



Introduction
The design of small molecules and molecular assemblies with predictable structures has enabled the construction of catalysts, pharmaceuticals, electronics, and smart materials. For example, organic chemists and coordination chemists can design small molecules with well-defined three-dimensional (3D) structures, dynamics, and reactivity. The design of proteins is a much higher order fundamental problem, but one with similarly important implications. It has long been appreciated that the properties of proteins depend on their intricately folded structures. However, we have only recently begun to be capable of designing proteins with predetermined structures. Indeed, 35 years ago it was considered inconceivable that it might ever be possible to design proteins with similar predictability and function.
As in other fields of chemistry, the progress from natural products to fully synthetic proteins has followed a multi-decade path. For example, in the 1950s to 1980s protein drugs, such as insulin and growth hormone were isolated from natural sources. More recently, it has been possible to tap into the immunological repertoire to discover novel antibodies and rationally vary their sequences to create drugs that are addressing multiple unmet medical needs. We are now entering an era in which it has become possible to design proteins with predetermined structures and functions ‘de novo’. This endeavor has already illuminated the principles of protein folding, and proteins are now being designed de novo to test and extend our understanding of binding and catalysis.
Here, we discuss the development of de novo protein design from its establishment and naming over 30 years ago to early 2019. Before the late 1980s the design of proteins appeared to be impossible. The thermodynamic stability of the native fold of a protein relative to the unfolded form is small and represents the difference between much larger favorable and unfavorable terms, making it very difficult to accurately predict stability. Moreover, the number of possible sequences for even a short protein of 100 residues (20100) is larger than the number of atoms in the universe, precluding the possibility of trying all possible sequences. Indeed, it would not be possible to find a specific sequence by a random search, even if a protein could be mutated every femtosecond for the age of the universe! Similarly, the number of possible backbone conformations for a protein of this size represents an astronomically large number (10100), indicating that folding cannot occur by a random search of conformational space (Levinthal, Reference Levinthal1969; Bryngelson et al., Reference Bryngelson, Onuchic, Socci and Wolynes1995).
Given the immense complexity of proteins and this prevailing viewpoint, the development of de novo protein design was hardly trivial. In its original conception, the de novo design of proteins referred to the design of proteins from scratch – rather than by modification of the sequence of naturally occurring proteins (DeGrado et al., Reference DeGrado, Regan and Ho1987; Regan and DeGrado, Reference Regan and DeGrado1988). It is somewhat surprising that the name has continued to the present, given that W. Feldberg's dictum that a scientist often ‘would rather use a colleague's toothbrush than his terminology!’ (Katz, Reference Katz1969). Instead, the meaning of de novo design has expanded slightly to include computational methods to redesign natural proteins. De novo design also includes sequence-directed approaches, for example, by introducing repeating patterns of apolar and polar residues (DeGrado and Lear, Reference DeGrado and Lear1985; Kamtekar et al., Reference Kamtekar, Schiffer, Xiong, Babik and Hecht1993).
The evolution of de novo design occurred in three distinct periods (Table 1). The first era of manual protein design using physical models spanned from the late 1970s to the early-1980s. During this period, solid-phase peptide synthesis enabled relatively routine synthesis of peptides up to about 30–50 residues in length. However, gene synthesis was not yet routine, limiting the size of proteins that could reliably be produced. The second wave, spanning from the mid-1980s to the early 2000s focused on computational design guided by fundamental physicochemical principles. Proteins were designed using mathematical equations to define the backbone conformations (DeGrado et al., Reference DeGrado, Regan and Ho1987; Regan and DeGrado, Reference Regan and DeGrado1988; Harbury et al., Reference Harbury, Tidor and Kim1995) and sidechain repacking algorithms to design the sequence (Ponder and Richards, Reference Ponder and Richards1987; Desjarlais and Handel, Reference Desjarlais and Handel1995; Dahiyat and Mayo, Reference Dahiyat and Mayo1996). This period also marked the first example of cooperatively folded proteins (DeGrado et al., Reference DeGrado, Regan and Ho1987; Regan and DeGrado, Reference Regan and DeGrado1988), the first computationally repacked natural protein domains (Dahiyat and Mayo, Reference Dahiyat and Mayo1997; Lazar et al., Reference Lazar, Desjarlais and Handel1997), and the first computationally designed completely de novo protein whose structure was fully verified (Walsh et al., Reference Walsh, Cheng, Bryson, Roder and DeGrado1999). The third wave began in the early 2000s as the expanding database of crystallographic structures enabled fragment-based and bioinformatically informed computational protein design. The Protein Data Bank (PDB) was deconstructed into a list of parts consisting of protein fragments, each with defined sequence preferences and interaction patterns that could be reassembled to create novel proteins (Kuhlman et al., Reference Kuhlman, Dantas, Ireton, Varani, Stoddard and Baker2003; Huang et al., Reference Huang, Boyken and Baker2016a).
Table 1. The formative first 20 years of de novo protein design 1983–2003
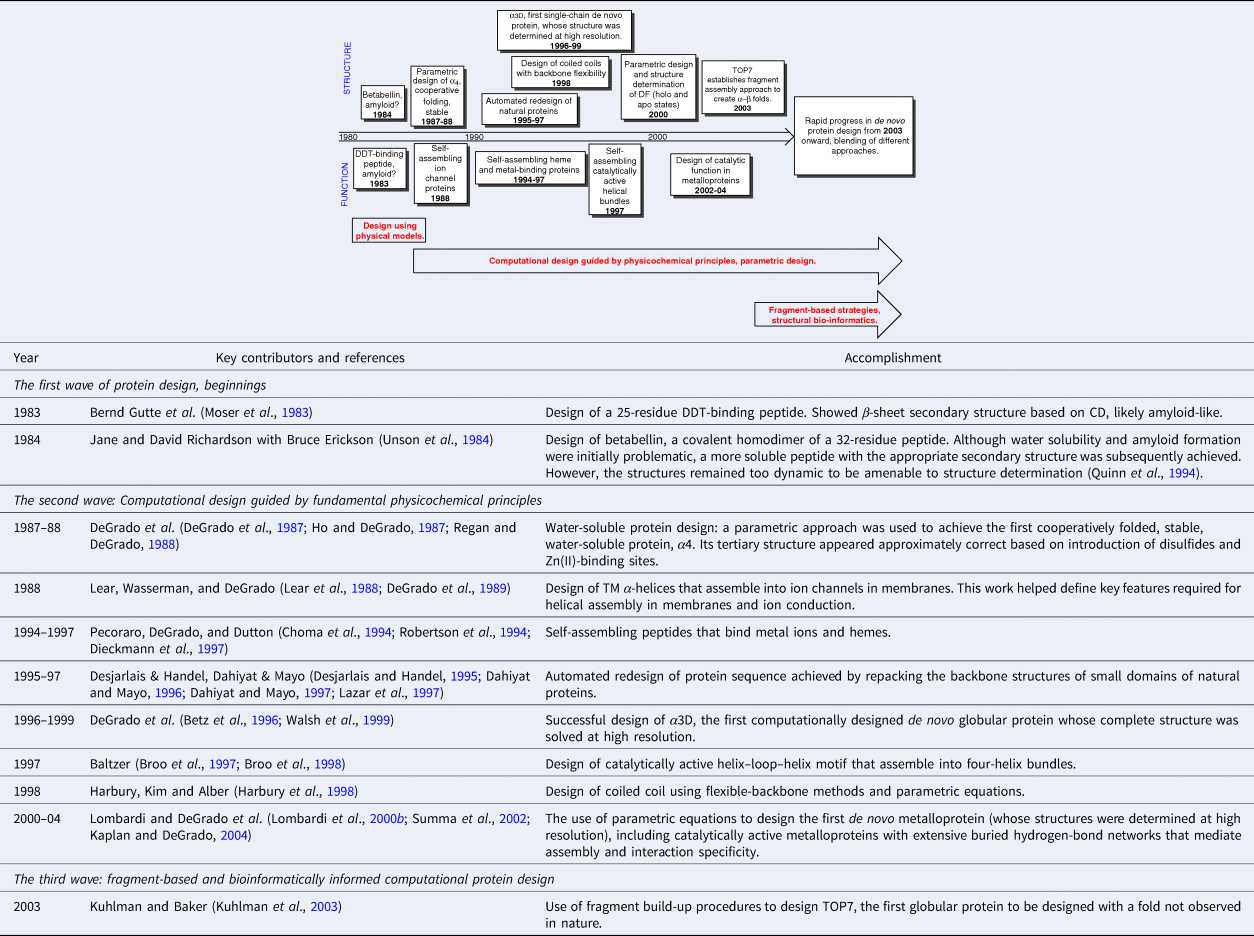
Table 1 highlights a number of key advances from the first 20 years, up to the development of fragment-based design of a protein designated TOP7, which was accomplished in 2003 (Kuhlman et al., Reference Kuhlman, Dantas, Ireton, Varani, Stoddard and Baker2003). Beyond this point, the field expanded rapidly, and the accomplishments are too many and varied to easily tabulate. Today, protein designers combine the essential tools from each of these periods. De novo design has already passed a number of milestones, the first of which was the construction of sequences that folded in water and membranes to adopt precisely predetermined folded conformations. Complex functions have also been achieved, ranging from binding and catalysis to transmembrane (TM) ion and electron transport. Here, we focus on the original question posed by the field of de novo design, is our knowledge of the principles of folding and function sufficient to design proteins from scratch. Therefore, we focus almost exclusively on de novo proteins whose structures and sequences have been designed using a mathematical parameterization or fragment assembly, rather than using the sequences or 3D structures of natural proteins as the starting point. To maintain this focus we do not discuss combinatorial sequence-based approaches such as binary patterning. We instead refer the reader to reviews of this outstanding work (Hecht et al., Reference Hecht, Das, Go, Bradley and Wei2004, Reference Hecht, Zarzhitsky, Karas and Chari2018). Also, wherever possible, we restrict our discussion to proteins whose structures and/or dynamics have been very extensively characterized by high-resolution methods.
Manual protein design
As early as 1979, Bernd Gutte used manual model building and physical models to design a 35-residue RNA-binding peptide (Gutte et al., Reference Gutte, Däumingen and Wittschieber1979), followed by a 25-residue peptide intended to bind dichlorodiphenyltrichloroethane (DDT) in 1983 (Moser et al., Reference Moser, Thomas and Gutte1983) (Fig. 1). While some binding was observed, solubility problems precluded determination of their structures. In the mid-1980s, Jane and David Richardson began their collaborative work with Bruce Erickson aimed at the design of ‘betabellins’ (and the related ‘betadoublets’), meant to mimic the structure of β-sandwich proteins (Richardson and Richardson, Reference Richardson and Richardson1989). In this case, computer graphics and secondary structure propensities gleaned from analysis of natural proteins were used to facilitate the design process. Again, poor solubility and aggregation proved to be problematic. Ultimately, Erickson demonstrated that at least one member of this class of designed proteins formed amyloid-like fibrils (Lim et al., Reference Lim, Saderholm, Makhov, Kroll, Yan, Perera, Griffith and Erickson1998). In retrospect, it is likely that the formation of amyloid-like structures explained the ability of Gutte's DTT-binding peptides to bind hydrophobic substances (West et al., Reference West, Wang, Patterson, Mancias, Beasley and Hecht1999). A variety of amyloids are well known to bind a variety of flat-aromatic molecules including amyloid dyes (West et al., Reference West, Wang, Patterson, Mancias, Beasley and Hecht1999). Attempts to increase solubility and decrease aggregation of the betabellin and betadoublet families of proteins led to derivatives with fluctuating structures that defied high-resolution structure determination (Quinn et al., Reference Quinn, Tweedy, Williams, Richardson and Richardson1994). The design of uniquely folded β-proteins continued to be challenging, and accurate design of such tertiary structures was achieved only in the last two years (Dou et al., Reference Dou, Vorobieva, Sheffler, Doyle, Park, Bick, Mao, Foight, Lee, Gagnon, Carter, Sankaran, Ovchinnikov, Marcos, Huang, Vaughan, Stoddard and Baker2018).
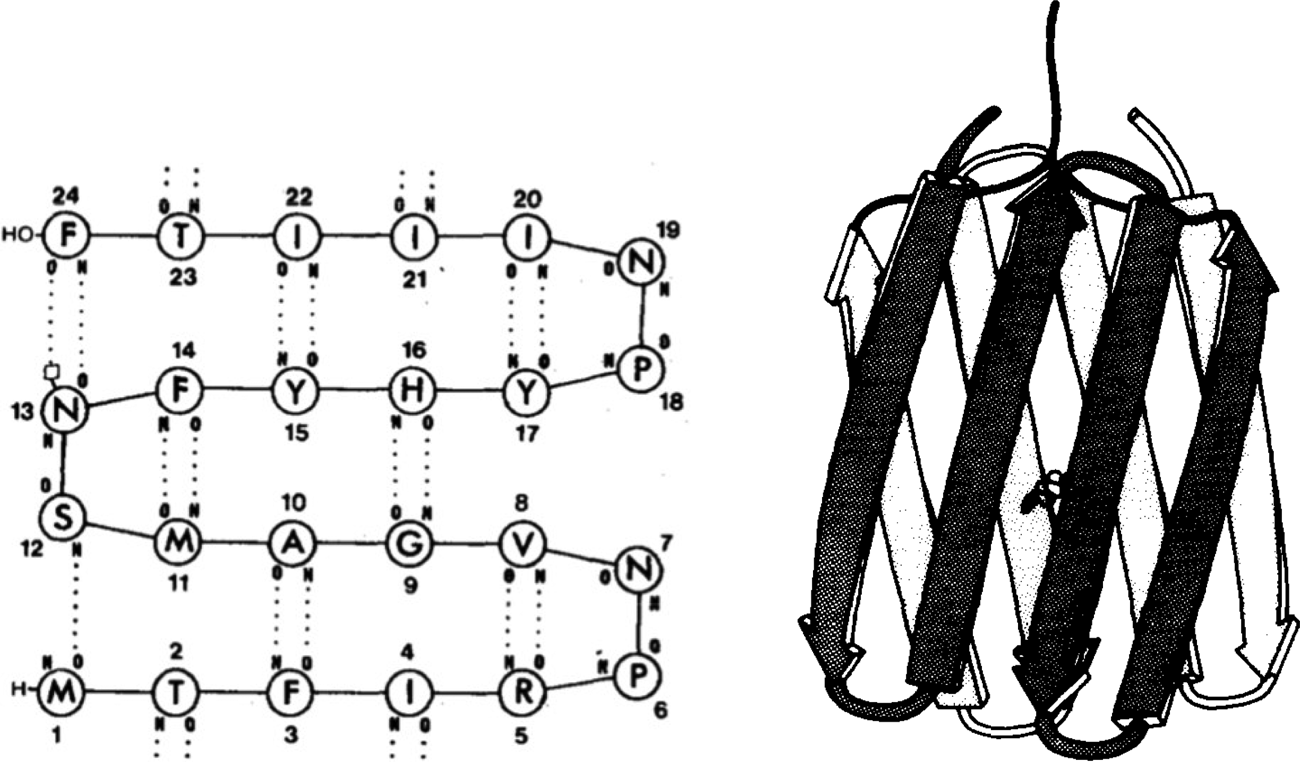
Fig. 1. (Left) Proposed secondary structure of a DDT-binding peptide (reproduced with permission from Moser et al. (Reference Moser, Thomas and Gutte1983)). (Right) Molecular model of a short segment of the amyloid fibril formed by betabellin (reproduced with permission from Richardson and Richardson (Reference Richardson and Richardson1989)).
Thus, by the mid-1980s, although there were sporadic attempts to design proteins with predetermined structures and functions, this goal had not been achieved. However, this was about to change due to a number of concurrent technical advances.
Computational design guided by fundamental physicochemical principles
Helical bundles, the first structurally defined proteins designed from scratch
In the 1970s and 1980s, a number of key advances made de novo protein design feasible for the first time. Methods of solid phase peptide synthesis had reached an advanced stage for the synthesis of sequences up to about 50 residues, and the synthesis of synthetic genes had become increasingly possible, allowing one to design larger novel proteins. Computer graphics coupled with methods of molecular mechanics and dynamics allowed one to work with highly complex structures, freeing the designer from working with cumbersome physical models. Crystallographic and nuclear magnetic resonance (NMR) methods were also rapidly improving. Finally, site-directed mutagenesis of natural proteins provided a better understanding of the energetics and kinetics of protein folding – and the contributions of individual sidechains to the process. It became generally accepted that the packing of hydrophobic residues in the solvent-accessible interiors of proteins contributed significantly to the driving force for water-soluble protein folding, and that polar interactions, although less favorable, often helped define the detailed geometries of protein structures (Fersht and Serrano, Reference Fersht and Serrano1993). Moreover, the preferences of amino acids for adopting specific secondary structures and rotamers enabled computational methods to select a sequence to stabilize a given fold (Box 1).
Box 1. Setting the stage for de novo protein design, sidechain packing algorithms, and automated sequence selection
Early studies showed that the sidechains in protein cores adopted low-energy conformations called rotamers (Janin et al., Reference Janin, Wodak, Levitt and Maigret1978), which were tightly packed with an efficiency similar to small molecule crystal lattices (Richards, Reference Richards1977). The distribution of each rotamer was subsequently shown to depend on secondary structure (McGregor et al., Reference McGregor, Islam and Sternberg1987; Dunbrack and Karplus, Reference Dunbrack and Karplus1993; Dunbrack and Cohen, Reference Dunbrack and Cohen1997). These findings led to a model in which sidechains were packed in protein cores as in a 3D jigsaw puzzle. These two requirements – that side chains form stable rotamers and that they be efficiently packed in protein interiors – provided two powerful restraints that define the interior-facing residues of uniquely folded globular proteins. The first cooperatively folded globular de novo proteins were designed following these imperatives by using minimal set of apolar and polar sidechains (DeGrado and Lear, Reference DeGrado and Lear1985; Eisenberg et al., Reference Eisenberg, Wilcox, Eshita, Pryciak, Ho and DeGrado1986; DeGrado et al., Reference DeGrado, Regan and Ho1987; Ho and DeGrado, Reference Ho and DeGrado1987).
