, Mitsui, Yokoyama and Hirao2012). In recent years, KlenTaq has become a favorite polymerase among X-ray crystallographers wishing to capture the structures of DNA polymerases synthesizing non-cognate and synthetic congeners of natural nucleotides (Betz et al., Reference Betz, Malyshev, Lavergne, Welte, Diederichs, Dwyer, Ordoukhanian, Romesberg and Marx2012, Reference Betz, Kimoto, Diederichs, Hirao and Marx2017; Singh et al., Reference Singh, Laos, Hoshika, Benner and Georgiadis2018).
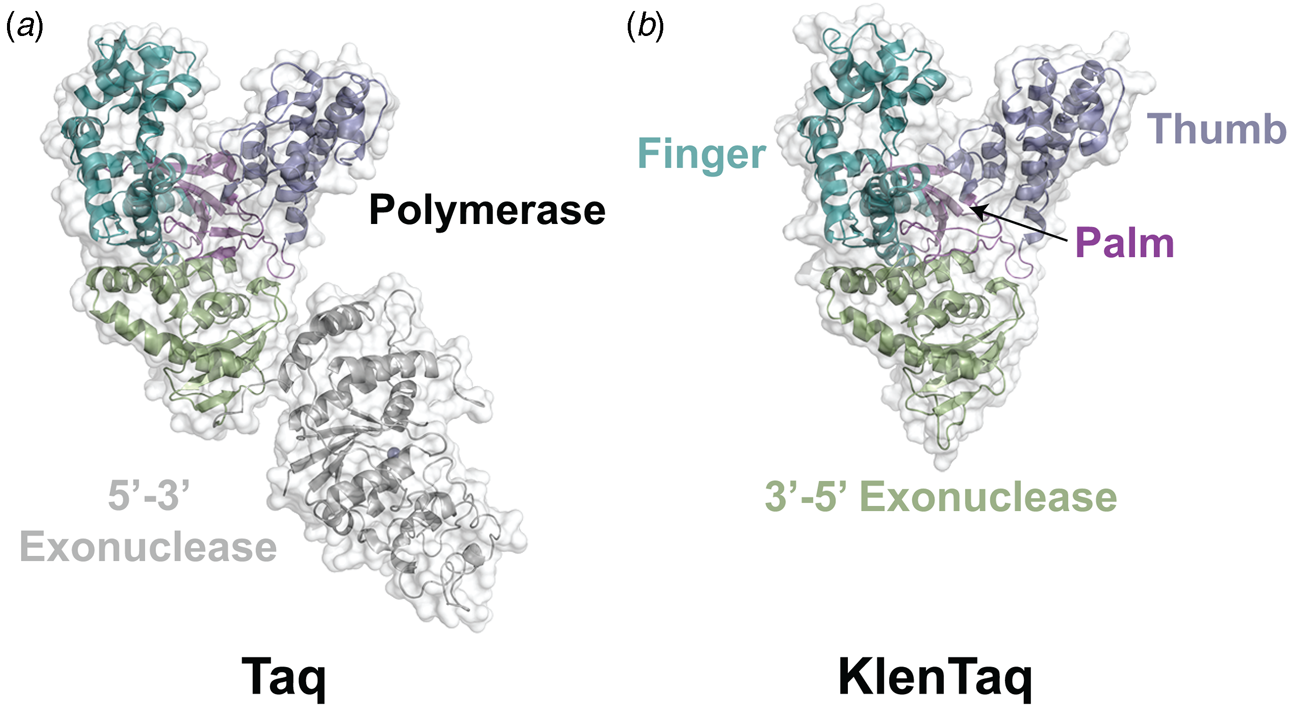
Fig. 15. Structural variants of Taq DNA polymerase. Crystal structures of (a) Taq (1TAQ) and (b) KlenTaq (1KTQ) DNA polymerase. KlenTaq is the Klenow DNA polymerase analog of Taq DNA polymerase in which the 5′-3′ exonuclease domain has been removed.
Exonuclease silencing
The 3′-5′ exonuclease proofreading activity associated with many DNA polymerases is designed to correct single-nucleotide mismatches that occur during the course of normal DNA synthesis. Mutations that silence this activity are often advantageous for synthetic biology applications that require polymerases to incorporate unnatural nucleotides into the growing strand. In the absence of these mutations, modified nucleotides are often difficult to incorporate as the rate of nucleotide addition must compete with the rate of DNA editing. Early attempts at silencing the 3′-5′ exonuclease domains led to the surprising discovery that certain exonuclease-silent (exo-) polymerases can function with enhanced activity. Tabor and Richardson, for example, discovered that T7 DNA polymerase (exo-) functions with ~10-fold higher activity than natural T7 DNA polymerase, which enables the enzyme to read through difficult hairpins (Tabor and Richardson, Reference Tabor and Richardson1989b). Similar activity silencing mutations led to the production of a Bst DNA polymerase variant that functions with elevated thermal stability (Riggs et al., Reference Riggs, Tudor, Sivaram and McDonough1996).
Accelerating DNA synthesis with non-specific DNA-binding domains
Improving the performance of thermophilic DNA polymerases that are capable of PCR amplification was an important early goal in molecular biology. Efforts to study this problem led to the realization that replicative DNA polymerases often use complicated mechanisms that cannot be applied in a general way to in vitro assays. For example, many replicative DNA polymerases rely on accessory proteins, such as thioredoxin (Das and Fujimura, Reference Das and Fujimura1979) or ring-shaped protein complexes that make up the ‘sliding clamp’ (Baker and Bell, Reference Baker and Bell1998), which are highly specific to individual polymerases. One exception is the double-stranded DNA-binding protein Sso7d isolated from Sulfolobus solfataricus, which provides general enzyme enhancing activity when fused to standard DNA polymerases (Wang et al., Reference Wang, Prosen, Mei, Sullivan, Finney and Vander Horn2004). Examples where DNA polymerases have been fused to the Sso7d DNA-binding domain include Taq and Stoffel (both A-family members) and Pfu, a hyperthermophilic archaeal B-family DNA polymerase isolated from P. furiosus. Activity assays show a ~5–20-fold increase in processivity for the three enzymes tested with the greatest increase observed for Stoffel (2.9 nt versus 51 nt per binding event). Importantly, addition of the Sso7d DNA-binding domain to the polymerase did not alter the catalytic properties of the enzyme, which is critical for high-fidelity DNA synthesis. Polymerases engineered with the Sso7d domain reduced the cycle times required for DNA amplification, generated amplicons of increased length, and provided increased tolerance against salt inhibition. Phusion DNA polymerase is an example of a DNA polymerase (Pfu fused to Sso7D) that was engineered for rapid, high-fidelity DNA synthesis of long amplicons.
Helix–hairpin–helix (HhH) motifs found in DNA modifying enzymes, including nucleases, ligases, polymerases, and helicases are a second example where a general DNA-binding motif has been used to enhance the activity of a DNA polymerase. In nature, two-thirds of DNA topoisomerase V is comprised of HhH motifs (Slesarev et al., Reference Slesarev, Stetter, Lake, Gellert, Krah and Kozyavkin1993). When these motifs are removed from the enzyme, topoisomerase retains activity but is more sensitive to salt inhibition than the full-length version. Guided by this observation, variants of Stoffel and Pfu DNA polymerases were constructed with HhH motifs fused to their N- and C-terminal regions (Pavlov et al., Reference Pavlov, Belova, Kozyavkin and Slesarev2002). The engineered polymerases exhibit increased resistance to inhibition under high salt conditions with more HhH motifs providing greater protection. As an example, an engineered Stoffel polymerase remains active in the presence of 250 mM NaCl, whereas the natural polymerase was found to be completely inactive. One hypothesis drawn from this result is that the lack of enzymatic activity observed under high salt conditions is not due to the presence of monovalent ions interacting with the enzyme active site, but rather an inability to form the complex needed for DNA synthesis.
The addition of helicase to the reaction mixture represents a third approach for improving polymerase activity. Helicase-dependent amplification (HDA) uses the energy from ATP and helicase to produce a single-stranded template that can be copied under ambient conditions (Vincent et al., Reference Vincent, Xu and Kong2004). As such, HDA has become an attractive technique for point of care diagnostics that require minimal instrumentation. Versions of HDA have been performed using UvrD helicase, Klenow DNA polymerase, MutL, and single-stranded binding protein (An et al., Reference An, Tang, Ranalli, Kim, Wytiaz and Kong2005). Thermophilic versions of this technique do not require accessory proteins but are limited to short amplicons of only 200 bp in length (An et al., Reference An, Tang, Ranalli, Kim, Wytiaz and Kong2005). To produce longer amplicons, a non-covalent system termed ‘helimerase’ was developed that relies on a coiled-coil motif to synchronize the activities of the helicase and polymerase (Motre et al., Reference Motre, Li and Kong2008). The complex forms in vitro as well as in vivo and can be used to produce amplicons that exceed 1 kb in length.