As computational power increased it became possible to consider the repacking protein of cores with the full set of natural amino acids. Here, one begins with a given backbone structure and explores large numbers of side chains that can fit together to stabilize the fold (Ponder and Richards, Reference Ponder and Richards1987). Ideally, each possible combination of sidechain and rotamer identities would be evaluated at each position, but the number of combinations rapidly becomes unmanageable without the use of computational algorithms, including genetic (Jones, Reference Jones1994; Willett, Reference Willett1995), Monte-Carlo (Metropolis et al., Reference Metropolis, Rosenbluth, Rosenbluth, Teller and Teller1953), and dead-end-elimination (Desmet et al., Reference Desmet, De Maeyer, Hazes and Lasters1992; Lasters et al., Reference Lasters, De Maeyer and Desmet1995; Gordon et al., Reference Gordon, Hom, Mayo and Pierce2003) algorithms. In 1987, Ponder and Richards introduced sidechain repacking algorithms to probe the combinatorics of packing in natural proteins (Ponder and Richards, Reference Ponder and Richards1987). In 1995, Desjarlais and Handel (Desjarlais and Handel, Reference Desjarlais and Handel1995; Johnson et al., Reference Johnson, Lazar, Desjarlais and Handel1999) used repacking algorithms together with a genetic algorithm to redesign the core of small natural protein domains. In a series of landmark papers (Dahiyat and Mayo, Reference Dahiyat and Mayo1996; Dahiyat and Mayo, Reference Dahiyat and Mayo1997), Mayo and coworkers expanded repacking algorithms to include selection of exterior sidechains, as well as the use of dead-end-elimination to facilitate the search. In 1997, Dahiyat and Mayo achieved the completely automated redesign of the sequence of a natural 28-residue Zn(II) finger motif peptide, starting with only the backbone structure of the second zinc finger module of the DNA binding protein Zif268. (Dahiyat and Mayo, Reference Dahiyat and Mayo1997). In the same year, Handel, Desjarlais, DeGrado, and coworkers introduced sidechain repacking algorithms to design a protein whose backbone was not taken from a natural protein. The structure of the resulting 73-residue protein, α3D was in excellent agreement with the design (Betz et al., Reference Betz, Bryson, Passador, Brown, O'Neil and DeGrado1996; Bryson et al., Reference Bryson, Desjarlais, Handel and DeGrado1998; Walsh et al., Reference Walsh, Cheng, Bryson, Roder and DeGrado1999). Today, sidechain repacking algorithms represent an important part of all fully atomistic computational approaches to protein design.
Thus, by the 1980s the stage was set for de novo protein design. Nevertheless, there was considerable skepticism that de novo design would be possible given the astronomical number of potential sequences and conformations for even a modestly long protein sequence. How then, might proteins have evolved within the first billion years after the formation of our planet? One attractive hypothesis was that modern-day proteins evolved from self-association of short peptides capable of forming secondary structural or other functional units. Dayhoff suggested that structures could be assembled through intermolecular association of multiple chains or by intramolecular association (folding) of proteins formed by duplicating of genes expressing for the primordial units (Eck and Dayhoff, Reference Eck and Dayhoff1966). DeGrado and Lear (Reference DeGrado and Lear1985) hypothesized that some of the first precursors to natural proteins were amphiphilic peptides, in which hydrophobic and polar residues segregate on opposite sides of an α-helix or β-sheet; assembly of the hydrophobic faces would drive folding in an aqueous environment. To test this hypothesis, they designed peptides composed of only Leu and Lys as hydrophobic and polar residues. When the polar and apolar residues were alternated in the sequence to match the geometric repeat of the β-sheet, the resulting peptide (LKLKLKL) assembled into a β-conformation in aqueous solution. However, when the polar and apolar residues were allowed to match that of an α-helix in (LKKLLKL)2, the peptides self-associated into tetrameric bundles of α-helices, which the authors speculated might have 222 symmetric structures similar to the recently recognized family of natural antiparallel four-helix bundle folding motif (Fig. 2a and d) (Argos et al., Reference Argos, Rossmann and Johnson1977; Weber and Salemme, Reference Weber and Salemme1980; Presnell and Cohen, Reference Presnell and Cohen1989; Beesley and Woolfson, Reference Beesley and Woolfson2019).
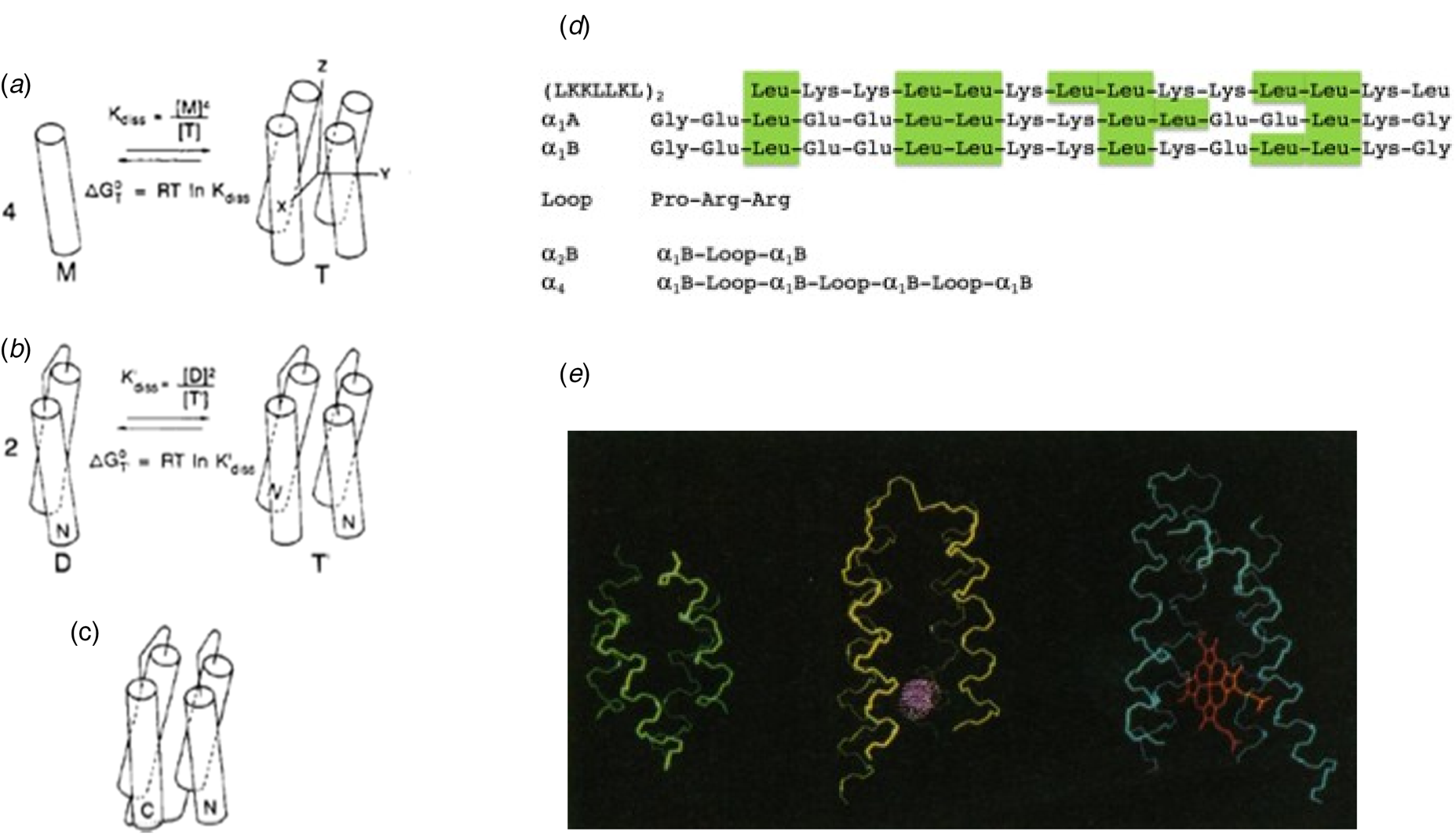
Fig. 2. Design of a four-helix bundle. (a) A peptide was designed, which self-associated to form an antiparallel helical bundle in solution. A loop sequence was next inserted (b) between two helices to create a dimeric four-helix bundle, and then three loops were inserted between four helices to create the full-length helical bundle. At each stage, the free energy of assembly or folding was determined, and used to evaluate possible sequences. In this way, the complex problem of protein design was cut into smaller separable pieces. For simplicity, the monomeric species in panels (a) and (b) are shown as helices, but they were actually only partially helical, as shown by CD. Panel (d) shows the sequences of the peptides and proteins discussed in the text. Panel (e) shows an early energy-minimized model of α4 (left) as compared to larger natural four-helix bundle proteins (myohemerythrin, middle) and cytochrome c′ (right). Panels (a–c) are reproduced with permission from Ho and DeGrado (Reference Ho and DeGrado1987). Copyright (2007) American Chemical Society, while panel (e) is reproduced with permission from DeGrado et al. (Reference DeGrado, Wasserman and Lear1989).
This investigation set off a series of studies that culminated in the design of large families of helical bundle proteins. Success in designing a protein that folded into a desired structure did not come immediately, but instead in stages, as we came to understand the requirements for secondary structure formation, folding into a globular thermodynamically stable ensemble of closely related proteins, and ultimately into a single well-folded protein structure. Early attempts to crystallize (LKKLLKL)2 in the lab of David Eisenberg were unsuccessful. Therefore, DeGrado and Eisenberg collaborated on the redesign of the sequence of (LKKLLKL)2 to better stabilize the desired antiparallel tetrameric structure.
The initial design idealized the approximate D 2 symmetry of natural four-helix bundles (Eisenberg et al., Reference Eisenberg, Wilcox, Eshita, Pryciak, Ho and DeGrado1986) (i.e. with two-fold rotational symmetry axes running down the bundle as well as between neighboring antiparallel helices, labeled Z, Y, and X in Fig. 2a) of natural four-helix bundle proteins. Internal symmetry reduced the size of the sequence space that needed to be considered, and it allowed the basic unit to be used repeatedly to build the entire four-helix bundle. Similar parametric models with minimal numbers of adjustable parameters (Salemme, Reference Salemme1983; Lasters et al., Reference Lasters, Wodak, Alard and van Cutsem1988; Betz and DeGrado, Reference Betz and DeGrado1996; Lombardi et al., Reference Lombardi, Summa, Geremia, Randaccio, Pavone and DeGrado2000b; North et al., Reference North, Summa, Ghirlanda and DeGrado2001; Offer et al., Reference Offer, Hicks and Woolfson2002; Emberly et al., Reference Emberly, Mukhopadhyay, Tang and Wingreen2004; Grigoryan and DeGrado, Reference Grigoryan and DeGrado2011) have since been used to build-up more complex tertiary structures including the rubredoxin fold (Nanda et al., Reference Nanda, Rosenblatt, Osyczka, Kono, Getahun, Dutton, Saven and Degrado2005), Triosephosphate isomerase (TIM) barrels (Huang et al., Reference Huang, Feldmeier, Parmeggiani, Velasco, Hocker and Baker2016b), β-barrels (Dou et al., Reference Dou, Vorobieva, Sheffler, Doyle, Park, Bick, Mao, Foight, Lee, Gagnon, Carter, Sankaran, Ovchinnikov, Marcos, Huang, Vaughan, Stoddard and Baker2018), β-propellers (Voet et al., Reference Voet, Noguchi, Addy, Simoncini, Terada, Unzai, Park, Zhang and Tame2014), coiled coils (Harbury et al., Reference Harbury, Plecs, Tidor, Alber and Kim1998; Huang et al., Reference Huang, Oberdorfer, Xu, Pei, Nannenga, Rogers, DiMaio, Gonen, Luisi and Baker2014; Thomson et al., Reference Thomson, Wood, Burton, Bartlett, Sessions, Brady and Woolfson2014), and repeat proteins (Brunette et al., Reference Brunette, Parmeggiani, Huang, Bhabha, Ekiert, Tsutakawa, Hura, Tainer and Baker2015), as discussed in subsequent sections. In parametric protein design, one begins with highly symmetrical backbones, to create a ‘draft’ of the desired structure and then lifts symmetry as needed to accommodate the asymmetric placement of loops and active sites (Lombardi et al., Reference Lombardi, Summa, Geremia, Randaccio, Pavone and DeGrado2000b; Huang et al., Reference Huang, Boyken and Baker2016a; Polizzi et al., Reference Polizzi, Wu, Lemmin, Maxwell, Zhang, Rawson, Beratan, Therien and DeGrado2017; Dou et al., Reference Dou, Vorobieva, Sheffler, Doyle, Park, Bick, Mao, Foight, Lee, Gagnon, Carter, Sankaran, Ovchinnikov, Marcos, Huang, Vaughan, Stoddard and Baker2018).
The designed self-associating tetrameric peptide, α1A was built manually using a set of physical ‘Kendrew’ models (Eisenberg et al., Reference Eisenberg, Wilcox, Eshita, Pryciak, Ho and DeGrado1986). Physicochemical principles guided all aspects of the design. Leu sidechains were chosen for the hydrophobic interior, where they were able to interdigitate in low-energy rotamers. Helix-promoting Glu and Lys residues were chosen for the exterior-facing residues, and they were arranged to form favorable electrostatic interactions. Although α1A failed to crystallize (Ho and DeGrado, Reference Ho and DeGrado1987), a short, 12-residue fragment of α1A (designated α1) isolated as a byproduct of the synthesis was crystallized. Too short to form the desired full-length bundle, this peptide assembled into multiple association states in solution and the solid state (Patterson et al., Reference Patterson, Anderson, DeGrado, Cascio and Eisenberg1999; Prive et al., Reference Prive, Anderson, Wesson, Cascio and Eisenberg1999).
Ho and DeGrado (Reference Ho and DeGrado1987) next used computer graphics and energy minimization to redesign the sequence, minimizing the exposure of apolar residues on the surface. In contrast to α1, the resulting full-length α1B peptide cooperatively assembled into a highly stable tetrameric four helix bundle (−22 kcal mol−1, 1 M standard state). The α1B tetramer was compact and globular, and detailed NMR investigations also showed that the helices began and ended precisely as in the design (Osterhout et al., Reference Osterhout, Handel, Na, Toumadje, Long, Connolly, Hoch, Johnson, Live and DeGrado1992). The first attempt to build loops between the helices revealed an important and previously unarticulated aspect of protein folding – the sequence of a protein must not only stabilize the desired fold. Instead it must destabilize all closely related folds while stabilizing the native structure (DeGrado et al., Reference DeGrado, Regan and Ho1987; Ho and DeGrado, Reference Ho and DeGrado1987).
The final α4 protein was 74 residues in length, and expressed well in bacteria. It represented the first example of a de novo designed protein with a cooperatively folded, globular conformation in aqueous solution (DeGrado et al., Reference DeGrado, Regan and Ho1987; Regan and DeGrado, Reference Regan and DeGrado1988). Furthermore, it was highly stable, with a cooperative equilibrium unfolding transition near 6 M guanidine hydrochloride. Clearly, the first milestones in de novo protein design had been passed. Furthermore, structure-stabilizing disulfides (Regan et al., Reference Regan, Rockwell, Wasserman and DeGrado1994) and metal-binding sites (Handel and DeGrado, Reference Handel and DeGrado1990; Regan and Clarke, Reference Regan and Clarke1990; Handel et al., Reference Handel, Williams and DeGrado1993) were successfully introduced into the tertiary structure, as confirmed by NMR (Handel and DeGrado, Reference Handel and DeGrado1990; Handel et al., Reference Handel, Williams and DeGrado1993). Thus, the Zn2+-binding derivatives of α4 indeed achieved the correct overall fold that positioned residues distant in sequence into close proximity to create the functional binding site. A second milestone was crossed.
Over the past few decades, studies of natural proteins have shown that they can natively achieve a wide-ranging spectrum of order, ranging from intrinsically disordered (random coil), to compact but flexible, to ones with well-packed cores. However, in the 1980s there was less understanding of this spectrum of native states, so there was considerable interest in determining the degree of structural uniqueness that could be achieved with a minimal protein such as α4. Solution NMR and fluorescence studies showed that the buried hydrophobic residues of α4 were conformationally more mobile than those of most crystallographically characterized proteins. Over the next decade, various groups attempted to address this issue, as reviewed previously (Bryson et al., Reference Bryson, Betz, Lu, Suich, Zhou, O'Neil and DeGrado1995; DeGrado et al., Reference DeGrado, Summa, Pavone, Nastri and Lombardi1999), and only a few early contributions will be mentioned here. Expecting that a more diverse sequence might lead to improved properties, Jane and David Richardson designed a protein, called FELIX, which incorporated all the natural amino acids (Hecht et al., Reference Hecht, Richardson, Richardson and Ogden1990). However, FELIX had very marginal stability (around −1 kcal mol−1versus −20 kcal mol−1 for α4), and subsequent studies by this group showed that it did not unfold in a cooperative transition – instead they concluded that FELIX adopted a ‘non-stable and non-unique tertiary structure’ (Gernert et al., Reference Gernert, Richardson and Richardson1993). Stroud et al. constructed a monomeric four-helix bundle by stitching loops between four identical helical peptides (Schafmeister et al., Reference Schafmeister, LaPorte, Miercke and Stroud1997) that had originally been designed to solubilize membrane proteins, but instead were found to self-associate into a tetrameric four-helix bundle (Fig. 3a). Although a crystal structure was determined, the loops were disordered, so it was not possible to determine the topology of the bundle. Finally, by introducing polar interactions and introducing geometric complementarity into the originally designed α2B scaffold, it was possible to design and structurally characterize uniquely folded dimeric four-helix bundles (Hill and DeGrado, Reference Hill and DeGrado1998, Reference Hill and DeGrado2000; Hill et al., Reference Hill, Hong and DeGrado1999, Reference Hill, Raleigh, Lombardi and DeGrado2000).

Fig. 3. (a) Crystal structure of a peptide that was designed to solubilize membrane proteins, but was serendipitously found to crystallize as a four helix coiled-coil bundle DHP1 (PDB: 4HB1). (b) NMR structure of α3D (PDB: 2A3D) is stabilized by a set of apolar sidechains that pack in a geometrically complementary manner, shown in ball-and-stick format. (c) The model of 3-His α3D based on EXAFS data and NMR structure of α3D (PDB: 2A3D).
A breakthrough in de novo design of uniquely folded proteins occurred with the ability to computationally ‘repack’ the hydrophobic core of designed backbones (Ponder and Richards, Reference Ponder and Richards1987). As mentioned above, Handel (Desjarlais and Handel, Reference Desjarlais and Handel1995) and Mayo (Dahiyat and Mayo, Reference Dahiyat and Mayo1997) demonstrated the use of these algorithms for repacking the core of small natural protein domains. DeGrado, Handel, and coworkers introduced the use of these algorithms to design of a de novo protein (Betz et al., Reference Betz, Bryson, Passador, Brown, O'Neil and DeGrado1996; Bryson et al., Reference Bryson, Desjarlais, Handel and DeGrado1998), rather than starting with the 3D structure of a natural protein. They designed a three-helix bundle, α3D, through sidechain repacking and energy minimization. The interior sidechains consisted of a diverse set of apolar residues that packed in a geometrically complementary manner. Interhelical electrostatic interactions at solvent-exposed positions were also used to specify a single topology. The NMR structure (Fig. 3b) (Walsh et al., Reference Walsh, Cheng, Bryson, Roder and DeGrado1999) was in close agreement with the design providing the first example of the de novo design of a globular protein with an accurately predetermined structure. Another important milestone in de novo design had been passed. Given its relatively simple but cooperatively folded globular structure, α3D quickly became a very widely studied protein for computational and experimental studies of protein folding (Zhu et al., Reference Zhu, Alonso, Maki, Huang, Lahr, Daggett, Roder, DeGrado and Gai2003; Park et al., Reference Park, Xu, Stowell, Gai, Saven and Boder2006; Liu et al., Reference Liu, Dumont, Zhu, DeGrado, Gai and Gruebele2009; Adhikari et al., Reference Adhikari, Freed and Sosnick2012; Shao, Reference Shao2014; Chung et al., Reference Chung, Piana-Agostinetti, Shaw and Eaton2015; Zeng et al., Reference Zeng, Jiang and Wu2016; Maruyama and Mitsutake, Reference Maruyama and Mitsutake2017; Walder et al., Reference Walder, LeBlanc, Van Patten, Edwards, Greenberg, Adhikari, Okoniewski, Sullan, Rabuka, Sousa and Perkins2017; Xiong et al., Reference Xiong, Mao and Gong2017; Jumper et al., Reference Jumper, Faruk, Freed and Sosnick2018; Koebke et al., Reference Koebke, Ruckthong, Meagher, Mathieu, Harland, Deb, Lehnert, Policar, Tard, Penner-Hahn, Stuckey and Pecoraro2018; Yoo et al., Reference Yoo, Louis, Gopich and Chung2018; Gadzala et al., Reference Gadzala, Dulak, Kalinowska, Baster, Brylinski, Konieczny, Banach and Roterman2019). Its folding kinetics are among the most extensively characterized of small cooperatively folded proteins (Chung et al., Reference Chung, Piana-Agostinetti, Shaw and Eaton2015). The protein α3D has also become as a template for the design of metalloproteins (Fig. 3c) (Chakraborty et al., Reference Chakraborty, Kravitz, Thulstrup, Hemmingsen, DeGrado and Pecoraro2011; Mocny and Pecoraro, Reference Mocny and Pecoraro2015; Tebo and Pecoraro, Reference Tebo and Pecoraro2015; Plegaria and Pecoraro, Reference Plegaria and Pecoraro2016). Many examples of functional helical bundles based on α3D and designed four-helix scaffolds were soon to follow, as discussed below.
The design of uniquely folded proteins also coincided in time with the understanding that proteins fold in a funnel-like manner, accruing increasing native tertiary structure as folding progresses. This smooth process is known as minimal frustration (for a review see, Wolynes (Reference Wolynes2015)). The final ensemble of states – whether it be a uniquely and tightly packed 3D structure or a more loosely folded ‘molten globule’ – depends on whether the sequence can assume one single backbone structure and sidechain packing arrangement or a more energetically diverse set of structures and packings. One of the surprises of protein design was that the folding landscape can so easily occur with minimal frustration, and that consideration of the native state frequently leads to a foldable sequence that does not get ‘stuck’ in numerous off-pathway solutions. The smoothness of the folding funnel for natural proteins has often been discussed in terms of evolution. We believe it is also an intrinsic propensity of the properties and geometry of the protein backbone and the reliance on the hydrophobic interaction to drive folding in nature. The need to tightly pack the amide backbone leads to highly compact secondary structures in which the polar amides form intramolecular hydrogen bonds to compensate for stabilizing interactions with water that occur in the unfolded state. Ultimately, the burial and favorable packing of apolar sidechains in the protein interior drives folding, while the requirement to maintain water-solubility dictates the predominant placement of polar residue on the exterior. Together, these restraints lead not only to a stable folded structure, but also to minimal frustration along the folding pathway. It is also interesting to note that the misfolding of the same protein sequences into amyloids (Chiti and Dobson, Reference Chiti and Dobson2009) generally occurs through aggregation, presumably on a much rougher landscape. The ability to fold rapidly along a smooth funnel (and hence kinetically escape amyloid formation in a non-equilibrium living system) must have been one of the earliest features in the molecular evolution of proteins.
Coiled coils
Coiled coils represent a special class of helical bundles, which have been particularly useful stepping stones in the development of de novo protein design. The α-helical coiled coil (Fig. 4) represents a structure of intermediate complexity, bridging the gap between simple monomeric helices and native proteins. The classical left-handed coiled-coil has a seven-residue geometric repeat labeled, ‘abcdefg’; ‘a’ and ‘d’ side-chains project toward the bundle core and are mostly hydrophobic whereas ‘e’ and ‘g’ residues face the inter-subunit interface and are generally more polar (Crick, Reference Crick1953). Hodges and co-workers used a sequence-based approach to design repeating heptapeptides as models for two-stranded coiled coils. In the prototype, (LeuaGlub,AlacLeudGlueGlyfLysg)n, apolar Leu residues at positions ‘a’ and ‘d’ of the heptad hydrophobically stabilize the structure (Lau et al., Reference Lau, Taneja and Hodges1984). This heptad repeat formed the basis for the design of a 29-residue peptide (O'Neil and DeGrado, Reference O'Neil and DeGrado1990) that was used to determine the helical propensities of various amino acids. Subsequent determination of the crystal structure of this peptide showed that it formed a trimeric antiparallel structure, rather than the expected parallel dimer. Shortly thereafter, studies on derivatives of the two-stranded coiled-coil domain of a yeast transcription factor, GCN4, further illustrated the role of polar and packing interactions in determining the stoichiometry and topology of coiled coils (Harbury et al., Reference Harbury, Zhang, Kim and Alber1993, Reference Harbury, Kim and Alber1994). Alber, Harbury, Kim, and coworkers showed that van der Waals (vdW) packing between buried residues at the ‘a’ and ‘d’ positions play critical roles in determining the stoichiometry and structure of coiled coils. Amino acid substitutions as subtle as Leu-to-Ile substitutions switch the assembly from favoring trimers to tetramers, and this switch could be understood and predicted based on simple packing arguments. Moreover, Alber, Harbury, and Kim introduced the use of flexible-backbone methods and parametric equations to design both right-handed and left-handed coiled coils (Harbury et al., Reference Harbury, Plecs, Tidor, Alber and Kim1998), representing another important milestone in de novo protein design.
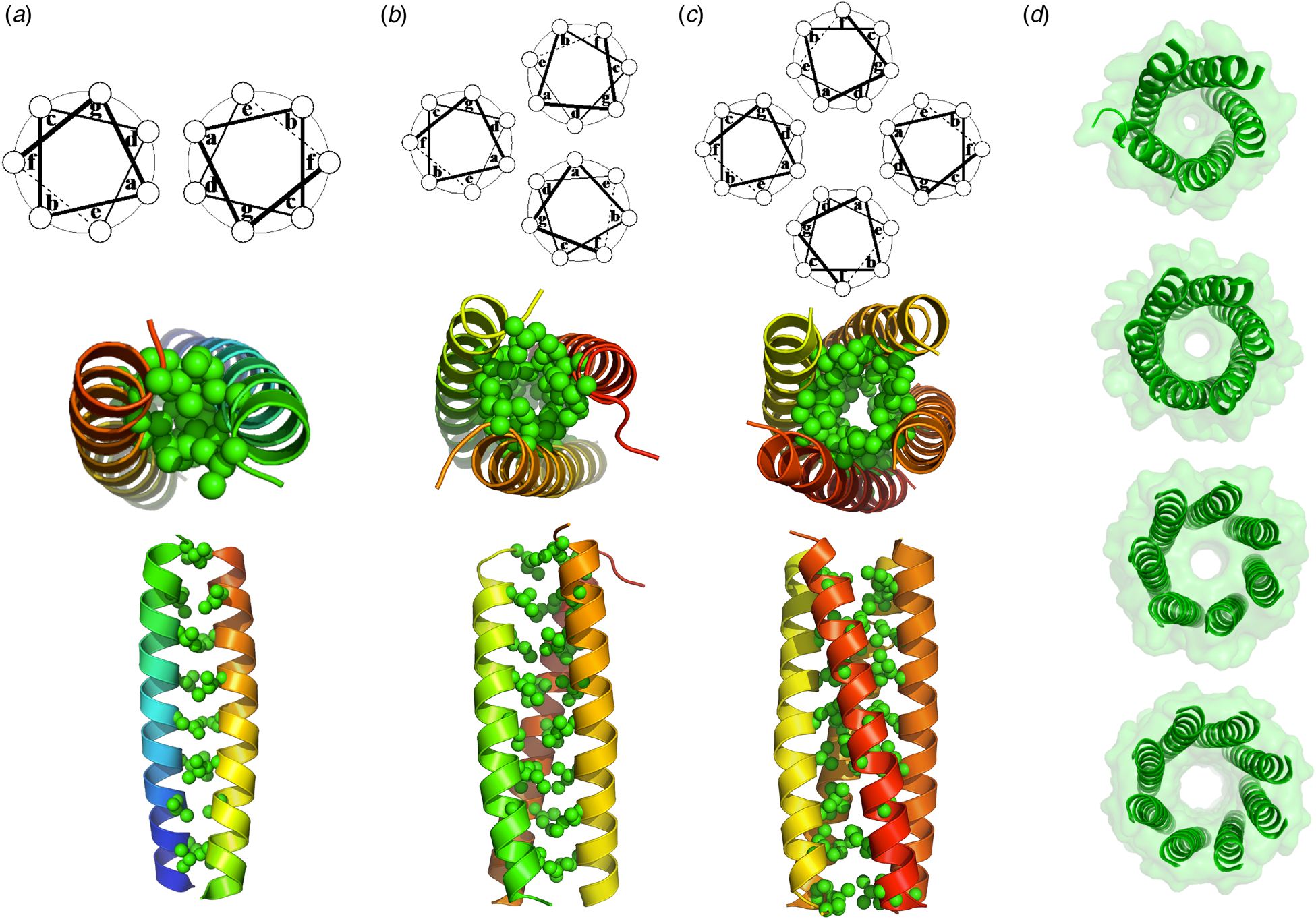
Fig. 4. (a) A crystal structure of a dimeric natural coiled-coil GCN4 interaction (PDB: 2ZTA) and the corresponding helical wheel. (b) A side on and end on views of the hydrophobic interior of a trimeric coiled-coil GCN4 derivative (PDB: 1GCM) along with the corresponding helical wheel. (c) A side on and end on views of the hydrophobic interior of a tetrameric GCN4 derivative (PDB: 1GCL) along with the corresponding helical wheel. (d) End on views of de novo designed penta-, hexa-, hepta-, and octameric bundles (PDB: 4PND, 4H8O, 5EZ8, 6G67).
More recently, Woolfson and coworkers extended these studies to the design 4- to 8-stranded bundles by manipulating the physicochemical and steric properties of the residues at the ‘e’ and ‘g’ positions (Fig. 4) (Thomson et al., Reference Thomson, Wood, Burton, Bartlett, Sessions, Brady and Woolfson2014). Importantly, coiled coils with some of these association states had never been characterized before – yet another milestone in de novo protein design. Moreover, Baker and coworkers extended the use of parametric equations to design regular bundles, with a variety of geometric repeats and stoichiometries (Huang et al., Reference Huang, Oberdorfer, Xu, Pei, Nannenga, Rogers, DiMaio, Gonen, Luisi and Baker2014). They also automated the process of searching for backbones that allow the formation of hydrogen-bond networks into homo- and hetero-dimeric coiled coils (Boyken et al., Reference Boyken, Chen, Groves, Langan, Oberdorfer, Ford, Gilmore, Xu, DiMaio, Pereira, Sankaran, Seelig, Zwart and Baker2016; Chen et al., Reference Chen, Boyken, Jia, Busch, Flores-Solis, Bick, Lu, VanAernum, Sahasrabuddhe, Langan, Bermeo, Brunette, Mulligan, Carter, DiMaio, Sgourakis, Wysocki and Baker2019). Today, the design of regular coiled coils of various sizes and shapes would appear to be a solved problem.
Functional de novo designed helical bundles
As the principles for designing structurally unique helical bundles became better understood, it also became possible to design functions. The interior of helical bundles can be elaborated to bind a variety of metal ions and small substrates. Much of this work predated the development of integrated packages for protein structure prediction and design such as Rosetta, and instead relied on physical principles and molecular mechanics force fields to guide the designs. More recently, Rosetta has brought most of the essential steps into a single framework, simplifying the overall process and allowing inclusion of structural bioinformatics data into the design process (Leaver-Fay et al., Reference Leaver-Fay, Tyka, Lewis, Lange, Thompson, Jacak, Kaufman, Renfrew, Smith, Sheffler, Davis, Cooper, Treuille, Mandell, Richter, Ban, Fleishman, Corn, Kim, Lyskov, Berrondo, Mentzer, Popović, Havranek, Karanicolas, Das, Meiler, Kortemme, Gray, Kuhlman, Baker and Bradley2011).
Overall strategy for building metal ion and cofactor-binding sites
Metal ion sites in proteins serve both structural and functional roles. Structural sites, such as in zinc fingers, tend to have common, coordinately saturated geometries that stabilize the folded conformation of the protein. In contrast, functional sites often have coordinately unsaturated in geometries that are enforced by the fold of the protein. Metalloproteins catalyze a remarkable array of reactions and a given metal ion such as manganese or iron can be used in different enzymes to catalyze a number of oxidative, reductive, and hydrolytic transformations (Yu et al., Reference Yu, Cangelosi, Zastrow, Tegoni, Plegaria, Tebo, Mocny, Ruckthong, Qayyum and Pecoraro2014). Thus, the activity of a given metalloprotein represents a partnership between the metal ion cofactor and the protein matrix: the metal ion brings non-discriminate chemical reactivity, while the protein stabilizes the metal ion in aqueous solution, fine tunes its reactivity, and binds substrates for catalysis. The protein also often positions hydrogen bond donors, acceptors and tunes the electrostatic environment for catalysis. De novo design allows us to probe and expand our understanding of these processes.
While it is possible to graft metal ion sites into existing proteins, in our approach to de novo design of metalloproteins the geometrically stringent requirements for metal ion and substrate binding instead dictate the backbone of the protein (Lombardi et al., Reference Lombardi, Summa, Geremia, Randaccio, Pavone and DeGrado2000b). The ligation geometry and the requirement that the ligating sidechains adopt energetically accessible conformations together provide powerful restraints that help define the overall fold and backbone structure. Second-shell hydrogen bonds to the primary ligands provide an additional restraint, which further restricts the possible backbone geometries. The function dictates the nature of the ligands (most commonly, Met, Cys, Asp/Glu, and His) employed in a given design. The nature of the ligands and their geometry help control the affinity and redox properties of the bound metal ion as well as its Lewis acidity. The availability of ligation sites for interaction with exogenous ligands, including water, O2, and organic substrates provides another important restraint. Finally, flexibility must be considered to stabilize multiple states as substrates come on and off, and, in some cases, the metal ions change oxidation state.
When the site is symmetrical this can facilitate parametric design of the protein backbone as illustrated in Fig. 5. The design is completed by introduction of loops, and sequence selection completed as in the above section. As described below, while the initial designs are often symmetrical, it is frequently necessary to lift the symmetry in subsequent designs as required for function.

Fig. 5. The desired geometry of the metal ion-binding site dictates the overall 3D structures during de novo protein design. In panel (a), a trigonal 3-Cys site dictates the backbone of a three-helix bundle in the TRI series of peptides (Dieckmann et al., Reference Dieckmann, McRorie, Tierney, Utschig, Singer, O'Halloran, Penner-Hahn, DeGrado and Pecoraro1997, Reference Dieckmann, McRorie, Lear, Sharp, DeGrado and Pecoraro1998; Mocny and Pecoraro, Reference Mocny and Pecoraro2015) (PDB: 2JGO). The structure is stabilized in the desired conformation by favorable vdW packing and the hydrophobic interactions between buried apolar residues (far right). In panel (b), a more complex C 2 symmetrical site is formed from 4-Glu and two-His residues, which bind to two transition metal ions in a four-helix bundle in the DF series of proteins (Lombardi et al., Reference Lombardi, Pirro, Maglio, Chino and DeGrado2019). The two-fold axis is denoted by an oval. A large number of second-shell hydrogen bonds were positioned to stabilize the ligands in the desired conformation, and the remaining interior residues chosen (not shown) were apolar sidechains that pack efficiently in the interior of the bundle.