Determinants of sugar recognition
Natural DNA polymerases are significantly less tolerant toward chemical modifications made to the sugar moiety than the nucleobase. One of the few early reports on sugar recognition is an acyclic peptide nucleic acid derivative that functions as a chain terminator of DNA synthesis (Martinez et al., Reference Martinez, Ansari, Gibbs and Burgess1997). However, despite facile preparation, this analog is less efficient compared to 2′,3′-dideoxyribonucleoside triphosphates, which is the standard reagent set used for Sanger sequencing (Sanger et al., Reference Sanger, Nicklen and Coulson1977). A slightly different example is C4′-acylated thymidine triphosphates developed to study DNA strand repair (Marx et al., Reference Marx, Macwilliams, Bickle, Schwitter and Giese1997, Reference Marx, Spichty, Amacker, Schwitter, Hubscher, Bickle, Maga and Giese1999). Other prominent examples where sugar modifications have been evaluated in DNA polymerase reactions include the recognition of: 2′,5′-isomeric DNA by Klenow and HIV RT (Sinha et al., Reference Sinha, Kim and Switzer2004); acyclic nucleotides by Vent (Gardner et al., Reference Gardner, Joyce and Jack2004); glucose nucleotides by Vent (Renders et al., Reference Renders, Abramov, Froeyen and Herdewijn2009); flexible nucleic acids by Klenow (Heuberger and Switzer, Reference Heuberger and Switzer2008); locked nucleic acid (LNA) by Superscript III (Crouzier et al., Reference Crouzier, Dubois, Edwards, Lauridsen, Wengel and Veedu2012); cyclohexynyl nucleic acid (CeNA) by HIV RT and Vent (Kempeneers et al., Reference Kempeneers, Renders, Froeyen and Herdewijn2005); hexose nucleic acid (HNA) by Klenow and Taq (Pochet et al., Reference Pochet, Kaminski, Van Aerschot, Herdewijn and Marliere2003); and TNA by Superscript II and MMLV RT (Chaput and Szostak, Reference Chaput and Szostak2003; Chaput et al., Reference Chaput, Ichida and Szostak2003). However, the activity observed with these substrates is significantly less than the wild type activity observed with natural substrates. In some cases, manganese ions are used to loosen the enzyme active site, which is a common technique for increasing the tolerance of a DNA polymerase for unnatural nucleoside triphosphates (Dube and Loeb, Reference Dube and Loeb1975; Tabor and Richardson, Reference Tabor and Richardson1989a). However, as noted previously, supplementing the reaction with manganese ions often leads to higher rates of nucleotide misincorporation.
Tabor and Richardson were among the first to explore the determinants of substrate specificity by a DNA polymerase (Tabor and Richardson, Reference Tabor and Richardson1995). Recognizing that bacteriophage T7 DNA polymerase incorporates chain terminating ddNTPs into DNA more efficiently than DNA polymerases from E. coli and Taq, polymerases bearing hybrid sequences in the enzyme active site were constructed and tested to determine the molecular basis of substrate specificity. The mutational study uncovered a single hydroxyl group on Tyr526 that was responsible for the observed substrate specificity. Substitution of Tyr526 in T7 DNA polymerase with phenylalanine increases the discrimination against ddNTPs by >2000-fold, while replacing the analogous Phe residue in either E. coli DNA polymerase I or Taq DNA polymerase with Tyr decreases discrimination against ddNTPs up to 8000-fold. Since E. coli DNA polymerase I binds ddTTP and dTTP with equal affinity, the source of discrimination likely occurs at a subsequent step in the catalytic cycle.
Related studies on Vent DNA polymerase (exo-) isolated from Thermococcus litoralis demonstrated that mutating the active site residue Ala488 to a larger side chain increases the incorporation of sugar-modified nucleotides, including ddNTPs, NTPs, and 3′-dNTPs (Cordycepin) (Gardner and Jack, Reference Gardner and Jack1999). The pattern of relaxed specificity at this position roughly correlates with the size of the amino acid substitution with larger residues showing a higher tolerance for sugar-modified substrates. Similar effects were observed when the Vent Ala488 mutation was transferred to other archaeal DNA polymerases, including Pfu (exo-) (Evans et al., Reference Evans, Fogg, Mamone, Davis, Pearl and Connolly2000) and Kod (exo-) (Hoshino et al., Reference Hoshino, Kasahara, Fujita, Kuwahara, Morihiro, Tsunoda and Obika2016). Addition of the Vent A488L mutation to 9°N produced a commercial enzyme known as Therminator polymerase, which found early widespread use as a research tool for DNA sequencing using acyclic nucleotide analogs (Gardner and Jack, Reference Gardner and Jack2002).
Since its discovery, Therminator DNA polymerase has become the most widely studied and experimentally utilized engineered polymerase for synthesizing modified nucleotides (Gardner et al., Reference Gardner, Jackson, Boyle, Buss, Potapov, Gehring, Zatopek, Correa, Ong and Jack2019). Derived from a hyperthermophilic euryarcheon Thermococcus sp. 9°N, this B-family polymerase carries an A485L mutation in the O-helix of the finger subdomain along with the 3′-5′ exonuclease silencing mutations D141A and E143A. Despite the fact that position 485 faces away from the polymerase active site and does not directly interact with the incoming nucleoside triphosphate, this mutation imparts strong gain-of-function activity for a wide variety of sugar, base, and backbone modified substrates (Bergen et al., Reference Bergen, Betz, Welte, Diederichs and Marx2013; Kropp et al., Reference Kropp, Betz, Wirth, Diederichs and Marx2017). This observation is thought to be due to a change in the dynamics between the open and closed state of the fingers, which increases the occupancy of the closed conformation necessary for chemical catalysis. This relatively straightforward mechanism could explain the ability for Therminator to accept a broad range of substrates, including noncognate substrates (NTPs) (McCullum and Chaput, Reference McCullum and Chaput2009), base-modified substrates (dN*TPs), sugar-modified substrates (xNTPs) (Lapa et al., Reference Lapa, Chudinov and Timofeev2016), unnatural base pair substrates (dXTP and dYTP) (Hwang and Romesberg, Reference Hwang and Romesberg2008), and terminator substrates (ddNTP, acyclic, 3′-blocked dNTP, and lightning terminators) (Gardner and Jack, Reference Gardner and Jack2002; Litosh et al., Reference Litosh, Wu, Stupi, Wang, Morris, Hersh and Metzker2011).
Recognizing the importance of the A485L mutation as a critical determinant of substrate specificity, a significant effort has been made to further improve the activity of this mutation through rational design (Gardner et al., Reference Gardner, Jackson, Boyle, Buss, Potapov, Gehring, Zatopek, Correa, Ong and Jack2019). In the case of RNA synthesis, combining the A485L mutation with Y409G and E664K, the steric gate and so-called second steric-gate, respectively, enabled Tgo DNA polymerase to synthesize RNA strands up to 1.7 kb in length (Cozens et al., Reference Cozens, Pinheiro, Vaisman, Woodgate and Holliger2012). The attachment of two biotinylated ‘peptide legs’ to Therminator led to a polymerase complex with streptavidin that increased the processivity of DNA synthesis from less than 20 nucleotides to several thousand nucleotides per binding event (Williams et al., Reference Williams, Steffens, Anderson, Urlacher, Lamb, Grone and Egelhoff2008). The A485L mutation has also been used to improve XNA synthesis wherein an engineered polymerase named TgoT (V93Q, D141A, E143A, and A485L) provided the backbone for generating new polymerase variants that can synthesize a variety of artificial nucleic acids, including CeNA, ANA, FANA, HNA, TNA, and LNA (Pinheiro et al., Reference Pinheiro, Taylor, Cozens, Abramov, Renders, Zhang, Chaput, Wengel, Peak-Chew, Mclaughlin, Herdewijn and Holliger2012). In the case of next-generation sequencing (NGS), Therminator was used as the starting point for generating a polymerase that facilitates the synthesis of fluorescently-tagged nucleotides (Gardner et al., Reference Gardner, Wang, Wu, Karouby, Li, Stupi, Jack, Hersh and Metzker2012). If the past is any indication of the future, it would seem likely that the next generation of engineered polymerases will benefit from further exploration of the Therminator position.