Di- and tetranuclear metal complexes
Dimetal (e.g. di-Co, di-Fe, and di-Mn) proteins catalyze a variety of hydrolytic and redox processes (Marsh and Waugh, Reference Marsh and Waugh2013; Wang et al., Reference Wang, Liang and Lippard2015; Jasniewski and Que, Reference Jasniewski and Que2018; Crichton, Reference Crichton and Crichton2019). Their metal-binding sites are rich in Glu/Asp and His ligands, and the metal ions are generally bridged by water (also OH− or O2−) and/or carboxylate-containing sidechains. We were particularly drawn to the O2− utilizing proteins, which include hydroxylases, fatty acid desaturases, radical-generating ribonucleotide reductases, catalases, ferritins, and aldehyde decarbonylases. Although the overall structures of these proteins are highly diverse, in each case the di-Mn or di-Fe sites of all these proteins are housed within an antiparallel four-helix bundle that is generally embedded into a much larger structure (Summa et al., Reference Summa, Lombardi, Lewis and DeGrado1999; Lombardi et al., Reference Lombardi, Summa, Geremia, Randaccio, Pavone and DeGrado2000b).
In 2000, Lombardi and DeGrado designed a minimal diiron protein (DF) (Lombardi et al., Reference Lombardi, Summa, Geremia, Randaccio, Pavone and DeGrado2000b), not by modification of the sequence of a natural diiron protein, but rather by starting from first-principles and using a set of equations to generate the fold of the structure. The backbone was a D 2-symmetric four-helix bundle – each helix donating a single Glu ligand. An additional His residue was placed on just two of the helices, leaving two free sites to interact with substrates such as O2. The final model was a two-fold symmetric dimer of helical hairpins, whose backbone structure was dictated by: (1) coordination requirements of the Glu4His2-diiron site; (2) suitable helical packing angles and distances; and (3) Asp and Tyr second-shell H-bonds to the coordinating His and Glu (Fig. 5). The core was packed using the algorithm of Desjarlais and Handel (Reference Desjarlais and Handel1995).
Remarkably, the first designed sequence folded into a very stable dimetal-binding protein; for the first time, a de novo metalloprotein showed a crystal structure in excellent agreement with the intended design (Lombardi et al., Reference Lombardi, Summa, Geremia, Randaccio, Pavone and DeGrado2000b). Both the backbone and the entire network of first- and second-shell ligands were realized precisely as in the intended design (Figs 6a and b). Moreover, the solution NMR structure of metal-free apo-DF1 was nearly identical to the holo-protein, indicating that the six coordinating and the four second-shell ligands were largely preorganized with Å-level accuracy even in the absence of the metal cofactor (Maglio et al., Reference Maglio, Nastri, Pavone, Lombardi and DeGrado2003). Thus, DF imposed its structure onto the metal cofactor rather than vice versa, demonstrating that a pre-organized binding site in the apolar core could be stabilized by a sufficient set of H-bonds and salt bridges.
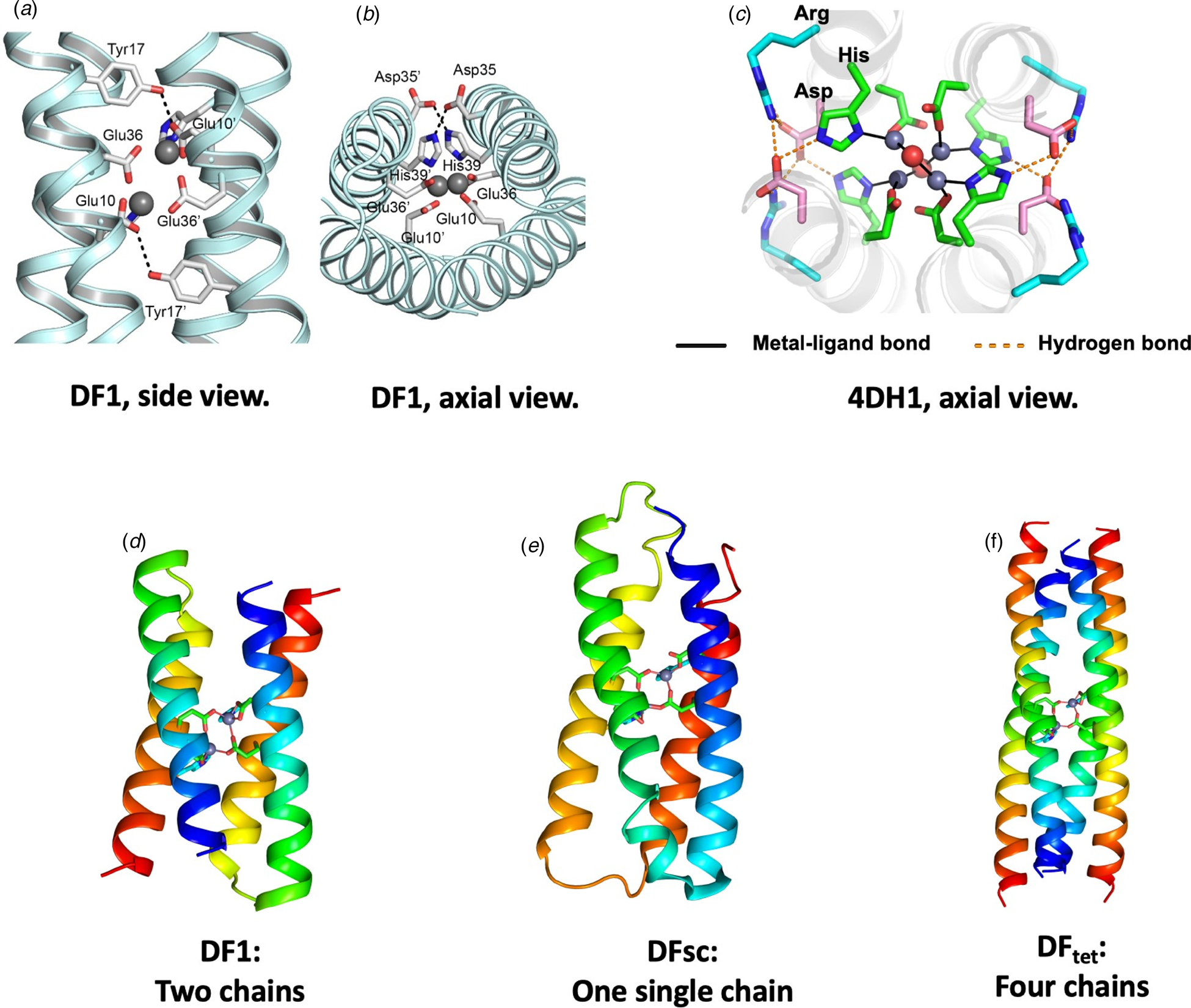
Fig. 6. Design of DF family proteins. Panels (a–c) show experimentally determined structures of extended metal-ligand and second-shell hydrogen-bonded networks in DF1 and related proteins. Two projections of DF1s metal-binding site are shown in (a) and (b) (PDB: 1EC5). Panel (c) shows an axial view of 4DH1 (PDB: 5WLL), a DF analog that binds four Zn(II) ions. An Asp residue forms a second-shell-hydrogen bond to a His ligand, and an Arg residue forms a third-shell hydrogen bond. Overall, the network includes four Zn, two waters, eight Asp, four His, and four Arg – all converging at the center of the bundle. Panels (d–f) illustrate how the backbone of DF (d) was elaborated to create a single chain (DFsc, PDB: 2HZ8) or a self-assembling tetramer (DFtet).
In subsequent work, DF1 was engineered to realize a number of binding and catalytic functions. Each step illustrated a tradeoff between protein stability and function (Shoichet et al., Reference Shoichet, Baase, Kuroki and Matthews1995). The desired changes were highly destabilizing, as they involved burial of additional polar groups (Reig et al., Reference Reig, Pires, Snyder, Wu, Jo, Kulp, Butch, Calhoun, Szyperski, Solomon and DeGrado2012) and removal of Leu sidechains to create an substrate-binding site proximal to the metal ions (DeGrado et al., Reference DeGrado, Di Costanzo, Geremia, Lombardi, Pavone and Randaccio2003; Maglio et al., Reference Maglio, Nastri, Pavone, Lombardi and DeGrado2003). To compensate, stabilizing substitutions were placed at positions distant from the active site, and an idealized αR-αL-β (Lahr et al., Reference Lahr, Engel, Stayrook, Maglio, North, Geremia, Lombardi and DeGrado2005) interhelical loop featuring a network of hydrogen-bonded sidechain/mainchain interactions was installed to favor the folded structure (Faiella et al., Reference Faiella, Andreozzi, de Rosales, Pavone, Maglio, Nastri, DeGrado and Lombardi2009). Also, the C 2 symmetry of the initial DF led to functional limitations that could be overcome by building a single-chain version of the protein (DFsc) (Calhoun et al., Reference Calhoun, Kono, Lahr, Wang, DeGrado and Saven2003) with three interhelical loops (Fig. 6e).
In an alternate approach, Summa et al. designed DFtet, which consisted of four disconnected helices that could be combinatorially assembled to facilitate evaluation of multiple sequence variants for catalytic functions (Fig. 6f) (Marsh and DeGrado, Reference Marsh and DeGrado2002; Summa et al., Reference Summa, Rosenblatt, Hong, Lear and DeGrado2002; Kaplan and DeGrado, Reference Kaplan and DeGrado2004). To increase stability, the helices of DFtet were extended to 33 residues, and the overall bundle was redesigned to conform to a left-handed coiled coil using an algorithm that incorporates the Crick equations. By engineering the electrostatic interaction at the helix–helix interfaces and an internal hydrogen-bond network, it was possible to design a uniquely folded two-component A2B2 tetramer (Summa et al., Reference Summa, Rosenblatt, Hong, Lear and DeGrado2002), as well as a three-component AA·AB·B2 heterotetramer (Marsh and DeGrado, Reference Marsh and DeGrado2002; Kaplan and DeGrado, Reference Kaplan and DeGrado2004). Both assembled with very high specificity. A Monte-Carlo algorithm that explicitly evaluated the electrostatic interactions in the desired heterotetramer, as well as other unwanted alternative topologies, facilitated the design. To the best of our knowledge, this was the first use of a computational algorithm to design a sequence that not only stabilized the desired structure (positive design), but also destabilized undesired outcomes (negative design). Since then, sophisticated methods that incorporate machine-learning have been developed for positive and negative design of coiled coils (Grigoryan et al., Reference Grigoryan, Reinke and Keating2009). Rosetta's H-bond network algorithm can also now facilitate the process of building hydrogen bond networks (Boyken et al., Reference Boyken, Chen, Groves, Langan, Oberdorfer, Ford, Gilmore, Xu, DiMaio, Pereira, Sankaran, Seelig, Zwart and Baker2016; Chen et al., Reference Chen, Boyken, Jia, Busch, Flores-Solis, Bick, Lu, VanAernum, Sahasrabuddhe, Langan, Bermeo, Brunette, Mulligan, Carter, DiMaio, Sgourakis, Wysocki and Baker2019).
A variety of catalytic and binding functions have been engineered into DF protein scaffolds (Lombardi et al., Reference Lombardi, Pirro, Maglio, Chino and DeGrado2019). Precisely as designed, the bespoke site presented unoccupied ligand-binding sites for water, O2, and organic substrates. By modifying the environment surrounding the diiron site it has been possible to design DF analogs that catalyze the O2-dependent oxidation of dihydroquinones (Faiella et al., Reference Faiella, Andreozzi, de Rosales, Pavone, Maglio, Nastri, DeGrado and Lombardi2009) and amino phenols (Kaplan and DeGrado, Reference Kaplan and DeGrado2004) at rates approaching that of the alternative oxidase enzyme. Furthermore, by asymmetrically introducing an additional His ligand (and additional second- and third-shell hydrogen bonding groups) the DF protein has been further engineered to catalyze aniline hydroxylase, mimicking a family of related non-heme enzymes (Reig et al., Reference Reig, Pires, Snyder, Wu, Jo, Kulp, Butch, Calhoun, Szyperski, Solomon and DeGrado2012; Snyder et al., Reference Snyder, Betzu, Butch, Reig, DeGrado and Solomon2015). Finally, a DFsc variant was designed to stabilize the radical semiquinone anion, which is otherwise unstable in aqueous solution (Ulas et al., Reference Ulas, Lemmin, Wu, Gassner and DeGrado2016). The protein stabilized the semiquinone by reducing the midpoint potential for its formation via the one-electron oxidation of the catechol by approximately 400 mV (9 kcal mol−1). Hence, the stability of a radical species was drastically stabilized by harnessing its binding energy to the metalloprotein.
Most recently, the design principles used in the construction of DF proteins have recently been extended to engineer tetranuclear Zn2+ clusters (Chino et al., Reference Chino, Zhang, Pirro, Leone, Maglio, Lombardi and DeGrado2018; Zhang et al., Reference Zhang, Chino, Liu, Tang, Hu, DeGrado and Lombardi2018a). The site included four bridging Asp and four terminal His ligands, as well as a total of 16 polar side chains in a fully connected hydrogen-bonded network (Fig. 6c). Similar to DFtet, the designed proteins have clusters of apolar sidechains above and below the binding site, which drive the assembly of the bundle. Solution NMR and crystallography confirmed that the desired structure, including a vast network of hydrogen-bonded interactions had indeed been achieved.
Trigonal binding sites in three-helix bundle
Many metal ions are bound in a trigonal geometry, for example, representing three vertices of a tetrahedron, a trigonal pyramid, an octahedron, or a trigonal planar arrangement. The three-helix bundle is particularly compatible with this geometry, and early work with template-assembled peptides using, for example, bi-pyridyl-metal ion interactions (Ghadiri and Case, Reference Ghadiri and Case1993), achieved this geometry.
In the 1990s, Pecoraro, DeGrado, and coworkers designed the first three-helical bundle metalloproteins, which interacted with Hg(II) in an unusual three-coordinate 3-Cys geometry (Fig. 5) (Dieckmann et al., Reference Dieckmann, McRorie, Tierney, Utschig, Singer, O'Halloran, Penner-Hahn, DeGrado and Pecoraro1997, Reference Dieckmann, McRorie, Lear, Sharp, DeGrado and Pecoraro1998). Building on this early success, the Pecoraro lab has greatly expanded the field of de novo designed metalloproteins. His group has generated a number of metal complexes that are not known in nature, but can be assembled through de novo protein design. The three-fold symmetry of the bundle is ideal for binding metal ions such as Zn(II), Hg(II), Cd(II), Pb(II), As(III), and Bi(III) that prefers lower coordination numbers. The metal binding sites were created by introducing cysteine residues in the ‘a’ position of the coiled-coil heptad at various locations in the bundle. The resulting proteins showed mid-nM affinities for cadmium, lead and mercury. Spectroscopic studies, including extended X-ray absorption fine structure (EXAFS), 113Cd, 207Pb, and 199Hg NMR as well 113mCd and 199mHg PAC helped elucidate fine structural details of the coordination sphere, which allowed for further fine-tuning of metal coordination sphere (Chakraborty et al., Reference Chakraborty, Touw, Peacock, Stuckey and Pecoraro2010, Reference Chakraborty, Kravitz, Thulstrup, Hemmingsen, DeGrado and Pecoraro2011; Iranzo et al., Reference Iranzo, Chakraborty, Hemmingsen and Pecoraro2011).
The formation of catalytically competent metal-binding sites in metalloproteins often requires the energetically unfavorable burial of a large number of polar residues in the hydrophobic interior. Pecoraro and coworkers reasoned that the structural stability imparted by the above-mentioned 3-Cys sites might be used to stabilize a second catalytically active metal-binding site within the same bundle. Using this principle they used the 3-Cys Hg(II)-binding site as a structural site to support a second catalytic three-His Zn-binding site. The resulting protein was a remarkably efficient catalyst of CO2 hydration (k cat/K M = 1.8 × 105 M−1 s−1 at pH 9.5) within 500-fold of carbonic anhydrase (Zastrow et al., Reference Zastrow, Peacock, Stuckey and Pecoraro2012). The designed zinc-based active site has also been transplanted into α3D (the anti-parallel 73-residue single-chain three-helical bundle protein discussed above) by mutating three of the core leucine residues to histidines (Fig. 3c) (Zastrow and Pecoraro, Reference Zastrow and Pecoraro2013a, Reference Zastrow and Pecoraro2013b). The single-chain antiparallel topology is inherently more stable than the self-assembled trimeric bundle; therefore the 3-Cys structural site is no longer necessary. The resulting metalloenzyme ZnIIα3DH3 efficiently promotes p-NPA hydrolysis and CO2 hydration. Its kinetic parameters are somewhat lower than those of the 3-chain predecessor; however, due to its single-chain topology, it can be improved using directed evolution.
To expand the repertoire of catalyzed chemical reactions by de novo designed trimeric coil coils enzymes to redox transformations, Pecoraro explored copper binding of TRIL23H, a close relative of metallohydrolase-supporting peptide TRIL9CL23H, but without the mercury structural site. TRIL23H binds Cu(II) with nM–μM affinity and Cu(I) with pM affinity fulfilling the key requirement for redox cycling. The copper ion in Cu(I/II)(TRIL23H)3 is bound by three histidine residues leaving two sites open to substrate/reductant coordination in a manner similar to that of the CuT2 center of copper nitrite reductase (Tegoni et al., Reference Tegoni, Yu, Bersellini, Penner-Hahn and Pecoraro2012). The designed metalloenzyme catalyzes reduction of nitrate to NO using ascorbate as the ultimate reductant for at least five turnovers.