Improving DNA polymerase performance for PCR
PCR has had a major impact on molecular biology by providing a simple method for amplifying DNA (Saiki et al., Reference Saiki, Gelfand, Stoffel, Scharf, Higuchi, Horn, Mullis and Erlich1988). Early experiments required fresh polymerase to be added during each extension cycle due to the high temperatures >95 °C required for denaturing the DNA strands prior to the start of another round of synthesis. This arduous task greatly reduced the speed of amplification, as it not only required the physical presence of a researcher to add new enzyme but also lowered the theoretical limit of DNA replication due to the presence of increasing quantities of inactive enzyme. A solution to this problem came when a thermophilic DNA polymerase was isolated from the bacterium species T. aquaticus (Chien et al., Reference Chien, Edgar and Trela1976). Taq DNA polymerase was harnessed for its intrinsic thermal stability, which allows for uninterrupted cycles of DNA replication. PCR has since found widespread use in DNA cloning, NGS, criminal forensics, molecular diagnostics, epigenetic mapping, and pathogen detection (Garibyan and Avashia, Reference Garibyan and Avashia2013). However, as the demand for PCR amplification has grown, so has the need for new variants that can function under more demanding conditions.
Genotyping biological samples require precise DNA amplification to distinguish single-nucleotide polymorphisms from random mutations. Recognizing that motif C in A- and B-family DNA polymerases may contribute to mismatch extension through indirect H-bonding between the minor groove and a histidine side chain (Franklin et al., Reference Franklin, Wang and Steitz2001), Marx and coworkers applied a structure-guided approach to identify variants of Taq DNA polymerase that function with increased fidelity. An automated fluorescent screen was established to evaluate 1316 variants of Klenow DNA polymerase (exo-) bearing mutations at positions 879–881 (Summerer et al., Reference Summerer, Rudinger, Detmer and Marx2005). Protein expression was conducted in 96-well plates and crude lysate was queried for activity in 384-well format. Fidelity values were assigned based on the ratio of extension from primers containing matched and mismatched 3′-terminal residues. A Klenow variant with LVL at positions 879–881 exhibited strong kinetic discrimination against mismatch extension. Transferring the LVL mutations to analogous positions in wild-type Taq DNA polymerase produced an engineered version of Taq DNA polymerase with increased discrimination against transitions and transversions (Summerer et al., Reference Summerer, Rudinger, Detmer and Marx2005).
Taq DNA polymerase is readily inactivated by hemoglobin and humic acid present in blood and soil samples used for DNA analysis. Surprisingly, Klentaq1, a truncated version of Taq DNA polymerase with a 278 aa N-terminal deletion, can amplify single-copy genomic DNA in the presence of 5–10% whole blood (Abu Al-Soud and Radstrom, Reference Abu Al-Soud and Radstrom1998; Abu Al-Soud and Radstrom, Reference Abu Al-Soud and Radstrom2000). To generate an enzyme with improved activity, Barnes and coworkers screened a library of 40 arbitrary but functional variants with mutations at positions 626 and 706–708 for improved PCR performance under increasing amounts of whole blood (Kermekchiev et al., Reference Kermekchiev, Kirilova, Vail and Barnes2009). The screen revealed that mutation of E708 to K, L, or W resulted in enhanced resistance to various inhibitors, including plasma, hemoglobin, lactoferrin, serum IgG, soil extracts, and humic acid. The resulting polymerase facilitates the amplification of single-copy human genomic targets from whole blood, which eliminates the need for a sample treatment step.
Archaeal B-family DNA polymerases are widely used enzymes for PCR because of their high thermal stability and presence of a strong 3′-5′ proofreading exonuclease domain. However, despite high sequence and structural homology, the Thermococcales order of archaeal DNA polymerases exhibits strikingly different kinetic properties that affect their PCR performance. Kod DNA polymerase, for example, possesses higher processivity (defined as the number of dNTP incorporations per binding event) than its related homologs but is 10 °C less stable (83 versus 93 °C) than Pfu DNA polymerase, which limits its utility as an enzyme for PCR. To improve the processivity of Pfu DNA polymerase, Connolly and coworkers transferred residues from the forked-point (polymerase junction between the template-binding and editing cleft consisting of seven arginine residues) and entire thumb regions of Kod DNA polymerase to Pfu (Elshawadfy et al., Reference Elshawadfy, Keith, Ee Ooi, Kinsman, Heslop and Connolly2014). The resulting polymerase with the combined forked-point and thumb regions from Kod DNA polymerase retained the high thermal stability of Pfu while gaining an increased capacity for PCR performance.
Similar efforts to explore the natural diversity of DNA polymerases were performed by recombining gene fragments of A-family DNA polymerases taken from soil samples of microorganisms found near thermal hot springs (Yamagami et al., Reference Yamagami, Ishino, Kawarabayasi and Ishino2014). Corresponding regions of the pol gene for Taq DNA polymerase were substituted with the amplified gene fragments and the chimeric variants were tested for activity. Biochemical analysis led to the identification of two mutations, E742R and A743R, that impart higher DNA-binding affinity and faster primer extension activity on Taq DNA polymerase. Both factors resulted in improved PCR performance, suggesting that natural diversity is a promising strategy for finding new amino acid positions with strong gain of function activity.
The ability to sequence epigenetic modifications is an important goal of genomic research. Of all possible epigenetic modifications, none is more prevalent than 5-methylcytosine (5mC). This subtle chemical change has far reaching implications for normal cellular growth and development as well as several neurological diseases and cancer (Allis and Jenuwein, Reference Allis and Jenuwein2016). Bisulfite treatment converts natural cytosine bases to a 5,6-dihydrouracil 6-sulfonate (dhU6S) intermediate that is subsequently hydrolyzed to deoxyuracil (dU). Because 5mC is resistant to bisulfite treatment, this approach can be used to identify 5mC epigenetic markers by mapping the conversion of bases that are read as dC before and after bisulfite treatment. Unfortunately, this approach leads to significant degradation of the genomic DNA sample, which hampers genome-wide association studies. Holliger and coworkers recently discovered that the engineered polymerase 5D4, previously developed to recognize hydrophobic base analogs, is able to amplify DNA carrying the bisulfite intermediate (Millar et al., Reference Millar, Christova and Holliger2015). This discovery greatly improves the workflow and sensitivity of 5mC detection in genomic DNA samples.
Mutagenic DNA polymerases that function with low fidelity have value as reagents for creating degenerate libraries for directed evolution studies and offer clues into the mechanistic underpinnings of substrate recognition during DNA synthesis. To investigate this phenomenon, Loeb and coworkers created a library of ~ 200 000 mutant Taq DNA polymerase variants comprising random mutations in the dNTP binding pocket of motif A (residues 605–617) (Patel et al., Reference Patel, Kawate, Adman, Ashbach and Loeb2001). The library was screened for activity using a temperature-sensitive complementation assay in E. coli and a subset of active variants were tested for fidelity using a mismatch primer extension assay. Taq polymerase variants with strong mismatch extension activity each contain substitutions at I614, indicating that a single, highly mutable, active amino acid is critical for DNA polymerase fidelity. A Taq DNA polymerase variant bearing the I614K mutation was shown to function with a 20-fold higher error rate than wild-type Taq DNA polymerase and can bypass damaged and abasic sites in DNA templates. This example provides an approach for producing polymerases that function with error-prone activity during PCR.
Engineering polymerases by directed evolution
In the last 20 years, the field of polymerase engineering has benefited from the growth of new technologies that make it possible to generate custom polymerases by directed evolution. Whether searching designer libraries that carefully sample all possible single-point mutations at defined positions or less sophisticated libraries that contain random mutations at unknown positions, the technologies available today allow users to rapidly search large combinatorial libraries (>107 unique members) in timeframes ranging from days to weeks. These efforts have been aided by the development of clever strategies for establishing genotype–phenotype linkages that make it possible to determine the sequence of active variants with valuable gain-of-function mutations. The most common approaches perform the activity step in vitro, which allows for greater control over the reaction conditions and substrate chemistries, including the use of synthetic congeners that bear little or no resemblance to natural nucleotides. In addition to establishing new enzymes with practical applications in biotechnology and medicine, these studies also provide a wealth of information about how polymerases function. As these studies continue, sufficient knowledge may be gained that will enable future generations to one day bypass the need for directed evolution and allow computational methods to predict individual sequences with desired activities. However, realizing these dreams will require a greater understanding of the determinants that govern substrate specificity, which is a major goal of most polymerase-engineering efforts.