The functional versatility of the trimeric coiled coils goes beyond catalysis. Peacock and co-workers have successfully utilized them to create magnetic resonance imaging probes with excellent relaxivity properties (Berwick et al., Reference Berwick, Lewis, Jones, Parslow, Dafforn, Cooper, Wilkie, Pikramenou, Britton and Peacock2014, Reference Berwick, Slope, Smith, King, Newton, Gillis, Adams, Rowe, Harding, Britton and Peacock2016). Tanaka and coworkers extended the trimeric helical Ile zipper peptides described by Alber and coworkers to create a 3-His site capable of binding transition metals with different geometries (Suzuki et al., Reference Suzuki, Hiroaki, Kohda and Tanaka1998; Kiyokawa et al., Reference Kiyokawa, Kanaori, Tajima, Koike, Mizuno, Oku and Tanaka2004; Tanaka et al., Reference Tanaka, Mizuno, Fukui, Hiroaki, Oku, Kanaori, Tajima and Shirakawa2004). The ability of the resulting peptides to oligomerize in a predicable manner was used to induce trimerization of DNA-binding domains of the heat shock proteins from Saccharomyces cerevisiae (Murase et al., Reference Murase, Ishino, Ishino and Tanaka2012). Fusing a variant of the green fluorescent protein to metal-binding coiled coils produced fluorescent sensors for metal ions (Murase et al., Reference Murase, Ishino, Ishino and Tanaka2012).
Directed evolution of the esterase activity of a Zn2+-binding helical bundle built on a natural protein scaffold
In the process of creating a metal-mediated protein–protein interface, Kuhlman and co-workers discovered MID1, a zinc-binding dimeric helix–loop–helix protein that can promote p-nitrophenol ester and phosphoester hydrolysis with reasonable catalytic efficiencies (Der et al., Reference Der, Machius, Miley, Mills, Szyperski and Kuhlman2012b). While this work involved modification of an existing natural protein rather than full de novo design, the fold used was of similar complexity to the de novo scaffolds discussed above, allowing comparison of the two approaches. The Rosetta Match algorithm was used to identify protein structures from the PDB that could form half of a tetrahedral Zn2+ binding site when His or Cys ligands were introduced at appropriate surface locations. In the design strategy, a complete tetrahedral site was formed when the proteins associated to form symmetrical homodimers. A total of 600 natural protein scaffolds were screened, resulting in 1.5 million design trajectories, which were evaluated over 25 000 cpu hours. Eight designs were experimentally evaluated, and one, designated MID1, was sufficiently well behaved to allow characterization. In the intended design, MID1 contains two symmetrically related Zn2+-consisting of His residues at positions i and i + 4 introduced along the surface of a small helix–loop–helix domain from rabenosyn. This arrangement had been used for many years to mediate Zn2+ binding in de novo designed peptides (Ghadiri and Choi, Reference Ghadiri and Choi1990; Ruan et al., Reference Ruan, Chen and Hopkins1990; Krantz and Sosnick, Reference Krantz and Sosnick2001; Tang et al., Reference Tang, Signarvic, DeGrado and Gai2007; Signarvic and DeGrado, Reference Signarvic and DeGrado2009) as well as Zn2+-mediated dimerization of de novo designed proteins (Handel and DeGrado, Reference Handel and DeGrado1990; Handel et al., Reference Handel, Williams and DeGrado1993) and natural proteins (Salgado et al., Reference Salgado, Faraone-Mennella and Tezcan2007, Reference Salgado, Radford and Tezcan2010).
NMR and crystallographic analysis of MID1 showed considerable plasticity, with both similarities as well as differences to the design. As in the design each i, i + 4 His residue ligated a single ion via the ε-nitrogen. However, the third His bound in an unexpected geometry via the δ nitrogen, and the fourth His did not ligate Zn2+ at all (Der et al., Reference Der, Machius, Miley, Mills, Szyperski and Kuhlman2012b).
These differences were surprising given the above-mentioned successes in de novo metalloprotein design, in which the functional requirements were used to define both the fold and the site. Moreover, small perturbations to MID1, such as single-site amino acid substitutions or changing the metal ion from Zn2+ to Co2+ caused large changes in the helix-packing geometry of MID1 (Fig. 7) (Der et al., Reference Der, Machius, Miley, Mills, Szyperski and Kuhlman2012b). Serendipitously, MID1 had a weak 4-nitrophenyl esterase activity associated with the unexpected 3-His binding geometry, which resulted in a free ligation site on the bound Zn2+ (Der et al., Reference Der, Edwards and Kuhlman2012a).

Fig. 7. Structural plasticity of MID1 (a and b). Two views of the crystal structures of di-zinc MID1 (PDB: 3V1C, blue ribbon), di-cobalt MID1 (PDB: 3V1D, magenta), di-zinc MID1-H12E (PDB: 3V1E, yellow), and di-zinc MID1-H35E (PDB: 3V1F, green) are shown with one of the two helix–loop–helix motifs superimposed. The overlay shows the variability in metal ion positions and ligand geometry, as well as variations in inter-subunit interactions. Panels (c) and (d) illustrate a similar superposition of di-zinc MID1 (PDB: 3V1C, blue ribbon, orange carbon atoms as sticks) with di-Zinc MID1sc10 (PDB: 5OD1, gray ribbon, magenta C atoms as sticks) showing a large rigid-body rotation of the helical hairpins, a shift in the primary ligand from His39 to His35, and a 7 Å shift of the metal ion. Panel (e) shows the substrates used to characterize the catalytic activity of MID1sc10.
Similarly, Song and Tezcan introduced zinc binding sites into cytochrome bc562 to promote controlled self-assembly into tetrameric species. The resulting assembly promotes hydrolysis of various substrates (Song and Tezcan, Reference Song and Tezcan2014).
The plasticity of the MID1 protein proved beneficial for in vitro evolution of a stereoselective metalloenzyme capable of hydrolysis of model fluorogenic substrates (Studer et al., Reference Studer, Hansen, Pianowski, Mittl, Debon, Guffy, Der, Kuhlman and Hilvert2018). A single-chain version of MID1, MID1sc with a single metal-binding site served as the starting point for in vitro evolution. In all five rounds of cassette mutagenesis, two rounds of random mutagenesis, and two rounds of DNA shuffling were employed. Ultimately, a catalytic efficiency of k cat/K M = 980 000 M−1 s−1 (k cat = 1.6 s−1; K M = 1.6 µM) was achieved, highlighting the power of directed evolution in combination with rational protein design (Studer et al., Reference Studer, Hansen, Pianowski, Mittl, Debon, Guffy, Der, Kuhlman and Hilvert2018). The crystallographic structure of the resulting protein, MID1sc10 showed that the protein had undergone a number of remarkable changes in the course of evolution. One of the His ligands was lost and another gained at a different location, resulting in a 7 Å translation of the metal-binding site. Moreover, a substrate-binding site was created by multiple substitutions as well as a large, rigid-body rotation of one helix–turn–helix motif (Fig. 7).
It is instructive to compare the contributions of the metal ion versus the protein to the esterase activity of MID1scversus some of the purposefully designed proteins discussed above. The value of k cat/k uncat for MID1sc is 1.6 × 105, while that of a designed amyloid-forming Zn2+-binding heptapeptide IHIHIQI is 100-fold lower (1.6 × 103) at the same pH (Rufo et al., Reference Rufo, Moroz, Moroz, Stohr, Smith, Hu, DeGrado and Korendovych2014). The heptapeptide has a similar 3-His active site capable of activating a water molecule for hydrolysis (Lee et al., Reference Lee, Wang, Makhlynets, Wu, Polizzi, Wu, Gosavi, Stohr, Korendovych, DeGrado and Hong2017), but lacks cavities to bind the substrates. By contrast, MID1sc has a deep pocket capable of stereospecific binding of the large hydrophobic substrate, 1 (Fig. 7) used in the directed evolution experiments. The substrate-binding interactions result in considerable stereospecificity for 1 and a relatively tight K M of 1.6 µM. By comparison, both MID1sc10 and IHIHIQI hydrolyze the minimal substrate, 2, with similar values of k cat/K M (32 M−1 s−1 for MID1 versus 62 M−1 s−1 for IHIHIQI), likely reflecting the contribution of the preorganized metal complex. The additional catalytic efficiency of MID1sc10 for substrate 1 likely reflects more precise positioning of the substrate for attack in the Michaelis complex. These studies show the power of directed evolution to create substrate-binding interactions that work in concert with a metal to produce significant rate enhancements.
Helical bundles as catalysts and inhibitors of protein–protein interactions
Four-helix bundles were also used to test concepts of catalysis and to design inhibitors of protein–protein interactions. Baltzer and co-workers employed this strategy to design catalytic proteins. A 42-residue peptide KO-42 assembles into an antiparallel four-helix bundle with catalytic sites engineered on the surface of the bundle as demonstrated by NMR, circular dichroism (CD) spectroscopy and ultracentrifugation, to catalyze hydrolysis of p-nitrophenyl esters with a rate enhancement of three orders of magnitude compared to the imidazole control (Broo et al., Reference Broo, Brive, Ahlberg and Baltzer1997). Subsequent rational improvement of the design allowed for introduction of enantioselective recognition of substrates (Broo et al., Reference Broo, Nilsson, Nilsson, Flodberg and Baltzer1998), a hallmark of natural proteins, and for elucidation of the role the pK a of the active residue as well as the geometry of the active site on catalysis (Broo et al., Reference Broo, Nilsson, Nilsson, Flodberg and Baltzer1998; Baltzer et al., Reference Baltzer, Broo, Nilsson and Nilsson1999). Expansion of the active site in the bundles to include additional residues to provide transition state stabilization allowed for hydrolysis of challenging phosphoester substrates, including uridine 3′−2,2,2-trichloroethylphosphate, a mimic of RNA (Razkin et al., Reference Razkin, Nilsson and Baltzer2007, Reference Razkin, Lindgren, Nilsson and Baltzer2008). The simple architecture of KO-42 is nonetheless amenable to introduction of binding sites for complex substrates, whose recognition relies on multiple substrate–protein interactions. In addition to a histidine-based active site to promote proton-transfer, KO-42 was modified to incorporate positively charged residues to stabilize negatively charged aldimine. The resulting peptide bundles T-4 and T-16 promote aldimine to ketamine conversion, emulating biosynthetic transamination reactions (Allert and Baltzer, Reference Allert and Baltzer2003). Finally, the graded reactivity of KO-42 has been used to allow the site-directed assembly of auxiliary binding groups, to create binders of protein surfaces with sub-nanomolar affinity for the proteins of interest (Baltzer, Reference Baltzer2011; Yang et al., Reference Yang, Gustavsson, Haraldsson, Karlsson, Norberg and Baltzer2017a, Reference Yang, Koruza, Fisher, Knecht and Baltzer2017b).
While the binding and catalytic sites of derivatives of KO-42 lie along the surface of the bundle, Woolfson and coworkers used the hollow surface of de novo designed proteins to create functional sites. They succeeded in building a catalytic dyad in a peptide that self-assembles into a heptameric coiled coil with no known natural analogs that promotes ester hydrolysis (Burton et al., Reference Burton, Thomson, Dawson, Brady and Woolfson2016).
Helical bundles have been designed or selected to bind to a variety of other protein surfaces, to create inhibitors of protein–protein interactions (Fujiwara and Fujii, Reference Fujiwara and Fujii2013; Fujiwara et al., Reference Fujiwara, Kitada, Oguri, Nishihara, Michigami, Shiraishi, Yuba, Nakase, Im, Cho, Joung, Kodama, Kono, Ham and Fujii2016). A recent example illustrates how far de novo protein design has progressed from the early days of parametric helical bundle design of proteins to incorporate the sophisticated computational design algorithms in Rosetta as well as directed evolution and sequence display of combinatorial libraries in the work flow. Baker and coworkers recently combined these technologies to design mimics of interleukin-2 (IL-2) that bind to the IL-2 receptor βγc heterodimer (IL-2Rβγc), but not to IL-2Rα or IL-15Rα. The designs used the natural four-helix bundle, IL-2, as a starting point. In a series of steps the IL-2 bundle was progressively idealized using parametric protein design, and its folding topology was simplified by introduction of short idealized loops. At each round of design, the sequences were experimentally evaluated and the affinity was enhanced by multiple rounds of display on yeast. Crystal structures of an optimized design protein alone and in complex with IL-2Rβγc, are very similar to the designed model. The family of designed proteins has superior therapeutic activity to IL-2 in mouse models of melanoma and colon cancer, with reduced toxicity and undetectable immunogenicity.
Helical bundles for binding complex cofactors
Dutton and DeGrado utilized a sequence-based approach to design heme-binding proteins designated ‘maquettes’ to probe the function of multi-heme proteins. A 31-residue long peptide designed to mimic the key structural features of cytochrome bc1 was shown to assemble in the presence of four hemin moieties to form a four-helix bundle. Introduction of a flexible Cys containing linker allowed for further stabilization of the structure, effectively creating a helix–loop–helix motif (Robertson et al., Reference Robertson, Farid, Moser, Urbauer, Mulholland, Pidikiti, Lear, Wand, DeGrado and Dutton1994). The original designs have been elaborated by Dutton, Moser, Gibney, Anderson, and coworkers to include complex single-chain topologies that allowed sequence diversification and recombinant expression (Grayson and Anderson, Reference Grayson and Anderson2018). The simple geometry of maquettes (Fig. 8a) allowed for direct elucidation of factors that define electrochemical properties of heme in metalloproteins and subsequent rational tuning of the redox potential of the cofactors (Kennedy and Gibney, Reference Kennedy and Gibney2001; Reedy and Gibney, Reference Reedy and Gibney2004). Subsequent studies show that the maquette architecture can support diverse protein functionalities ranging from light capture to catalysis (Koder et al., Reference Koder, Anderson, Solomon, Reddy, Moser and Dutton2009; Lichtenstein et al., Reference Lichtenstein, Farid, Kodali, Solomon, Anderson, Sheehan, Ennist, Fry, Chobot, Bialas, Mancini, Armstrong, Zhao, Esipova, Snell, Vinogradov, Discher, Moser and Dutton2012; Kodali et al., Reference Kodali, Mancini, Solomon, Episova, Roach, Hobbs, Wagner, Mass, Aravindu, Barnsley, Gordon, Officer, Dutton and Moser2017; Watkins et al., Reference Watkins, Jenkins, Grayson, Wood, Steventon, Le Vay, Goodwin, Mullen, Bailey, Crump, MacMillan, Mulholland, Cameron, Sessions, Mann and Anderson2017). The malleable, dynamic maquette scaffolds bind cofactors with high affinity and serve as starting points for further improvement supporting the notion that substantial initial level of functionality is fairly easy to achieve in de novo designed proteins. Nevertheless, it did not prove to be possible to solve solution NMR or crystallographic structures of the family of maquettes with their cofactors bound. One structure was solved for an apo-structure, but the structure was not compatible with the requirements of binding heme (Huang et al., Reference Huang, Koder, Lewis, Wand and Dutton2004).

Fig. 8. Cofactor-binding helical bundles. Panel (a) shows a model of a two-porphyrin maquette. High-resolution structures have not been published for cofactor-bound maquettes, likely due to dynamic properties (Koder et al., Reference Koder, Anderson, Solomon, Reddy, Moser and Dutton2009; Lichtenstein et al., Reference Lichtenstein, Farid, Kodali, Solomon, Anderson, Sheehan, Ennist, Fry, Chobot, Bialas, Mancini, Armstrong, Zhao, Esipova, Snell, Vinogradov, Discher, Moser and Dutton2012; Kodali et al., Reference Kodali, Mancini, Solomon, Episova, Roach, Hobbs, Wagner, Mass, Aravindu, Barnsley, Gordon, Officer, Dutton and Moser2017; Watkins et al., Reference Watkins, Jenkins, Grayson, Wood, Steventon, Le Vay, Goodwin, Mullen, Bailey, Crump, MacMillan, Mulholland, Cameron, Sessions, Mann and Anderson2017). However, recent work on other de novo proteins including PS1 indicates that it is possible to design uniquely structured porphyrin-binding proteins (Polizzi et al., Reference Polizzi, Wu, Lemmin, Maxwell, Zhang, Rawson, Beratan, Therien and DeGrado2017). Panels (b) and (c) illustrate PS1, a porphyrin-binding protein, that was instead computationally designed to carefully optimize the packing of the core as well as the packing of the cofactor (Polizzi et al., Reference Polizzi, Wu, Lemmin, Maxwell, Zhang, Rawson, Beratan, Therien and DeGrado2017). The high-resolution solution structure of the apo-state has two conformations that appear to facilitate binding of the porphyrin. Both conformers have well-packed hydrophobic core, but differ in the orientation of the helices in the binding site. Binding of the porphyrin results in ordering of the entire protein.
Multiheme-binding helical bundles can also be designed completely de novo based on parameterized backbones, the first being closely related to α4 (Choma et al., Reference Choma, Lear, Nelson, Dutton, Robertson and DeGrado1994). Subsequent parameterizations were based on positioning keystone residues for first- and second-shell ligation as well as steric packing. This approach was expanded to enable design of a variety of cofactors that contain various metals (Bender et al., Reference Bender, Lehmann, Zou, Cheng, Fry, Engel, Therien, Blasie, Roder, Saven and DeGrado2007; Fry et al., Reference Fry, Lehmann, Saven, DeGrado and Therien2010, Reference Fry, Lehmann, Sinks, Asselberghs, Tronin, Krishnan, Blasie, Clays, DeGrado, Saven and Therien2013; Korendovych et al., Reference Korendovych, Senes, Kim, Lear, Fry, Therien, Blasie, Walker and DeGrado2010).
Only recently has the successful design of a porphyrin-binding protein with sub-Ångstrom accuracy been accomplished as verified by high-resolution structure determination. The key was to consider what had traditionally been considered as separate sectors – the hydrophobic core and ligand-binding site – inseparable units (Figs 8b and c) (Polizzi et al., Reference Polizzi, Wu, Lemmin, Maxwell, Zhang, Rawson, Beratan, Therien and DeGrado2017). Flexible backbone design of a parametrically defined protein template allows to simultaneously pack both the protein interior both proximal to and remote from the ligand-binding site. Thus, tight interdigitation of core side chains quite removed from the binding site structurally can cooperate to restrain and stabilize the first- and second-shell packing around the ligand. The resulting protein, PS1, bound an electron-deficient, non-natural porphyrin at temperatures up to 100 °C, and its structure was in sub-Ångstrom agreement with the design. These results illustrated the unification of core packing and binding site definition as a central principle of ligand-binding protein design. It also bodes well for the design of ‘maquettes’ that are uniquely structured, rather than multi-conformational in nature.