Phage display
Phage display is one of the oldest and most successful methods for evolving peptides and proteins with ligand binding activity (Smith and Petrenko, Reference Smith and Petrenko1997). With this technique, a gene encoding a protein of interest is inserted into a phage coat protein gene, which causes the phage to display the protein on its outside surface while retaining the encoding genetic information inside the bacteriophage. A modified version of phage display was originally developed by Jestin, and subsequently refined by Romesberg, to facilitate the evolution of polymerases with new activities (Jestin et al., Reference Jestin, Kristensen and Winter1999; Xia et al., Reference Xia, Chen, Sera, Fa, Schultz and Romesberg2002). In this method, phage particles are engineered to display the DNA primer–template duplex and polymerase variant in close proximity. The polymerase library is expressed as an N-terminal fusion of the minor M13 phage coat protein pIII in such a way that the phage surface contains one copy of the polymerase and four copies of a short acidic peptide. Separately, a complementary basic peptide is conjugated to the DNA primer, annealed to a DNA template, and combined with the phage particle to form a coiled-coil linking the DNA primer–template duplex to the phage surface. Activity screens are then performed in-cis by enriching for polymerase variants that can incorporate a biotin-tagged nucleotide into the growing DNA strand, which is used to capture the phage particle on streptavidin-coated beads (Fig. 16). The beads are washed to remove inactive variants and the genes encoding functional polymerases are recovered by eluting the bacteriophage with DNase I. The population of enriched phage particles is then amplified by infecting a fresh E. coli culture. Recently, the technique has been improved by incorporating p-azidophenylalanine into the pIII protein, which allows for an alkynyl-modified primer–template duplex to be conjugated to the phage surface using click-chemistry (Chen et al., Reference Chen, Hongdilokkul, Liu, Adhikary, Tsuen and Romesberg2016). The revised protocol avoids the need to synthesize and purify peptide–DNA conjugates comprising the basic peptide and DNA primer.

Fig. 16. Phage display. Bacteriophage particles are constructed using a proximity strategy that places the polymerase and DNA primer–template duplex in close proximity on the phage surface. Activity screening leads to the identification of polymerase variants that incorporate a biotin-tagged substrate that is captured on streptavidin-coated beads. Functional variants are recovered by eluting the beads with DNase and amplified by infecting a fresh E. coli culture.
Phage display was used by Jestin and coworkers to evolve a population of Taq DNA polymerase variants with thermostable reverse transcriptase activity (Vichier-Guerre et al., Reference Vichier-Guerre, Ferris, Auberger, Mahiddine and Jestin2006). Romesberg and coworkers have used this technique to identify variants of the Stoffel fragment (SF) of Taq DNA polymerase that function with improved activity for ribonucleoside triphosphates (Xia et al., Reference Xia, Chen, Sera, Fa, Schultz and Romesberg2002), 2′-methoxy (OCH3) nucleoside triphosphates (Fa et al., Reference Fa, Radeghieri, Henry and Romesberg2004), and the unnatural PICS:PICS self-pair (Leconte et al., Reference Leconte, Chen and Romesberg2005). Further characterization of the polymerase with 2′ OCH3 activity revealed that this variant (SM19) could also recognize substrates with 2′-fluoro (F), 2′-azido (N3), and 2′-amino (NH2) modifications (Schultz et al., Reference Schultz, Gochi, Chia, Ogonowsky, Chiang, Filipovic, Weiden, Hadley, Gabriel and Leconte2015). Using the click-chemistry version of phage display, SM19 was evolved to yield SM4-9, which is a thermostable polymerase able to PCR amplify DNA containing the 2′-OCH3 and 2′-F modifications on pyrimidine residues (Chen et al., Reference Chen, Hongdilokkul, Liu, Adhikary, Tsuen and Romesberg2016).
The major benefit of the phage display approach is the ability to detect a single nucleotide incorporation event using biotinylated substrates. Anticipated weaknesses include complications of phage particle assembly, the potential for low multiple turnover activity caused by the in-cis selection strategy, and the possibility for high background due to non-specific binding to the solid support. In the case of SM4-9, for example, the selection required the screening of 500–1000 individual variants between each of the four rounds of selection (Chen et al., Reference Chen, Hongdilokkul, Liu, Adhikary, Tsuen and Romesberg2016).
Compartmentalized self-replication
In 2001, Holliger and coworkers developed a polymerase evolution strategy called compartmentalized self-replication (CSR) that is based on a simple feedback loop in which a polymerase replicates its own gene by PCR (Ghadessy et al., Reference Ghadessy, Ong and Holliger2001). With this technique (Fig. 17), a population of E. coli expressing different polymerase variants is encapsulated along with the reaction buffer, dNTPs, and primers into emulsions that are produced by vigorous bulk mixing of aqueous and organic phases. During thermocycling, E. coli lysis occurs, releasing the polymerase and encoding plasmid into the surrounding solution. The emulsion serves as a barrier separating each polymerase extension assay into an individual reaction compartment. If the polymerase is able to amplify its own gene using the gene-specific primers supplied in the aqueous phase, then adaptive gains are made that directly and proportionately translate to an increase in the number of amplicons present that encode the active polymerase variant. Through iterative rounds of selective amplification, active polymerases will outcompete the inactive variants.
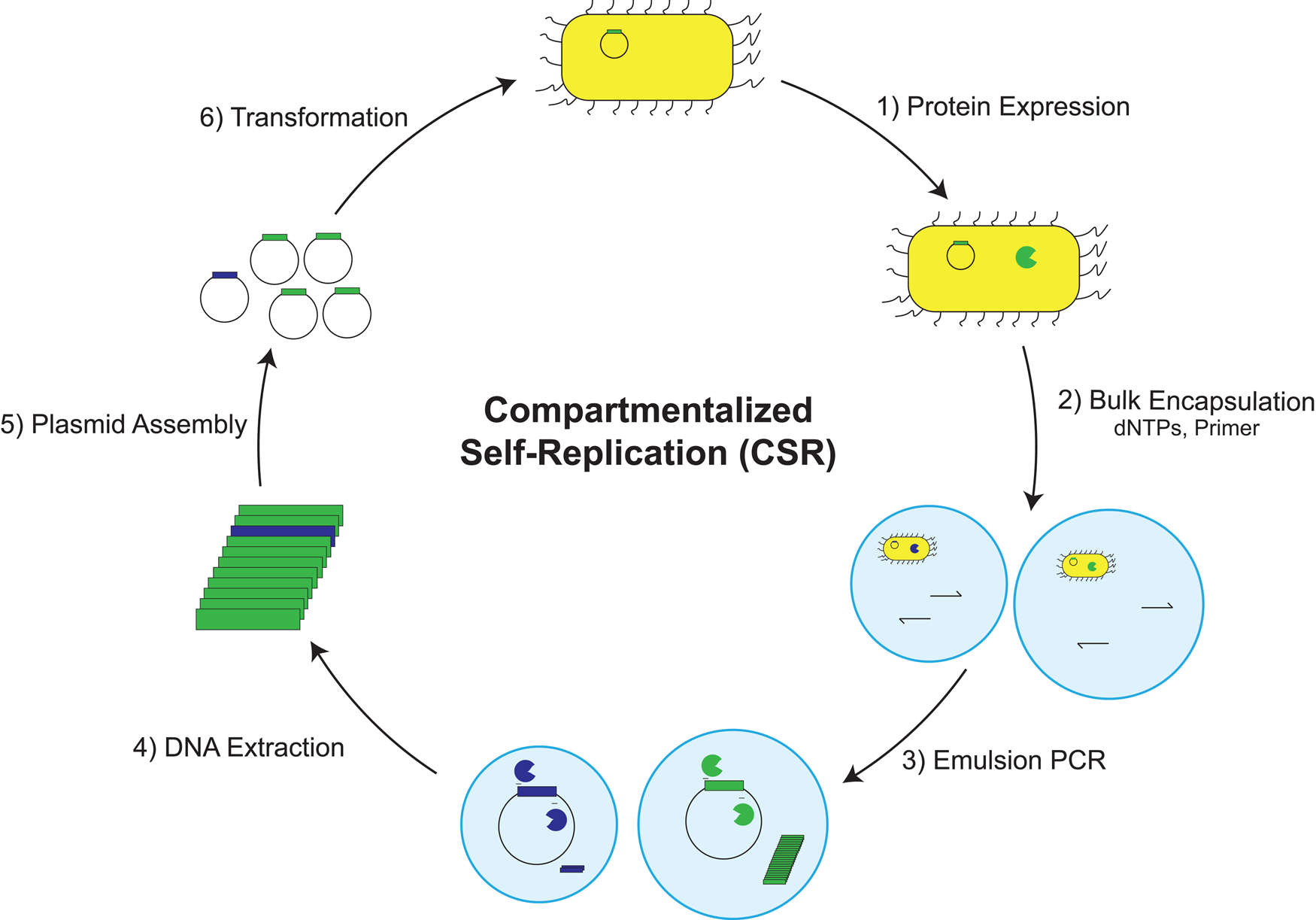
Fig. 17. Compartmentalized self-replication. A library of polymerase genes expressed in E. coli is encapsulated in bulk emulsions. Following PCR amplification inside the droplet, active polymerases generate multiple copies of their own gene, while inactive variants fail to replicate the gene. The degree of amplification is directly proportional to the activity of the polymerase. Through iterative rounds of selective amplification, polymerases with desired activity outcompete the population of inactive variants.
CSR has proven useful for generating polymerases with enhanced thermostability and increased resistance to a range of blood and other environmental inhibitors that prevent DNA samples from being amplified using natural polymerases (Ghadessy et al., Reference Ghadessy, Ong and Holliger2001; Baar et al., Reference Baar, d'Abbadie, Vaisman, Arana, Hofreiter, Woodgate, Kunkel and Holliger2011). Molecular breeding experiments performed on thermophilic polymerases led to the isolation of a chimeric polymerase with an increased ability to amplify DNA from ice-age specimens (d'Abbadie et al., Reference d'Abbadie, Hofreiter, Vaisman, Loakes, Gasparutto, Cadet, Woodgate, Paabo and Holliger2007). CSR has been used to generate polymerases that can recognize a broad range of nucleoside triphosphates, including α-phosphorothioate dNTPs (Ghadessy et al., Reference Ghadessy, Ramsay, Boudsocq, Loakes, Brown, Iwai, Vaisman, Woodgate and Holliger2004), dNTPs with hydrophobic base analogs (Loakes et al., Reference Loakes, Gallego, Pinheiro, Kool and Holliger2009), and γ-modified dNTPs for sequencing and kinetic assays (Hansen et al., Reference Hansen, Wu, Fox, Arezi and Hogrefe2011). More recently, Benner and coworkers used CSR to evolve a polymerase that could amplify DNA with a six-letter genetic alphabet that includes the unnatural base pair P:Z (Laos et al., Reference Laos, Shaw, Leal, Gaucher and Benner2013).
Modified versions of CSR have been developed to reduce the adaptive burden of amplifying the entire polymerase gene (>2 kb). The first modified version, termed short-patch CSR (spCSR), focuses the amplification step on a narrow segment of the polymerase gene, which is then incorporated into the full-length gene when the plasmid is reconstructed between rounds of selection (Ong et al., Reference Ong, Loakes, Jaroslawski, Too and Holliger2006). spCSR enables the isolation of Taq DNA polymerase variants with enhanced activity for 2′-modified nucleotides including NTPs (Ong et al., Reference Ong, Loakes, Jaroslawski, Too and Holliger2006) as well as Pfu variants capable of replacing dCTP with fluorescent Cy3- and Cy5-labeled dCTP substrates in PCR reactions (Ramsay et al., Reference Ramsay, Jemth, Brown, Crampton, Dear and Holliger2010). Ellington and coworkers developed another version of CSR called reverse-transcription CSR (RT-CSR), which enables the screening of up to 109 polymerase variants for RT activity (Ellefson et al., Reference Ellefson, Gollihar, Shroff, Shivram, Iyer and Ellington2016). RT-CSR was used to produce a thermostable polymerase that actively proofreads DNA synthesis during RT-PCR.
CSR benefits from a strong feedback loop that enables the identification of new polymerase variants that are capable of PCR. However, the PCR reactions take place in polydisperse droplets, which could lead to uneven levels of PCR amplification. CSR is also limited to the range of polymerase functions that promote DNA, or RNA in the case of RT-CSR, templated synthesis.
Compartmentalized self-tagging
Efforts to establish engineered polymerases with increased tolerance for challenging substrates with highly modified sugars led to the development of compartmentalized self-tagging (CST) (Pinheiro et al., Reference Pinheiro, Taylor, Cozens, Abramov, Renders, Zhang, Chaput, Wengel, Peak-Chew, Mclaughlin, Herdewijn and Holliger2012). CST is based on a positive selection loop where a polymerase tags its encoding DNA plasmid with a biotinylated primer that hybridizes to a complementary region of the plasmid (Fig. 18). The initial primer–plasmid complex is a weak affinity interaction that becomes stabilized when the primer is extended by the polymerase. After extension, the primer–plasmid complexes are captured on streptavidin-coated beads, which are washed with mild denaturants to remove the unextended primer–plasmid pairs. Plasmids encoding active library members are then recovered from the beads, PCR amplified, and used to initiate another round of selection and amplification.

Fig. 18. Compartmentalized self-tagging. E. coli cells expressing different polymerase variants are encapsulated in bulk emulsions. Following E. coli lysis, the polymerase is challenged to extend a biotinylated primer annealed to the plasmid. Active polymerases that extend the primer increase the stability of the primer–plasmid complex. After disruption of the emulsion, the primer–plasmid complexes are captured on streptavidin beads, and plasmids annealed to unextended primers are removed with washing. Plasmids annealed to extended primers are recovered, PCR amplified, and used to initiate another round of selection.
CST enabled the discovery of engineered polymerases that could synthesize XNA polymers with backbone structures that are distinct from those found in DNA and RNA (Pinheiro et al., Reference Pinheiro, Taylor, Cozens, Abramov, Renders, Zhang, Chaput, Wengel, Peak-Chew, Mclaughlin, Herdewijn and Holliger2012). By exploring diverse library repertoires of Tgo DNA polymerase that sampled mutations within a 10 Å shell of the polymerase active site, novel polymerase variants were identified that could copy DNA templates into HNA, CeNA, TNA, FANA, and ANA. In this same study, a statistical coupling analysis was used to identify polymerases that could copy the XNA strands back into DNA. Together, these polymerase pairs demonstrate the capacity for artificial genetic polymers to replicate using engineered polymerases to facilitate the passage of genetic information back and forth between DNA and XNA. CST is widely recognized as a major advance in synthetic genetics, a field which aims to explore the structural and functional properties of XNA by in vitro selection (Joyce, Reference Joyce2012).
The major advantage of CST is that it allows for the evolution of polymerases that can synthesize nucleic acid polymers with diverse sugar-phosphate backbones. However, the range of functions is limited to DNA-templated reactions (i.e. DNA-dependent XNA polymerases), as the selection strategy uses the plasmid DNA as the template for the primer-extension reaction. CST also requires affinity purification on a solid support matrix, which lowers the partitioning efficiency of the selection due to unwanted non-specific binding of DNA to the matrix. Finally, the reliance on a metastable primer–plasmid complex requires fine-tuning of the denaturing conditions to ensure proper separation of the plasmids encoding active and inactive variants.
Droplet-based optical polymerase sorting
To overcome some of the weaknesses of previous in vitro selection technologies, our laboratory established a general strategy for evolving new polymerase functions called droplet-based optical polymerase sorting (DrOPS) (Larsen et al., Reference Larsen, Dunn, Hatch, Sau, Youngbull and Chaput2016). DrOPS is a high-throughput approach that combines the ultrafast screening power of microfluidics with the high sensitivity of optical sorting. With this technique, a library of polymerase variants is expressed in E. coli and single cells are encapsulated in microfluidic droplets containing a fluorescent sensor that is responsive to polymerase activity (Fig. 19). As with CSR and CST, the surrounding oil acts as a barrier preventing the contents of one droplet from mixing with the contents of another droplet. However, unlike CSR and CST, microfluidic devices are used to generate a uniform population of droplets. The latest microfluidic designs are capable of generating 18 μm droplets at a rate of 30 000 per second, which allows for the production of >108 droplets in 1 h (Vallejo et al., Reference Vallejo, Nikoomanzar, Paegel and Chaput2019). Following droplet production, the polymerase and encoding plasmid are released into the droplet by lysing the E. coli with heat. Polymerases that successfully copy the template into full-length product produce a fluorescent signal by disrupting a donor–quencher pair located at the 5′-end of the template strand. The population of droplets can then either be sorted directly using a custom microfluidic fluorescence-activated droplet sorting (FADS) device or converted to double emulsion droplets that are compatible with a traditional fluorescence-activated cell sorting instrument (Vallejo et al., Reference Vallejo, Nikoomanzar, Paegel and Chaput2019).

Fig. 19. Droplet-based optical polymerase sorting. E. coli cells expressing different library members are encapsulated in water-in-oil droplets using a microfluidic device. The droplets are collected and lysed off-chip to release the polymerase and encoding plasmid into the solution. Polymerases that extend the primer to full-length product trigger a fluorescent sensor by disrupting a fluorescent donor–quencher pair. Fluorescent droplets are sorted using a custom FADS device. Recovered DNA is PCR amplified and used to initiate another round of selection.