Beyond helical bundles
By 2000, the accurate de novo design of homo-oligomeric coiled coils (Harbury et al., Reference Harbury, Tidor and Kim1995, Reference Harbury, Plecs, Tidor, Alber and Kim1998; Ogihara et al., Reference Ogihara, Weiss, DeGrado and Eisenberg1997) and helical bundles such as α3D and DF (Lombardi et al., Reference Lombardi, Summa, Geremia, Randaccio, Pavone and DeGrado2000b) had been accomplished. By contrast, the design and structure determination of uniquely folded globular proteins containing β-structure remained problematic. Early attempts to design an all-β protein called betabellin resulted in structures with poor solubility (Richardson and Richardson, Reference Richardson and Richardson1989), likely due to amyloid formation (Lim et al., Reference Lim, Saderholm, Makhov, Kroll, Yan, Perera, Griffith and Erickson1998). Analysis of the failures, however, led to important insights (Richardson and Richardson, Reference Richardson and Richardson2002). The edges of β-sheets are sticky sites that can engage in aggregation and amyloid formation. In natural proteins, such aggregation is minimized by decreasing the length of edge strands and endowing them with Pro residues or polar groups that decrease inter-chain hydrogen-bonding and hydrophobic interactions that can lead to oligomerization.
Nevertheless, in the 1990s significant progress was made toward the design of peptides that form β-hairpins, including the Trp zipper peptides that displayed well-defined β-hairpin conformations stabilized by cross-strand pairs of indole rings (Cochran et al., Reference Cochran, Skelton and Starovasnik2001). Also, by 1998, several groups had demonstrated the design of three-stranded β-sheets, with varying degrees of water-solubility and stability (Das et al., Reference Das, Raghothama and Balaram1998; Kortemme et al., Reference Kortemme, Ramirez-Alvarado and Serrano1998; Schenck and Gellman, Reference Schenck and Gellman1998; Sharman and Searle, Reference Sharman and Searle1998). As mentioned above, Dahiyat and Mayo had also succeeded in the fully automated redesign of the sequence of a zinc finger peptide, resulting in a peptide that folded into a structure consisting of an α-helix packed against an antiparallel β-hairpin (Richardson and Richardson, Reference Richardson and Richardson2002). Imperiali's group also redesigned a similar zinc finger to produce a peptide that folded in the absence of metal ions (Struthers et al., Reference Struthers, Cheng and Imperiali1996a, Reference Struthers, Cheng and Imperiali1996b).
The fundamental parameterization approach described above is by no means limited to helical bundles. Lombardi and coworkers built METP, a β-hairpin miniaturized electron transfer protein, by parameterizing the metal-binding site of a natural rubredoxin (Lombardi et al., Reference Lombardi, Marasco, Maglio, Di Costanzo, Nastri and Pavone2000a). Nanda, DeGrado and coworkers designed RM1, a stable minimalist protein that folds in a β-sheet structure both in the presence and in the absence of iron (Fig. 9). RM1s design was based on a simple dimeric sheet–turn–sheet secondary motif. RM1 binds iron to form a stable, redox-active four-cysteine thiolate iron site that is structurally and functionally analogous to that of rubredoxin (Nanda et al., Reference Nanda, Rosenblatt, Osyczka, Kono, Getahun, Dutton, Saven and Degrado2005). Recently, Nanda, Fialkowski, and coworkers have been able to design ambidoxin, a 12-residue peptide with alternating D- and L-amino acid residues to stabilize a functional 4Fe–4S cubane cluster through metal–side chain interactions and an intricate network of hydrogen bonds (Kang et al., Reference Kang, Kim, Ko, Kim, Cho, Huh, Kim, Nam, Thach, Youn, Kim, Yun, DeGrado, Kim, Hammond, Lee, Kwon, Ha and Kim2018).

Fig. 9. (Left) RM1 design cycle: (a) three-stranded sheet topology of natural rubredoxin, (b) C 2 symmetry, (c) active-site geometry, (d) miniRM dimer, and (e) RM1 with Trpzip linker shown in red. Reproduced with permission from Nanda et al. (Reference Nanda, Rosenblatt, Osyczka, Kono, Getahun, Dutton, Saven and Degrado2005). Copyright (2005) American Chemical Society. (Right) Computational model of ambidoxin.
Membrane protein design
There are two structural classes of TM proteins: β-barrels that are found in the outer membranes of bacteria and mitochondria and the helical bundles, which are found in cytoplasmic and organelle membranes. Given the greater functional diversity of the helical bundle class of membrane proteins, most work in de novo design has focused on this class of membrane proteins. De novo membrane protein design has contributed significantly to understanding fundamental principles by which membrane proteins achieve their folded conformations and functions such as active ion transport.
Understanding the rules of membrane protein folding, stability, and assembly
Helical membrane proteins begin folding as they exit the translocon, completing the process in the membrane environment (Engelman et al., Reference Engelman, Chen, Chin, Curran, Dixon, Dupuy, Lee, Lehnert, Matthews, Reshetnyak, Senes and Popot2003; White and von Heijne, Reference White and von Heijne2008). The folding of membrane proteins thus can be minimally approximated by a two-stage process involving the biosynthetic or physical insertion of TM helices into membranes followed by their subsequent assembly to form native structures. The features required for insertion are well understood from elegant studies of von Heijne, White, and others who examined the sequence-dependence of helix insertion into membranes via the translocon (Hessa et al., Reference Hessa, Kim, Bihlmaier, Lundin, Boekel, Andersson, Nilsson, White and von Heijne2005; White and von Heijne, Reference White and von Heijne2005, Reference White and von Heijne2008). The resulting ‘biological hydrophobicity scale’ was in good agreement with those obtained from model compounds as well as scales derived from structural informatics of membrane proteins (Senes et al., Reference Senes, Chadi, Law, Walters, Nanda and Degrado2007; Schramm et al., Reference Schramm, Hannigan, Donald, Keasar, Saven, Degrado and Samish2012). Such information has long provided restraints for design of monomeric helical peptides that insert into membranes (Ren et al., Reference Ren, Lew, Wang and London1999; Morein et al., Reference Morein, Koeppe, Lindblom, de Kruijff and Killian2000; Caputo and London, Reference Caputo and London2003), and has been incorporated into programs for membrane protein design, such as Rosetta Membrane (Elazar et al., Reference Elazar, Weinstein, Biran, Fridman, Bibi and Fleishman2016; Koehler Leman et al., Reference Koehler Leman, Mueller and Gray2017; Duran and Meiler, Reference Duran and Meiler2018).
De novo design has contributed to understanding the next key step in membrane protein folding when helices laterally associate to form an inter- or intra-molecular TM bundle. Much of the work has focused on engineering assemblies of TM α-helices from single-spanning membrane proteins, chosen for their biological relevance and technical advantages. Over 50% of all membrane proteins are single-spanning, yet they are the least structurally characterized class of MPs. Their lateral TM helix interactions play vital roles in signaling, complex formation, and ion conduction (Kirrbach et al., Reference Kirrbach, Krugliak, Ried, Pagel, Arkin and Langosch2013; Lomize et al., Reference Lomize, Lomize, Krolicki and Pogozheva2017). Aberrant folding or assembly is also involved in devastating diseases from cancer to Alzheimer's disease (Partridge et al., Reference Partridge, Therien and Deber2004; Schlebach and Sanders, Reference Schlebach and Sanders2015). Additionally, unlike complex multi-pass proteins, single-span TM bundles allow investigation of inter-helical interactions with a clear unfolded state – a monomeric α-helix – free of extracellular domains or loops that cloud interpretation. Moreover, conformational specificity and folding can be simply evaluated by determining whether a single oligomeric state is formed.
Small residue motifs that stabilize TM helix–helix-packing interactions
Some of the earliest studies on TM helix–helix interactions focused on the identification of sequence motifs, such as the GX3G, found in glycophorin A. GX3G, or more generally the Small-X3-Small (in which Small is Gly, Ala, or Ser) motif is involved in both intramolecular folding as well as intermolecular assembly of TM helices (Langosch et al., Reference Langosch, Brosig, Kolmar and Fritz1996; Brosig and Langosch, Reference Brosig and Langosch1998; Senes et al., Reference Senes, Gerstein and Engelman2000, Reference Senes, Ubarretxena-Belandia and Engelman2001). The small residues line along one face of the helix and mediate a very close approach of the backbones of two helices, which interact with a right-handed crossing angle of near 40° (MacKenzie et al., Reference MacKenzie, Prestegard and Engelman1997). The interface is stabilized through extensive vdW interactions (Duong et al., Reference Duong, Jaszewski, Fleming and MacKenzie2007; Mueller et al., Reference Mueller, Subramaniam and Senes2014) and CH hydrogen bonds between the backbone Cα–H and the carbonyl oxygen of neighboring helices (Senes et al., Reference Senes, Ubarretxena-Belandia and Engelman2001; Arbely and Arkin, Reference Arbely and Arkin2004; Mueller et al., Reference Mueller, Subramaniam and Senes2014). The stability of the Small-X3-Small motif depends critically on the position in the membrane as well as the sequence context surrounding the two small residues (Duong et al., Reference Duong, Jaszewski, Fleming and MacKenzie2007; Unterreitmeier et al., Reference Unterreitmeier, Fuchs, Schaffler, Heym, Frishman and Langosch2007; MacKenzie and Fleming, Reference MacKenzie and Fleming2008; Langosch and Arkin, Reference Langosch and Arkin2009).
A second motif that has been used extensively in membrane protein design is an antiparallel zipper-like packing with a Gly, Ala, or Ser in a (Small-X6)n motif (Adamian and Liang, Reference Adamian and Liang2002; Walters and DeGrado, Reference Walters and DeGrado2006). This sequence motif specifies folding into a structure similar to the alanine-coil seen water-soluble proteins (Gernert et al., Reference Gernert, Surles, Labean, Richardson and Richardson1995), with a left-handed crossing angle near −10° to −20°. The presence of a single small residue per heptad enables intimate packing. Computational design of model TM coiled-coil peptides (designated MS1 peptides) with various residues at the ‘a’ position showed association strengths in the order: Gly > Ala > Val > Ile. Moreover, MS1-Gly has a strong tendency to form antiparallel dimers, MS1-Ala formed a mixture of parallel and antiparallel dimers, while MS1-Val and MS1-Ile have a preference to form very weakly associating parallel dimers. Calculations based on exhaustive conformational searching and rotamer optimization were in excellent agreement with experiments, in terms of the overall stability of the structures and the preference for parallel versus antiparallel packing. These studies demonstrated that vdW interactions and electrostatic interactions contribute to the stability and topological preferences of the dimers.
Hydrogen-bonded interactions can stabilize membrane proteins
Hydrogen-bonds between polar sidechains can also contribute to the stability of membrane proteins. The introduction of strongly polar residues, including Asp, Asn, Glu, and Gln can lead to association of designed TM peptides (Choma et al., Reference Choma, Gratkowski, Lear and DeGrado2000; Zhou et al., Reference Zhou, Cocco, Russ, Brunger and Engelman2000, Reference Zhou, Merianos, Brunger and Engelman2001; Gratkowski et al., Reference Gratkowski, Lear and DeGrado2001). The energetics of the interaction depends on environment, ranging from very stabilizing near the middle of the TM helix (−2.0 kcal mol−1 per Asn side chain) to very weak (0 ± 0.5 kcal mol−1) near the ends of the helix, which locate to the headgroup region (Lear et al., Reference Lear, Gratkowski, Adamian, Liang and DeGrado2003). These data are consistent with the expectation that sidechain hydrogen bonding will contribute to stability in the relatively dry region of a membrane, but not in regions where water can compete for hydrogen bonds in the monomeric state.
Both the thermodynamics and geometric specificity of association of TM helices can be enhanced through the design of an extensive hydrogen-bonded network, as shown in work in which three Asn and three Thr sidechains were engineered to interact in a three-helix bundle (Tatko et al., Reference Tatko, Nanda, Lear and Degrado2006). More recently, Baker and coworkers have designed elongated membrane-spanning helical bundles, which contain hydrogen-bonded networks built using the HB-net module of Rosetta (Lu et al., Reference Lu, Min, DiMaio, Wei, Vahey, Boyken, Chen, Fallas, Ueda, Sheffler, Mulligan, Xu, Bowie and Baker2018). The designs included chains with two TM helices, representing the first examples of the de novo design of multi-pass membrane proteins, whose crystallographic structures were determined at high resolution.
Contribution of packing of large apolar residues to the stability of membrane proteins
All of the above designed membrane proteins relied on either polar interactions to drive assembly in the membrane, or small residues at appropriate spacings to drive folding through close contacts between the backbones of helices. However, such motifs, although not uncommon, are not a general feature of the interhelical packing seen in natural membrane proteins. Instead, helix–helix packings are stabilized by interactions of apolar sidechains, similar to that in water-soluble proteins. The hydrophobic effect, which represents the predominant driving force for protein folding in water, is negligible in lipid membranes. Thus, in membrane proteins it was unclear whether analogous side-chain packing in the native state provides significant structural stabilization. On the one hand, the same apolar moieties pack similarly with lipid tails in the exposed unfolded state. On the other hand, side-chains pack slightly more efficiently in membrane proteins (Eilers et al., Reference Eilers, Shekar, Shieh, Smith and Fleming2000; Adamian and Liang, Reference Adamian and Liang2001; Oberai et al., Reference Oberai, Joh, Pettit and Bowie2009; Zhang et al., Reference Zhang, Kulp, Schramm, Mravic, Samish and DeGrado2015), and hence might stabilize folding via favorable vdW interactions and possibly also lipid-specific effects such as ‘solvophobic’ exclusion (Langosch and Heringa, Reference Langosch and Heringa1998; Joh et al., Reference Joh, Oberai, Yang, Whitelegge and Bowie2009; Hong, Reference Hong2014; Anderson et al., Reference Anderson, Mueller, Lange and Senes2017). Mutations to membrane proteins that strongly disrupt vdW packing in the protein interior, either by introducing voids or steric clashes, have been shown to destabilize their native state to various degrees (Doura et al., Reference Doura, Kobus, Dubrovsky, Hibbard and Fleming2004; Joh et al., Reference Joh, Oberai, Yang, Whitelegge and Bowie2009; Baker and Urban, Reference Baker and Urban2012; Guo et al., Reference Guo, Gaffney, Yang, Kim, Sungsuwan, Huang, Hubbell and Hong2016). Nevertheless, it was less clear whether apolar packing can play a dominant role in membrane protein folding, or whether this feature is secondary to other more stabilizing interactions discussed above such as hydrogen bonding and weakly polar C–H-hydrogen bonds. If apolar packing contributed largely to the stabilization of membrane proteins it should be possible to design them based on this feature alone. However, for a number of years this proved to be very difficult (Whitley et al., Reference Whitley, Nilsson and von Heijne1994; Gurezka et al., Reference Gurezka, Laage, Brosig and Langosch1999; Choma et al., Reference Choma, Gratkowski, Lear and DeGrado2000; Zhou et al., Reference Zhou, Merianos, Brunger and Engelman2001; Yano et al., Reference Yano, Takemoto, Kobayashi, Yasui, Sakurai, Ohashi, Niwa, Futaki, Sugiura and Matsuzaki2002; Johnson et al., Reference Johnson, Heslop and Deber2004).
Building on the design principles discovered by Woolfson et al., in the construction of multistranded water-soluble coiled coils, Mravic et al. recently designed a homo-pentameric TM five-helix bundle stabilized by apolar packing in the membrane-spanning region alone (Mravic et al., Reference Mravic, Thomaston, Tucker, Solomon, Liu and DeGrado2019). Successful design required consideration of not only the ‘a’ and ‘d’ residues in the core, but also the more interfacial ‘e’ and ‘g’ residues. The resulting pentamers were remarkably stable, even at boiling temperatures in sodium dodecylsulfate. In spite of this extraordinary stability, the steric complementarity required for their folding was shown to be remarkably stringent when compared to helix–helix packings of water-soluble proteins. Thus, substitutions of Leu to Ile entirely disrupted any association of the helices. A strong hydrophobic driving force dominates folding in water, so natural proteins need not achieve stringent packing to fold. Without a hydrophobic force in bilayers, it appears geometric complementarity must be more strictly optimized to achieve folding in membrane proteins. Structural informatics shows that the designed packing motif recurs across the TM proteome, emphasizing a significant role for precise apolar packing in membrane protein folding and stabilization.