Despite being a relatively new technique for polymerase engineering, DrOPS has been used to evolve polymerase variants that can synthesize TNA, an artificial genetic polymer in which the natural ribose sugar found in RNA has been replaced with an unnatural threose sugar (Schöning et al., Reference Schöning, Scholz, Guntha, Wu, Krishnamurthy and Eschenmoser2000). In its first demonstration, DrOPS was used to identify a manganese-independent TNA polymerase from a site-saturation library of 8000 unique variants after a single round of high-throughput screening (Larsen et al., Reference Larsen, Dunn, Hatch, Sau, Youngbull and Chaput2016). More recently, DrOPS was combined with the protein-engineering approach of deep mutational scanning (Araya and Fowler, Reference Araya and Fowler2011), to map the sequence function relationships of a replicative DNA polymerase, Kod, isolated from the thermophilic archeae T. kodakarensis (Nikoomanzar et al., Reference Nikoomanzar, Vallejo and Chaput2019). The resulting enrichment profile provided an unbiased view of the ability of each single-point mutant to synthesize TNA. From a single high-throughput screen, two cases of epistasis were discovered, where double-mutant variants functioned with higher activity than the sum of the contributions from either of the individual mutations. This new polymerase, termed Kod-RS, recognizes TNA substrates with nearly the same efficiency as DNA substrates, suggesting that the mutations are beginning to reshape the enzyme active site. An engineered variant with even greater TNA polymerase activity was discovered by performing deep mutational scanning across the entire polymerase domain (Nikoomanzar et al., Reference Nikoomanzar, Vallejo, Yik and Chaput2020).
The DrOPS technique compares favorably with other polymerase-engineering technologies in several important ways. First, it provides enormous control over the composition of the primer, template, and nucleoside triphosphates, which should make it possible to select for any type of polymerase activity (i.e. transcription, reverse transcription, and replication). Second, it relies on physical methods for identifying and sorting individual droplets with active polymerases, which greatly increases the partitioning efficiency of the selection and reduces the occurrence of background DNA contamination relative to bead binding assays. Third, microfluidic approaches provide a more economical approach to library screening by allowing researchers to screen ~108 variants per day using ~106-fold less sample volume than is typically required for automated screening approaches (Price and Paegel, Reference Price and Paegel2016). The economy of scale is especially important when using unnatural nucleic acid substrates that can only be obtained by chemical synthesis and are not readily available from a commercial supplier.
Applications in synthetic biology
The slow but growing availability of engineered polymerases that can synthesize artificial genetic polymers (XNAs) with high efficiency and fidelity has already started to make an impact on applications in synthetic biology, biotechnology, and medicine. The following section summarizes major achievements that have been accredited to the discovery of engineered polymerases. Most notably, these examples focus on the generation of biologically stable versions of synthetic antibodies (aptamers) and catalytic enzymes that are composed entirely of XNA. Such efforts have made it possible to bypass the arduous task of introducing modifications post-selection in which medicinal chemists painstakingly modify the backbone structure for improved biological stability while carefully avoiding chemical changes that lead to losses in activity. Since many XNAs are resistant, if not recalcitrant to nuclease digestion, research efforts have focused on establishing methods for the discovery of XNAs with desired functional properties (Culbertson et al., Reference Culbertson, Temburnikar, Sau, Liao, Bala and Chaput2016). Mastering the production of these reagents by in vitro selection will lead to a new generation of diagnostic and therapeutic agents for the detection and treatment of human diseases.
Synthetic antibodies
Aptamers are nucleic acid molecules that mimic antibodies by folding into tertiary structures that can bind to a broad range of targets from ions and small molecules to proteins and whole cells (Dunn et al., Reference Dunn, Jimenez and Chaput2017). Although some aptamers exist naturally as the binding domain of riboswitches (Doudna and Cech, Reference Doudna and Cech2002), most are generated by in vitro selection or SELEX (systematic evolution of ligands by exponential enrichment) (Wilson and Szostak, Reference Wilson and Szostak1999). Similar to natural selection, in vitro selection is a Darwinian evolution process in which a large population of nucleic acid molecules (typically >1014 unique sequences) is challenged to bind a target (Joyce, Reference Joyce2004). Molecules that bind to the target are recovered and amplified to generate a new population of molecules that has become enriched in members with the desired activity. The process of selection and amplification is continued until the pool becomes dominated by members that bind the target with high affinity. The ability to amplify individual molecules with desired properties and to optimize their functions by directed evolution is a defining feature that separates nucleic acid molecules from other types of organic compounds that cannot replicate because they lack a genotype–phenotype connection (Szostak, Reference Szostak1992).
Aptamers are often compared to antibodies due to their ability to function with high ligand binding affinity and specificity (Jayasena, Reference Jayasena1999). However, unlike antibodies, aptamers do not require animals for their production, thus freeing them from the constraints of cellular biology and allowing for greater flexibility in their evolution under in vitro conditions. Once discovered, aptamers are produced through a chemical process rather than a biological process, which avoids the problem of viral or bacterial contamination and greatly reduces the potential for batch-to-batch variation. Aptamers developed for therapeutic purposes generally exhibit a lower immune response than proteins, and their small size (<30 versus ~150 kDa) provides access to biological areas that are inaccessible to antibodies (Nimjee et al., Reference Nimjee, Rusconi and Sullenger2005; Keefe et al., Reference Keefe, Pai and Ellington2010). Aptamers are able to fold reversibly, which overcomes the cold-chain problem that limits the shelf-life, reproducibility, and performance of antibodies. Therapeutic aptamers can also be deactivated with antisense oligonucleotides that recognize the binding domain, thereby providing a valuable antidote that can alleviate unwanted symptoms (Rusconi et al., Reference Rusconi, Roberts, Pitoc, Nimjee, White, Quick, Scardino, Fay and Sullenger2004). Finally, because aptamers are nucleic acid molecules, they can be seamlessly integrated into sensors, actuators, and other devices that are central to emerging technologies (Cho et al., Reference Cho, Lee and Ellington2009).
Despite the many benefits of aptamers relative to antibodies, aptamers composed of natural DNA and RNA are poor candidates for diagnostic and therapeutic applications, as these molecules are rapidly degraded by nucleases present in biological samples. In one case, an unmodified DNA aptamer developed as an inhibitor of α-thrombin exhibited an in vivo half-life of <2 min when assayed in a primate animal model (Griffin et al., Reference Griffin, Tidmarsh, Bock, Toole and Leung1993). Overcoming this problem led to the use of chemical modifications that protect the 2′-OH group against nucleases that utilize this position for cleavage of the phosphodiester bond. In particular, substitution of the 2′-OH group with amino (NH2), fluoro (F), and methoxy (OCH3) groups has led to enhanced nuclease stability (Keefe and Cload, Reference Keefe and Cload2008). For example, Macugen, the first FDA-approved aptamer, is an RNA sequence in which most of its 2′-OH groups have been replaced with 2′-F and 2′-OCH3 groups (Ng et al., Reference Ng, Shima, Calias, Cunningham, Guyer and Adamis2006). However, it is important to note that these modifications are often still prone to nuclease digestion (Cummins et al., Reference Cummins, Owens, Risen, Lesnik, Freier, Mcgee, Guinosso and Cook1995; Noronha et al., Reference Noronha, Wilds, Lok, Viazovkina, Arion, Parniak and Damha2000).
While numerous examples of 2′-modified aptamers have been described in the literature (Keefe and Cload, Reference Keefe and Cload2008), most were generated by transcription using T7 RNA polymerase. Recently, Romesberg and colleagues reported the directed evolution of variants of the Stoffel fragment of Taq DNA polymerase that accepts a broad range of 2′-modified substrates (Chen et al., Reference Chen, Hongdilokkul, Liu, Adhikary, Tsuen and Romesberg2016). One variant, SFM4-3, was found to PCR amplify substrates with 2′-F and 2′-azido (N3) groups (Chen et al., Reference Chen, Hongdilokkul, Liu, Adhikary, Tsuen and Romesberg2016). This engineered polymerase was subsequently used to evolve aptamers that bind to human neutrophil elastase (HNE), a serine protease associated with inflammatory diseases, using libraries that are partially substituted with 2′-modified nucleotides (Thirunavukarasu et al., Reference Thirunavukarasu, Chen, Liu, Hongdilokkul and Romesberg2017; Shao et al., Reference Shao, Chen, Sheng, Liu, Zhang and Romesberg2020). In a related study, Romesberg used the engineered polymerases, SFM4-6 and SFM4-9, to evolve fully 2′-OCH3 modified aptamers to HNE (Liu et al., Reference Liu, Chen and Romesberg2017). Structure–activity assays reveal that the 2′ modifications are necessary for aptamer activity.
A fundamentally different approach to nuclease stability involves the use of XNAs, which are artificial genetic polymers in which the ribose and deoxyribose sugars found in RNA and DNA have been replaced with a different sugar moiety (Chaput and Herdewijn, Reference Chaput and Herdewijn2019). TNA and HNA are particularly interesting as their backbone structures are recalcitrant to nuclease digestion, making them valuable systems for diagnostic and therapeutic applications (Hendrix et al., Reference Hendrix, Rosemeyer, Verheggen, Seela, Van Aerschot and Herdewijn1997; Culbertson et al., Reference Culbertson, Temburnikar, Sau, Liao, Bala and Chaput2016). The first XNA aptamers appeared in 2012 with the evolution of TNA sequences that can bind to human α-thrombin and HNA sequences having affinity for the HIV trans-activating response element and hen egg lysozyme (Pinheiro et al., Reference Pinheiro, Taylor, Cozens, Abramov, Renders, Zhang, Chaput, Wengel, Peak-Chew, Mclaughlin, Herdewijn and Holliger2012; Yu et al., Reference Yu, Zhang and Chaput2012). These results were viewed as a milestone in synthetic biology, as they demonstrated that heredity and evolution are no longer limited to DNA and RNA (Joyce, Reference Joyce2012).
The last few years have witnessed tremendous growth in the field of XNA aptamer research, with improved enzymes and selection techniques having given rise to higher quality aptamers whose oligonucleotide sequences derive entirely from building blocks with sugar moieties unrelated to those found in nature. Defestano and colleagues, for example, have developed a FANA aptamer that binds HIV RT with low picomolar affinity (Alves Ferreira-Bravo et al., Reference Alves Ferreira-Bravo, Cozens, Holliger and Destefano2015). Similarly, Herdewijn and colleagues evolved an HNA aptamer to rat vascular endothelial growth factor 164 (VEGF164) that distinguishes VEGF164 from the VEGF120 isoform (Eremeeva et al., Reference Eremeeva, Fikatas, Margamuljana, Abramov, Schols, Groaz and Herdewijn2019). In addition, new TNA aptamers have been discovered with affinity to the small molecule target ochratoxin A and the proteins thrombin and HIV RT (Dunn and Chaput, Reference Dunn and Chaput2016; Mei et al., Reference Mei, Liao, Jimenez, Wang, Bala, Mccloskey, Switzer and Chaput2018; Rangel et al., Reference Rangel, Chen, Ayele and Heemstra2018). More recently, our lab has developed a DNA-display strategy for evolving XNA aptamers in which each XNA strand is physically linked to its encoding DNA template (Dunn et al., Reference Dunn, McCloskey, Buckley, Rhea and Chaput2020). This strategy is analogous to protein display technologies, such as mRNA display that provide a covalent link between the encoding mRNA and translated protein (Roberts and Szostak, Reference Roberts and Szostak1997). This approach is generalizable to any XNA system where an XNA polymerase is available to copy DNA templates into XNA. It also avoids the need for an XNA reverse transcriptase, which improves the recovery of functional sequences that are present in low abundance after stringent washing has been performed to remove weaker affinity binders. Using this strategy, a TNA aptamer to HIV RT was produced that rivals the best monoclonal antibodies in terms of binding affinity and thermal stability (Dunn et al., Reference Dunn, McCloskey, Buckley, Rhea and Chaput2020). As these studies continue, it will be interesting to see how effective XNA aptamers will be at disrupting extracellular targets, such as the interaction of the viral spike protein of SARS-CoV-2 with the ACE2 receptor of human lung cells.
Catalysts for RNA modifying reactions
Nucleic acid enzymes provide powerful tools for precision medicine by allowing viral or disease-associated RNAs to be cleaved at specific nucleotide positions. The most widely studied member of this family of enzymes is DNAzyme 10-23 (Santoro and Joyce, Reference Santoro and Joyce1997), which has been evaluated in phases I and II clinical trials for a variety of diseases ranging from basal cell carcinoma to bronchial asthma (Fokina et al., Reference Fokina, Stetsenko and Francois2015). 10-23 is a magnesium-dependent enzyme that catalyzes the hydrolysis of a phosphodiester bond at a specific dinucleotide junction in the RNA substrate (Santoro and Joyce, Reference Santoro and Joyce1998). Unlike other gene-silencing technologies (e.g. antisense, siRNA, and CRISPR), DNAzymes benefit from a mechanism that does not require the recruitment of endogenous enzymes. Instead, Watson–Crick base pairing directs the enzyme to a cleavage site that is cut via an in-line attack by a deprotonated form of the 2′-OH group to produce an upstream cleavage product carrying a 2′,3′-cyclic phosphate and a downstream strand with a 5′-OH group. The enzyme is made generalizable by designing the substrate binding arms to be complementary to the cleavage site. This property of chemical simplicity, coupled with its ease of synthesis, has allowed 10-23 to become a popular tool for clinical and basic research. Over the years, numerous chemical modifications have been made to protect 10-23 from nuclease digestion and increase its efficacy in vivo (Fokina et al., Reference Fokina, Chelobanov, Fujii and Stetsenko2017). These include the introduction of an inverted 3′-3′ nucleotide and substitution of the deoxyribose sugar for other sugar moieties (Fokina et al., Reference Fokina, Chelobanov, Fujii and Stetsenko2017).
Although 10-23 is known to function with high activity (k cat/K m ~ 109 M−1 min−1) under optimized in vitro conditions, biological assays show that its capacity for RNA cleavage activity is greatly diminished in cellular environments where Mg2+ ions are present in lower abundance (Young et al., Reference Young, Lively and Deiters2010). One approach to solving this problem involves developing catalysts that carry functional groups that augment the chemical functionality of natural bases. Perrin and colleagues, for example, have evolved a divalent-metal independent DNAzyme that cleaves RNA with multiple turnover activity using imidazole side chains that mimic the mechanism of RNase A (Wang et al., Reference Wang, Liu, Lam and Perrin2018a). Another approach is to evolve XNA enzymes (XNAzymes) that are able to bind divalent metal ions with higher affinity. In 2015, Holliger and colleagues described the first examples of XNA catalysts with RNA cleavage and ligation activity (Taylor et al., Reference Taylor, Pinheiro, Smola, Morgunov, Peak-Chew, Cozens, Weeks, Herdewijn and Holliger2015). Using engineered polymerases, XNAzymes were isolated from four different backbone chemistries: FANA, ANA, CeNA, and HNA. However, despite the presence of high concentrations of Mg2+ ions, the XNAzymes produced from this study functioned with relatively weak activity. More recently, we have discovered a FANAzyme, termed FANAzyme 12-7, by in vitro evolution that functions at a rate enhancement of ~106-fold over the uncatalyzed reaction and exhibits substrate saturation kinetics typical of most enzymes (Wang et al., Reference Wang, Ngor, Nikoomanzar and Chaput2018b). Remarkably, FANAzyme 12-7 cleaves chimeric DNA substrates (DNA substrates having a riboG residue at the cleavage site) under physiological conditions with an activity rivaling that of known DNAzymes that were intentionally selected to recognize such substrates (Wang et al., Reference Wang, Vorperian, Shehabat and Chaput2020).
Future directions
Many synthetic biology applications are currently limited by a lack of polymerases that are available to perform a specific function with optimal activity. However, with the advent of new polymerase-engineering technologies, we anticipate that many of these limitations will likely be overcome in the near future. The following section provides some examples where polymerase-engineering technologies could help drive future innovations in synthetic biology, biotechnology, and medicine. Of course, many other examples are possible, including those that have not yet been envisioned.
Next-generation sequencing. Next-generation DNA-sequencing technologies that follow a sequencing-by-synthesis strategy require a DNA polymerase that can facilitate the incorporation of chemically modified nucleotides (Goodwin et al., Reference Goodwin, Mcpherson and Mccombie2016). Illumina technology, for example, utilizes dNTP substrates that carry a fluorescent dye and reversible 3′-terminator that are removed following nucleotide incorporation (Chen, Reference Chen2014). However, the removal chemistry leaves behind a portion of the linker connecting the fluorescent dye to the base, commonly referred to as a scar that can reduce the efficiency of polymerase-mediated primer extension. Evolving polymerases that can function with higher activity in the presence of scarred nucleotides may improve NGS technology by allowing for longer read lengths, faster turn-around times, and higher quality reads. A similar case exists for RNA-Seq applications where polymerases continually struggle to read through complex RNA structures. Polymerase evolution could overcome this problem by providing thermophilic reverse transcriptases that function at higher temperatures where larger RNA structures denature into single-stranded form.