Design of functional membrane proteins
Design of TM proteins capable of proton, metal ion, and electron transfer
Given recent progress in designing membrane proteins with predetermined structures, it should be increasingly possible to design function as well. In fact, there have already been some significant accomplishments in the de novo design of proteins capable of transporting protons, ions, and electrons. The first TM helical bundles were designed in the late 1980s as functional models for proton channels and ion channels – significantly before the first high-resolution structures of proteins of this class had been determined (Lear et al., Reference Lear, Wasserman and DeGrado1988; DeGrado et al., Reference DeGrado, Wasserman and Lear1989). To gain insight into the mechanisms by which α-helices in channels associate and conduct ions, several peptides containing only Leu and Ser residues were designed and computationally modeled. A 21-residue peptide (Leu-Ser-Ser-Leu-Leu-Ser-Leu)3, formed well-defined ion channels with single-channel conductance characteristics resembling the acetylcholine receptor. A second peptide (Leu-Ser-Leu-Leu-Leu-Ser-Leu)3, in which one Ser per heptad repeat was replaced by Leu, produced proton-selective channels. Computer graphics and energy minimization were used to create molecular models that were consistent with the observed properties of the channels. The deduced structures were helical bundles with left-handed crossing angles between the helices. The packing of small Ser residues in a zipper-like manner dictated a tetrameric arrangement for the proton channel (Leu-Ser-Leu-Leu-Leu-Ser-Leu)3 (Fig. 10a) and a hexameric channel for the ion channel forming (Leu-Ser-Ser-Leu-Leu-Ser-Leu)3. The hydroxyl sidechains of the Ser residues interacted with water to create a pore large enough to accommodate a solvated ion in the hexameric bundle. The tetrameric bundle was more tightly packed but had voids large enough to accommodate water molecules that appeared to form a proton conduction pathway via a water-hopping mechanism. While crystallographic structures were not available at the time, a large body of subsequent data supported the underlying hypothetical structures and conduction model (DeGrado et al., Reference DeGrado, Wasserman and Lear1989; Åkerfeldt et al., Reference Åkerfeldt, Kim, Camac, Groves, Lear and DeGrado1992, Reference Åkerfeldt, Lear, Waserman, Chung and DeGrado1993; Zhong et al., Reference Zhong, Jiang, Moore, Newns and Klein1998; Dieckmann et al., Reference Dieckmann, Lear, Zhong, Klein, DeGrado and Sharp1999; Randa et al., Reference Randa, Forrest, Voth and Sansom1999; Nguyen et al., Reference Nguyen, Liu and Moore2013).
Fig. 10. (a, b) Top and side views of computational models of de novo designed ion pores LS2 and PRIME, respectively. In panel (a), the Ser sidechains of LS2 are shown in ball-and-stick models. Leu residues that are important for packing interactions that stabilize the tetramer of LS2 are shown in green sticks. In panel (b), the carbon atoms of the porphyrin cofactor are shown in purple. (c) Rocker, a de novo designed zinc transporter, showing configurations that were used for positive (+) and negative (−) design.
The most ambitious functional membrane protein designed to date is a TM four-helix bundle, Rocker (Fig. 10c), that transports first-row transition metal ions Zn2+ in exchange for protons (Joh et al., Reference Joh, Wang, Bhate, Acharya, Wu, Grabe, Hong, Grigoryan and DeGrado2014, Reference Joh, Grigoryan, Wu and DeGrado2017). The design of a Zn2+/proton transporter presented several grand challenges: the first was the design of a membrane protein with a predetermined structure, and the determination of its structure and dynamics at high resolution (which had not yet been accomplished for a de novo membrane protein). Next, the design should precisely position polar ionizable Zn2+ ligands, which ordinarily are excluded from a membrane environment. Furthermore, to achieve antiporting, it was important to thermodynamically link the binding of protons to changes in the affinity for metal ions. Finally, it was important to anticipate and orchestrate dynamics to facilitate transport of an ion through the channel.
Joh, Grigoryan, and DeGrado designed Rocker using four helices that present metal-binding sites similar to those used in the water-soluble DF proteins discussed above. Previously, Pasternak et al. had found that the Glu sidechains in a 4Glu-2His di-Zn2+-binding DF protein were largely protonated in the metal-free apo state, due to the energetic cost of burying negatively-charged sidechains within the interior of a protein (Pasternak et al., Reference Pasternak, Kaplan, Lear and Degrado2001). Binding of Zn2+ displaces these protons, providing a means to achieve the desired thermodynamic coupling. A computational design algorithm was next used to stabilize two energetically degenerate asymmetric states of the protein while destabilizing a competing fully symmetrical state which might otherwise bind metal ions too tightly and impede motions required for ion transport. The computed TM bundle formed a dimer of dimers with two non-equivalent helix–helix interfaces (Fig. 10c); a ‘tight interface’ had a small inter-helical distance (8.9 Å) stabilized by efficient packing of small, Ala residues. The ‘loose interface’ had a larger interhelical distance of 12.0 Å and was less well packed. The resulting membrane-spanning four-helical bundle transported first-row transition metal ions Zn2+ and Co2+, but not Ca2+ across membranes. X-ray crystallography and solid-state and solution NMR confirmed that the overall helical bundle was composed of two tightly interacting pairs of helices, which interacted along the more dynamic interface. Vesicle flux experiments show that as Zn2+ ions diffuse down their concentration gradients, protons were antiported. These experiments illustrate the feasibility of designing membrane proteins with predefined structural and dynamic properties.
TM electron transfer is a critical part of the bioenergetic processes that power life. Electrons are transmitted across membranes by hopping between redox-active cofactors. Discher, Dutton, and co-workers have utilized the maquette scaffold to create helical bundles that contain both soluble and TM domains for light harvesting and electron transfer (Ye et al., Reference Ye, Discher, Strzalka, Xu, Wu, Noy, Kuzmenko, Gog, Therien, Dutton and Blasie2005; Goparaju et al., Reference Goparaju, Fry, Chobot, Wiedman, Moser, Leslie Dutton and Discher2016). Korendovych and coworkers designed a TM four-helix bundle PRIME that bound two iron-porphyrin cofactors in a bis-His geometry. The resulting protein is perfectly suited to catalyze the transfer of electrons across phospholipid bilayers (Fig. 10b) (Korendovych et al., Reference Korendovych, Senes, Kim, Lear, Fry, Therien, Blasie, Walker and DeGrado2010). Analytical ultracentrifugation, EPR, redox potentiometry and UV-visible CD spectroscopy showed that the desired complex had been formed. Moreover, the protein bound the targeted di-phenyl-porphyrin derivative with high affinity and high specificity relative to other porphyrin or heme derivatives. Thus, both cofactor binding and TM electron transfer were realized for the first time in a de novo TM bundle.
De novo design of TM peptides that recognize the TM helices of natural proteins
While there are a large number of reagents such as antibodies that are capable of recognizing water-soluble proteins or the extra-membrane regions of membrane proteins, there is a great need to develop equivalent reagents to target the membrane-spanning regions of TM proteins. Such reagents could be used to interrogate the interactions between TM helices in natural proteins. The Small-X3-Small motif has been used to design peptides that specifically recognize the TM domains of two different integrins. Integrins are heterodimers with single-TM helices that tightly interact in the resting state, but separate in the activated state. Yin et al. achieved the computation design of peptides that specifically recognize the TM helices of two closely related integrins (α IIbβ 3 and α Vβ 3) in micelles, bacterial membranes, and mammalian cells (Yin et al., Reference Yin, Slusky, Berger, Walters, Vilaire, Litvinov, Lear, Caputo, Bennett and DeGrado2007; Caputo et al., Reference Caputo, Litvinov, Li, Bennett, DeGrado and Yin2008). The peptides competed for the endogenous helix–helix interactions and hence activated the integrins in a sequence-specific manner. These data showed that sequence-specific recognition of helices in TM proteins can be achieved through optimization of the geometric complementarity of the target-host complex. Less sequence specificity was observed in more recently designed peptides that target β1 integrins. Nevertheless, very useful reagents were obtained to target and activate this class of proteins (Mravic et al., Reference Mravic, Hu, Lu, Bennett, Sanders, Orr and DeGrado2018).
Fragment-based and bioinformatically informed computational protein design
Backbone fragments and sequence statistics broaden the scope of protein design
Despite the success described above in the sections on parametric design of water-soluble proteins, the de novo design of larger cooperatively folded proteins rich in β-sheets remained problematic until recently (Hecht, Reference Hecht1994; Quinn et al., Reference Quinn, Tweedy, Williams, Richardson and Richardson1994; Yan and Erickson, Reference Yan and Erickson1994). It was therefore of great interest when Kuhlman and Baker described the design of TOP7 (Fig. 11a), a protein that was both rich in β structure and also had a fold not previously seen in nature.

Fig. 11. Representative examples of de novo designed protein scaffolds. (a) TOP7, a de novo designed fold with no natural analogs (PDB: 1QYS). (b) A computationally designed TIM barrel (PDB: 5BVL). (c) A de novo designed mini protein (PDB: 5TX8). (d). Pizza6, a de novo designed fold with no natural analogs (PDB: 6F0Q). (e) A de novo designed β-barrel (PDB: 6D0T).
The successful design of TOP7 (Kuhlman et al., Reference Kuhlman, Dantas, Ireton, Varani, Stoddard and Baker2003) introduced an exciting new chapter, in which backbone fragment libraries derived from the PDB were used to build-up backbones of de novo proteins. This approach provided a solution to what was largely a chicken-egg problem in protein design. In de novo design, one needs a backbone structure to design a sequence, but it is hard to specify the precise backbone structure without first specifying the sequence. Thus, the designer is set with the task of designing a ‘designable’ backbone structure (i.e. one that can be stabilized by a sequence composed of the 20 commonly occurring amino acids in this case). Today, one typically tests on the order of 103 to 105 backbones to see which are designable. For each possible backbone, one uses sidechain repacking and other sequence-design algorithms to determine whether it can be outfitted with a sequence that satisfies the physical restraints required for folding. In rotamer-based approaches to protein sequence selection, a Monte-Carlo algorithm is used to discover sequences that are predicted to fold into the desired structure using a pairwise decomposable potential function that allows an efficient search through amino acid sequence and rotamer space for any given backbone.
The question then is how one specifies a foldable backbone. While significant success was obtained using helical bundles that were specified using a set of algebraic equations, many protein folds are too asymmetric to describe using reduced-parameter models. Kuhlman, Baker, and coworkers introduced an approach that circumvented this problem (Kuhlman et al., Reference Kuhlman, Dantas, Ireton, Varani, Stoddard and Baker2003). In their approach, one first defines a coarse-grained graph of the desired protein, which contains information such as the positions of secondary structure and inter-residue contacts. This blueprint defines the target fold and guides the search for a foldable backbone that satisfies the design restraints. Mainchain fragments from crystallographic structures are then combined and spliced together to create physically reasonable backbones that also conform to the guiding restraints. Sequences are then designed based on this initial draft of a backbone. In the next step structures are predicted for the designed sequences – again using backbone fragment assembly together with conformational energy minimization to facilitate the backbone search. The design then proceeds through repeated cycles of structure prediction for a given designed sequence and sequence redesign of the resulting predicted structures. Thus, through repeated cycles of sequence design and structure prediction the computation converges on a highly designable structure-backbone combination. Using this approach Kuhlman and Baker designed TOP7, which was highly stable and showed all the characteristics of a native-folded protein. Most importantly, its crystallographic structure was in outstanding agreement (1.2 Å backbone root mean square deviation) with the design. A major milestone in de novo protein design had been crossed, with significant implications for the design of proteins with a variety of folds (Box 2).
Box 2. Structural bioinformatics, sequence propensities, and fragment-based strategies for de novo protein design
Although the first protein design algorithms were based on molecular mechanics force fields, over the years, the scoring and search algorithms have evolved to greater complexity. Modern scoring functions, such as that used in Rosetta now include contributions from physical molecular mechanics force fields, terms to approximate the hydrophobic effect, and residue-specific and sequence-specific mainchain statistics. Additionally, Rosetta uses fragments of up to 15 residues in length to build protein structures, as well as the underlying sequence probabilities to score them energetically. While absent from early approaches to de novo protein design, statistical terms and fragment libraries have become increasingly important to enable design of ever-more complex structures. Thus, in modern approaches to protein design, fragments from the PDB are clustered based on 3D similarity and used in assembly procedures for protein structure prediction and design (Leaver-Fay et al., Reference Leaver-Fay, Tyka, Lewis, Lange, Thompson, Jacak, Kaufman, Renfrew, Smith, Sheffler, Davis, Cooper, Treuille, Mandell, Richter, Ban, Fleishman, Corn, Kim, Lyskov, Berrondo, Mentzer, Popović, Havranek, Karanicolas, Das, Meiler, Kortemme, Gray, Kuhlman, Baker and Bradley2011; Marcos et al., Reference Marcos, Basanta, Chidyausiku, Tang, Oberdorfer, Liu, Swapna, Guan, Silva, Dou, Pereira, Xiao, Sankaran, Zwart, Montelione and Baker2017).
The underlying principles that are encoded in structural informatics can be understood and reconciled to physical principles. For example, it has long been known that different amino acid residues have distinct propensities for adopting a given secondary structure of being found in a given environment (e.g. buried versus exposed), and the underlying energetics can be roughly approximated through the Boltzmann distribution (Chou and Fasman, Reference Chou and Fasman1978). The derived pseudo-energies are generally in good agreement with more direct experiments (Miller et al., Reference Miller, Janin, Lesk and Chothia1987). Rotameric preferences are in agreement with torsional potentials from molecular mechanics (Dunbrack and Karplus, Reference Dunbrack and Karplus1993; Dunbrack and Karplus, Reference Dunbrack and Karplus1994) and hence can be extended to design of non-natural foldamers (Shandler et al., Reference Shandler, Shapovalov, Dunbrack and DeGrado2010). Also, the sequence-specific positional preferences for forming β turns or capping helices can be reconciled to first-principles (Wilmot and Thornton, Reference Wilmot and Thornton1988; Efimov, Reference Efimov1993). Similarly, the more residue-specific sequence preferences used in modern design algorithms likely reflect the sequence/energy landscape for a given substructure.
The extension of the success of TOP7 to other folds required a general platform for inputting blueprints for design and implementing them as restraints in flexible backbone design. This was realized in RosettaRemodel (Huang et al., Reference Huang, Ban, Richter, Andre, Vernon, Schief and Baker2011), which provided the framework for a wide range of design problems including: the insertion, deletion, and remodeling of loops; design of disulfides; input of symmetry operators, and various other aspects of de novo design. Importantly, parametric approaches to backbone design could be flexibly incorporated into the blueprint to facilitate the entire design process. This framework led to many successes in de novo design over the last half decade. Given that this work has been recently reviewed (Huang et al., Reference Huang, Boyken and Baker2016a) we will discuss it only briefly here, and focus on a few outstanding examples of Rosetta designs that have appeared in the last 2 years. The design of a number of α-β folds, including rubredoxin, P-loop, and Rossman folds have been achieved (Lin et al., Reference Lin, Koga, Tatsumi-Koga, Liu, Clouser, Montelione and Baker2015; Huang et al., Reference Huang, Boyken and Baker2016a; Marcos et al., Reference Marcos, Basanta, Chidyausiku, Tang, Oberdorfer, Liu, Swapna, Guan, Silva, Dou, Pereira, Xiao, Sankaran, Zwart, Montelione and Baker2017). Moreover, repeat proteins (discussed below), mini-proteins, and cyclic peptides including ones with D-amino acids or unnatural crosslinks have been prepared and structurally verified (Bhardwaj et al., Reference Bhardwaj, Mulligan, Bahl, Gilmore, Harvey, Cheneval, Buchko, Pulavarti, Kaas, Eletsky, Huang, Johnsen, Greisen, Rocklin, Song, Linsky, Watkins, Rettie, Xu, Carter, Bonneau, Olson, Coutsias, Correnti, Szyperski, Craik and Baker2016; Chevalier et al., Reference Chevalier, Silva, Rocklin, Hicks, Vergara, Murapa, Bernard, Zhang, Lam, Yao, Bahl, Miyashita, Goreshnik, Fuller, Koday, Jenkins, Colvin, Carter, Bohn, Bryan, Fernandez-Velasco, Stewart, Dong, Huang, Jin, Wilson, Fuller and Baker2017; Dang et al., Reference Dang, Wu, Mulligan, Mravic, Wu, Lemmin, Ford, Silva, Baker and DeGrado2017; Marcos et al., Reference Marcos, Chidyausiku, McShan, Evangelidis, Nerli, Carter, Nivon, Davis, Oberdorfer, Tripsianes, Sgourakis and Baker2018) (Fig. 11).
A number of long-standing problems in de novo protein design, including the design of stable all-β proteins have been solved in recent years. In ground-breaking work, Huang et al. (Reference Huang, Feldmeier, Parmeggiani, Velasco, Hocker and Baker2016b) solved a classical problem (Goraj et al., Reference Goraj, Renard and Martial1990; Tanaka et al., Reference Tanaka, Kimura, Hayashi, Fujiyoshi, Fukuhara and Nakamura1994; Houbrechts et al., Reference Houbrechts, Moreau, Abagyan, Mainfroid, Preaux, Lamproye, Poncin, Goormaghtigh, Ruysschaert, Martial and Goraj1995; Figueroa et al., Reference Figueroa, Oliveira, Lejeune, Kaufmann, Dorr, Matagne, Martial, Meiler and Van de Weerdt2013) of designing a TIM α-β barrel fold (Fig. 11b). The pseudo-symmetry of this fold was idealized, resulting in a protein with approximate four-fold symmetry. Next, in 2018 Marcos et al. described the principles for controlling the curvature of β-sheets, and applied them to the design of a series of proteins with curved β-sheets topped with α-helices (Marcos et al., Reference Marcos, Basanta, Chidyausiku, Tang, Oberdorfer, Liu, Swapna, Guan, Silva, Dou, Pereira, Xiao, Sankaran, Zwart, Montelione and Baker2017). Finally, in 2018 the design of a structurally well-defined all-β barrel protein was reported (Lu et al., Reference Lu, Min, DiMaio, Wei, Vahey, Boyken, Chen, Fallas, Ueda, Sheffler, Mulligan, Xu, Bowie and Baker2018). The successful design focused on a roughly four-fold symmetrical eight-stranded β-barrel, with the overall shape of a hyperboloid. An initial set of 41 designs were constructed using a set of parametric equations, but all were unsuccessful. A careful analysis of the failed designs showed the accumulation of strain along residues that interact across the strands. More nuanced, symmetry-breaking geometries lead to the successful design, whose structure was in good agreement with the design. Next, a site was introduced to bind a fluorophore in a flat planar geometry. This was not accomplished in a single step, but rather after several cycles of computation, experimental evaluation, generation of combinatorial libraries, and experimental screening. Thus, while it was not possible to design the ligand-binding site by computation alone, it was possible to reach this objective through iterative cycles of experiment and computation.