Oligonucleotide synthesis. Solid-phase DNA synthesis based on phosphoramidite chemistry has driven major advances in the biological sciences by providing easy access to synthetic oligonucleotides (Caruthers, Reference Caruthers1985). Examples where this technology has made a major impact include DNA nanotechnology (Seeman and Sleiman, Reference Seeman and Sleiman2018), digital data archiving (Ceze et al., Reference Ceze, Nivala and Strauss2019), genome synthesis (Hutchison et al., Reference Hutchison, Chuang, Noskov, Assad-Garcia, Deerinck, Ellisman, Gill, Kannan, Karas, Ma, Pelletier, Qi, Richter, Strychalski, Sun, Suzuki, Tsvetanova, Wise, Smith, Glass, Merryman, Gibson and Venter2016), PCR (Saiki et al., Reference Saiki, Gelfand, Stoffel, Scharf, Higuchi, Horn, Mullis and Erlich1988), and SELEX (Wilson and Szostak, Reference Wilson and Szostak1999). However, the process of industrial scale DNA synthesis produces large quantities of hazardous waste that require appropriate disposal mechanisms. Moving to an enzymatic DNA synthesis platform would eliminate or greatly reduce this problem by allowing the reactions to proceed in an aqueous environment. Toward this goal, several groups are working to develop terminal deoxynucleotide transferase (TdT) as a possible paradigm for template-independent enzymatic DNA synthesis (Palluk et al., Reference Palluk, Arlow, De Rond, Barthel, Kang, Bector, Baghdassarian, Truong, Kim, Singh, Hillson and Keasling2018; Lee et al., Reference Lee, Kalhor, Goela, Bolot and Church2019). One could imagine that such approaches would benefit from polymerase engineering by providing access to new TdT variants that allow for longer DNA synthesis lengths, higher yields, and access to diverse nucleotide chemistries. The application of this approach to XNA, for example, could provide access to synthetic XNAs that are difficult to generate by conventional solid-phase synthesis due to low nucleotide coupling yields.
Information storage. In an age of ever-increasing data, new mechanisms for data storage are in short supply. One paradigm that has attracted significant attention involves using DNA as a soft material for low energy, high-density information storage of digital data (Ceze et al., Reference Ceze, Nivala and Strauss2019). At its maximum, 1 g of DNA can store 455 exabytes of information, which vastly exceeds the largest conventional devices (Church et al., Reference Church, Gao and Kosuri2012). Information storage occurs in four main steps that involve encoding digital information (e.g. text, pictures, and movies) in DNA, writing the information by massively parallel DNA synthesis, reading the information by NGS analysis, and decoding the information back into digital format. However, because DNA is a naturally occurring molecule that is prone to nuclease digestion, information stored in DNA could be unintentionally lost through an accidental encounter with nucleases present in the environment. XNAs that are recalcitrant to nuclease digestion offer a solution to this problem by providing a biologically stable alternative to DNA (Culbertson et al., Reference Culbertson, Temburnikar, Sau, Liao, Bala and Chaput2016). For this to be possible, XNA polymerases will likely need to be optimized for higher fidelity and lower template-sequence bias, which could be achieved by directed evolution.
Engineering bacteria. One recent exciting example relevant to the field of synthetic biology is the generation of mutant E. coli strains derived from engineered bacteria that contain significant (~40–50%) ribonucleotide content in their genome (Mehta et al., Reference Mehta, Wang, Reed, Supekova, Javahishvili, Chaput and Schultz2018). These systems have the potential to provide new insight into the origin of life by offering a better understanding of the transition from the RNA to the DNA world. Such studies would evaluate the relevance of chimeric DNA–RNA genomes with respect to the processes of replication, transcription, and repair. In this area of research, engineered polymerases will almost certainly be required to produce new mutant E. coli strains that contain >50% RNA content in their genomes. Although a daunting task, establishing an E. coli strain with an entirely RNA-derived genome would herald a new advance in synthetic biology. Other related areas where polymerase engineering could contribute to the development of engineered bacterial strains involves ongoing efforts to create bacterial cells in which all synthetic biology information is stored in XNA polymers (Schmidt, Reference Schmidt2010). This field of study, commonly referred to as xenobiology, relies on the concept of genetic orthogonality in which synthetic biology information is stored in XNA chromosomes that are made to replicate independently from the natural host genome (Chaput et al., Reference Chaput, Herdewijn and Hollenstein2020). In this way, the growing field of xenobiology promises to make synthetically engineered organisms safer by establishing a genetic firewall between natural biology and synthetic biology (Herdewijn and Marliere, Reference Herdewijn and Marliere2009).
Conclusion
In summary, we provide a comprehensive review of polymerase engineering that travels the path from early exploratory studies to modern enzyme-engineering technologies where variants are sampled with incredible speed and accuracy. Though not explicitly discussed in this review, it should be noted that these endeavors have been supported by equally significant advances in nucleic acid chemistry, which provide access to chemical building blocks with new physical and chemical properties. This combination of nucleic acid chemistry with enzyme engineering will uniquely drive new applications in synthetic biology, medicine, and biotechnology.
Acknowledgement
We wish to thank members of the Chaput lab for helpful discussions and critical reading of the manuscript.
Financial support
This work was supported by the National Science Foundation (MCB: 1946312) and the W.M. Keck Foundation.
Conflict of interest
The authors declare no conflicts of interest.
















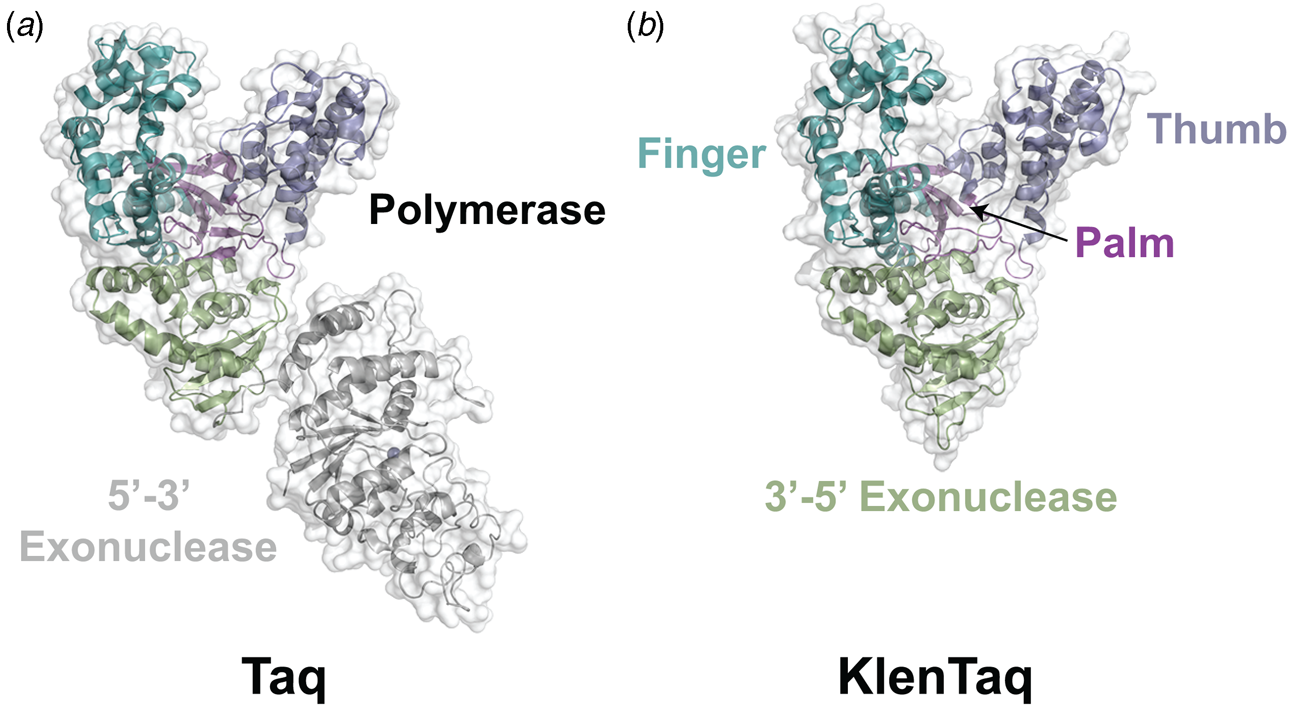





 Open access
Open access