Combining computational design with experimental library screening to achieve function
In another pioneering contribution, Baker and coworkers (Chevalier et al., Reference Chevalier, Silva, Rocklin, Hicks, Vergara, Murapa, Bernard, Zhang, Lam, Yao, Bahl, Miyashita, Goreshnik, Fuller, Koday, Jenkins, Colvin, Carter, Bohn, Bryan, Fernandez-Velasco, Stewart, Dong, Huang, Jin, Wilson, Fuller and Baker2017) integrated large-scale computational design, parallel oligonucleotide synthesis, yeast display screening, and next-generation sequencing to create libraries of approximately 40-residue mini-proteins that bind influenza hemagglutinin (HA), a protein located on a surface of the influenza virus, and botulinum neurotoxin (BoNT), the most acutely poisonous toxin known. They screened a library of ~4000 backbone geometries representing five well-defined miniprotein folds. ‘Hotspot’ residues, identified from previous crystal structures of HA and BoNT complexes with different binders, were then grafted onto the mini-protein scaffolds and the rest of sequence was computationally optimized to improve binding and stability. The resulting mini-protein binders were scored based on predicted binding energies and genes encoding ~10 500 of them were synthesized for yeast display. From this pool fluorescence-activated cell sorting enrichment identified 57 and 29 distinct mini-proteins that bound BoNT and HA, respectively. Interestingly, several peptides from a pool of ~6000 scrambled control sequences also showed high affinity for the targets, highlighting the power of naïve libraries in producing strong binders using de novo designed scaffolds (Cherny et al., Reference Cherny, Korolev, Koehler and Hecht2012). Importantly, the frequency of finding binders was significantly lower in the randomized sequences. Subsequent refinement of the design model and affinity maturation produced three HA binders and two BoNT binders that displayed excellent affinity (<10 and <1 nM, respectively) for their targets. These K ds are on par with those of scFv's (~200 pM) derived for the same target and just three orders of magnitude less potent than the corresponding monoclonal antibodies (~320 fM) (Kalb et al., Reference Kalb, Garcia-Rodriguez, Lou, Baudys, Smith, Marks, Smith, Pirkle and Barr2010). It is noteworthy that, given the small size of the mini-proteins, in practical terms the same dose by mass results in a comparable therapeutic effect. HB1.6928.2.3, one of the mini-protein binders of HA, provided full protection of mice from influenza before exposure to a lethal dose of the virus and 100% survival after intranasal administration of a single-therapeutic dose 24 h after the exposure.
The multistep approach developed by Baker and coworkers (Fig. 12) that employs protein design to combine the recognition power of ‘antibody-like’ protein–protein interactions with the stability and ease of production of small, potentially non-immunogenic, mini-proteins allows high-throughput screening to reach its full potential. The demonstrated success rate of finding a strong binder in a computationally designed library (~1%) is sufficient for identification of hits using high-throughput screening.

Fig. 12. Overview of the computational design and high-throughput screening of mini-protein binders. Reproduced with permission from Makhlynets and Korendovych (Reference Makhlynets and Korendovych2017). Copyright (2017) American Chemical Society.
Design of protein assemblies
Nature has evolved proteins that form structurally complex and functionally rich supramolecular assemblies. Given the wide array of structures that proteins can adopt, targeted design of protein-based supramolecular assemblies can provide a path to novel functional materials and nanostructures. De novo design of complex assemblies is rapidly expanding in manifold directions, and several recent reviews of the area are available (De Santis and Ryadnov, Reference De Santis and Ryadnov2015; Norn and Andre, Reference Norn and Andre2016; Kobayashi and Arai, Reference Kobayashi and Arai2017; Yeates, Reference Yeates2017; Beesley and Woolfson, Reference Beesley and Woolfson2019). Furthermore, the pallet for design has expanded to include a variety of materials including aromatic peptides and collagen triple helical peptides, which are beyond the scope of the current review. Here, we focus primarily on structures in which designed de novo are realized by expansion of the fragment assembly and parametric approaches discussed above.
Elongation in one dimension: superhelical assemblies with translational and screw symmetries
Advances in protein design have enabled the design of complex linear arrays of proteins with diverse morphologies, including fibers and nanotubes that can be used for molecular nanocompartment encapsulation, drug delivery, tissue engineering, and catalysis both in vivo and in vitro. Work in this field has exploded in recent years, and has been extensively reviewed elsewhere (Zhang et al., Reference Zhang, Yan, Altman, Lassle, Nugent, Frankel, Lauffenburger, Whitesides and Rich1999; Gazit, Reference Gazit2007; Childers et al., Reference Childers, Ni, Mehta and Lynn2009; Aida et al., Reference Aida, Meijer and Stupp2012; Webber et al., Reference Webber, Appel, Meijer and Langer2016; Kumar et al., Reference Kumar, Ing, Narang, Wijerathne, Hochbaum and Ulijn2018; Nguyen and Ueno, Reference Nguyen and Ueno2018). Here, we attempt to highlight a few classical, enabling studies as well as recent work, focusing on the principles of design and assembly.
One of the simplest high-order motifs in proteins is the cross-β structure, in which β-strands run perpendicular to the fibril axis, forming infinite parallel or antiparallel sheets (Pauling and Corey, Reference Pauling and Corey1951). The sheets often pair along an apolar interface, which further stabilizes the structure (Eisenberg and Jucker, Reference Eisenberg and Jucker2012) (Fig. 12). As early as the 1980s (DeGrado and Lear, Reference DeGrado and Lear1985; Osterman and Kaiser, Reference Osterman and Kaiser1985), simple hydrophobic/hydrophilic patterning was used to design heptapeptides (e.g. LKLKLKL, as discussed above) that can assemble on apolar interfaces into well-defined β-sheet assemblies. These minimalist principles were later extended to design of graphene-binding protofibrils that orient with their main axes along one of six directions defined by graphene's six-fold symmetry (Mustata et al., Reference Mustata, Kim, Zhang, DeGrado, Grigoryan and Wanunu2016). Moreover, Zn2+-dependent catalysts with efficiencies that rival those of natural enzymes by weight have also been designed starting from minimalist principles (Friedmann et al., Reference Friedmann, Torbeev, Zelenay, Sobol, Greenwald and Riek2015; Al-Garawi et al., Reference Al-Garawi, McIntosh, Neill-Hall, Hatimy, Sweet, Bagley and Serpell2017; Zozulia et al., Reference Zozulia, Dolan and Korendovych2018), and the structure of one of these assemblies has been determined by solid state NMR (Fig. 13c) (Lee et al., Reference Lee, Wang, Makhlynets, Wu, Polizzi, Wu, Gosavi, Stohr, Korendovych, DeGrado and Hong2017).
Fig. 13. Structures of amyloid fibrils. (a) Strands align perpendicular to the main fibril axis (indicated by a black line) in a structure of MAX1, a strand-turn-strand peptide designed by Schneider and coworkers (PDB: 2N1E). (b) Structure of MAX1, with polar Lys residues (blue sticks) on the solvent-exposed surface and apolar Val residues (green ball and sticks) forming a water-free interface. (c) Structure of a catalytic Zn2+-binding amyloid (PDB: 5UGK), showing a network of 3-His Zn2+ ion coordination, and an H-bonded zipper of Gln sidechains. (d) Structure of an α-amyloid assembly, αAmS (Zhang et al., Reference Zhang, Huang, Yang, Kratochvil, Lolicato, Liu, Shu, Liu and DeGrado2018b) (PDB: 6C4Z) the N- and C-termini of the individual helices are designated in blue and red, respectively.
Alex Rich and Shuguang Zhang were the first to recognize the potential of amphiphilic β-peptides (Zhang et al., Reference Zhang, Holmes, Lockshin and Rich1993) to form nanofiber scaffolds and membranous structures. Zhang developed such peptides for myriad applications including controlled drug delivery, tissue regeneration, and accelerated wound healing (Zhang, Reference Zhang2017). Similarly, Hamachi has designed a variety of remarkable self-assembling hydrogels that respond to a diverse array of environmental stimuli (Shigemitsu and Hamachi, Reference Shigemitsu and Hamachi2017), and Lynn has used peptide design to explore the possible role of amyloids in early evolution of life (Childers et al., Reference Childers, Ni, Mehta and Lynn2009).
Building-up one step in complexity, Schneider designed and determined high-resolution solid-state NMR structures (Nagy-Smith et al., Reference Nagy-Smith, Moore, Schneider and Tycko2015) of fibril-forming peptides consisting of a strand-turn-strand motif (Figs 13a and b). Members of the MAX1 series of peptides have a range of interesting properties ranging from antimicrobial materials to easily processed hydrogels with finely tuned mechanical properties (Schneider et al., Reference Schneider, Pochan, Ozbas, Rajagopal, Pakstis and Kretsinger2002). Shimon, Gazit, and coworkers have built assemblies with similar scaffolds, and characterized their structures by high-resolution X-ray crystallography (Pellach et al., Reference Pellach, Mondal, Harlos, Mance, Baldus, Gazit and Shimon2017). While the turns in these assemblies connect hydrogen-bonded strands, in other structures such as solenoids and larger amyloids, the turns often connect chains across β-sheets, and these motifs have been successfully used in design of fibrillary assemblies (Pellach et al., Reference Pellach, Mondal, Harlos, Mance, Baldus, Gazit and Shimon2017).
It is also possible to build fibrous structures from helices rather than β-strands (Fairman and Akerfeldt, Reference Fairman and Akerfeldt2005). In early work, pioneered by Woolfson, fibrils were built based on staggered pairing interactions between helical coiled-coil peptides; the resulting ‘sticky ends’ mediated assembly of the peptides into highly elongated coiled coils (Pandya et al., Reference Pandya, Spooner, Sunde, Thorpe, Rodger and Woolfson2000). By introducing kinks or branches they were able to engineer a variety of architectures. Fairman and coworkers adopted a related strategy to induce self-assembly (Fairman and Akerfeldt, Reference Fairman and Akerfeldt2005; Wagner et al., Reference Wagner, Phillips, Ali, Nybakken, Crawford, Schwab, Smith and Fairman2005). These workers designed coiled-coil peptides with insertions that caused the hydrophobic faces to misalign, resulting in a staggered, infinite assembly. More recently, Woolfson have built an orthogonal set of rotationally symmetric coiled coils, ranging from dimers to heptamers, which can be used as building blocks to create a variety of assemblies (Woolfson et al., Reference Woolfson, Bartlett, Burton, Heal, Niitsu, Thomson and Wood2015). By introducing favorable electrostatic, hydrophobic interactions or metal–ligand interactions (Nambiar et al., Reference Nambiar, Wang, Rotello and Chmielewski2018) near the ends of these coiled coils it is possible to induce assembly into infinite super-helical assemblies with the central axis of the individual coiled coils aligning along the fibril axis (Burgess et al., Reference Burgess, Sharp, Thomas, Wood, Thomson, Zaccai, Brady, Serpell and Woolfson2015). When the end-to-end interactions are designed to be highly favorable, the bundles assemble in solution to form elongated fibers (Fig. 14a); weaker end-to-end interactions can be used to induce intermonomer contacts in crystals (Ogihara et al., Reference Ogihara, Weiss, DeGrado and Eisenberg1997; Lanci et al., Reference Lanci, MacDermaid, Kang, Acharya, North, Yang, Qiu, DeGrado and Saven2012; Zhang et al., Reference Zhang, Huang, Yang, Kratochvil, Lolicato, Liu, Shu, Liu and DeGrado2018b)
Fig. 14. Structural assemblies of designed proteins. Proteins that assemble in one dimension to form fibers and tubes are shown in panels (a–d). Panel (a) shows the structure of a hexameric bundle designed (PDB: 4H8M), that has been engineered to assemble into stacked bundles (structure inferred by EM). Panel (b) illustrates a dimeric three-helix bundle assembled from helix–loop–helix motifs (PDB: 1G6U) consisting of one short and one long helix. The sequence was designed to cause the units to assemble with the loops on opposite sides of the bundle in an ‘up-down’ orientation to give a domain-swapped dimer. In a second design, the sequence was designed to cause the loops to align in an ‘up-up’ orientation that induced fibril formation. Panel (d) illustrates larger-diameter nano-pores composed of helix–loop–helix motifs (PDB: 6MK1), and panel (e) shows the assembly scheme for TET12SN family peptides that spontaneously assemble into a tetrahedral cages (reproduced from Lapenta (Reference Lapenta, Aupič, Strmšek and Jerala2018) 351 – Published by The Royal Society of Chemistry). Panel (f) illustrates a tetrahedral protein cage created by computationally designing protein–protein interfaces (PDB: 4NWR), and panel (g) illustrates a computationally designed protein crystal (PDB: 4H8M).
In other examples of helical fibril engineering, the individual helices are designed to form lateral assemblies that run nearly perpendicular to the fibril axis. Conticello and coworkers have designed assemblies of peptides based on a heptad repeat, in which two hydrophobic faces, (a/d) and (c/f), are separated by polar residues. Cryo-electron microscopy (EM) revealed that the peptides assemble into wide tubes that can encapsulate small molecules (Xu et al., Reference Xu, Liu, Mehta, Guerrero-Ferreira, Wright, Dunin-Horkawicz, Morris, Serpell, Zuo, Wall and Conticello2013; Shen et al., Reference Shen, Fallas, Lynch, Sheffler, Parry, Jannetty, Decarreau, Wagenbach, Vicente, Chen, Wang, Dowling, Oberdorfer, Stewart, Wordeman, De Yoreo, Jacobs-Wagner, Kollman and Baker2018). Additionally, peptides have been designed to assemble with their axes perpendicular to the fibril axis precisely as in twisted cross-β structures, forming very long-twisted ‘cross-α’ fibrils as seen in crystallographic structures of the assemblies (Zhang et al., Reference Zhang, Huang, Yang, Kratochvil, Lolicato, Liu, Shu, Liu and DeGrado2018b) (Fig. 13d). These peptides were used to direct assembly of fused florescent proteins in mammalian cells, and by varying the sequence it was possible to modulate the structure and assembly/disassembly kinetics. Cross-α structures are also of current interest, because they have been discovered in toxic peptides (Tayeb-Fligelman et al., Reference Tayeb-Fligelman, Tabachnikov, Moshe, Goldshmidt-Tran, Sawaya, Coquelle, Colletier and Landau2017). It will be interesting to see how wide-spread this assembly motif might be.
In each of the above examples of coiled-coil assemblies, the monomeric unit was a single helical peptide. Alternatively, more complex units can be used to create structures with greater structural diversity. The earliest example involved the design of fibrils assembled from domain-swapped versions of a three-helix bundle (Ogihara et al., Reference Ogihara, Ghirlanda, Bryson, Gingery, DeGrado and Eisenberg2001) related to α3D (Bryson et al., Reference Bryson, Betz, Lu, Suich, Zhou, O'Neil and DeGrado1995, Reference Bryson, Desjarlais, Handel and DeGrado1998). The basic design unit consisted of a hairpin consisting of one long and one short helix designed to assemble into three-helix structures (Figs 14b and c). Electrostatic interactions were manipulated to allow the unit to assemble into a closed, domain-swapped dimer a fibrillar array, depending on whether the helix–loop–helix motifs assembled with the loops in an anti (Fig. 14b) or a cis orientation. X-ray crystallography and EM confirmed the structure of the domain-swapped dimer and fibril, respectively (Ogihara et al., Reference Ogihara, Ghirlanda, Bryson, Gingery, DeGrado and Eisenberg2001). Furthermore, analysis of the crystallographic structure of a domain-swapped dimer illustrated principles for design of antiparallel six-helix bundles (Ghirlanda et al., Reference Ghirlanda, Lear, Ogihara, Eisenberg and DeGrado2002). Finally, by redesigning the hydrophobic core of the hexameric bundles, Grigoryan et al. engineered bundles that selectively solubilized only a single form of carbon nanotubes (Grigoryan et al., Reference Grigoryan, Kim, Acharya, Axelrod, Jain, Willis, Drndic, Kikkawa and DeGrado2011).
In nature, covalently assembled superhelical repeat proteins are often assembled by repeating simple motifs such as helix–loop–helix motifs with intervening tight loops or turns (Kobe and Kajava, Reference Kobe and Kajava2000; Kajava, Reference Kajava2012). Consensus sequence motifs have been generated for repeat proteins, and used to create robust scaffolds for selection of peptide binding proteins (Kajander et al., Reference Kajander, Cortajarena and Regan2006; Pluckthun, Reference Pluckthun2015). A number of repeats are assembled into a single-protein chain, and N- and C-terminal ‘capping motifs’ are also included to avoid run-away non-covalent assembly into fibrils. Taking a different approach, Conticello and coworkers used non-covalent assembly of peptides patterned after the helix–loop–helix motifs of thermophilic HEAT and leucine-rich variant motifs (Fig. 14c). Cryo-EM structures at near-atomic resolution demonstrated the formation of tubes with outer radii of 70 or 80 Å (Hughes et al., Reference Hughes, Wang, Wang, Kreutzberger, Osinski, Orlova, Wall, Zuo, Egelman and Conticello2019).
Recently, André and coworkers took a structure-based approach to design repeat proteins based on the leucine-rich repeat. They built on the known structures of natural proteins to design repeats with predefined shapes, which are then assembled to create helical arrays with predetermined super-helical geometries (Ramisch et al., Reference Ramisch, Weininger, Martinsson, Akke and Andre2014). ElGamacy, Lupas, and coworkers used an interface-directed strategy to design less regular solenoid-like proteins, in which the helix–loop–helix motifs alternating the handedness rather than repeating with exact symmetry (ElGamacy et al., Reference ElGamacy, Coles, Ernst, Zhu, Hartmann, Pluckthun and Lupas2018).
Baker and coworkers have developed generalized computational methods to engineer cyclic and superhelical arrays formed from helix–loop–helix motifs (Brunette et al., Reference Brunette, Parmeggiani, Huang, Bhabha, Ekiert, Tsutakawa, Hura, Tainer and Baker2015; Doyle et al., Reference Doyle, Hallinan, Bolduc, Parmeggiani, Baker, Stoddard and Bradley2015). The Rosetta program is used to build repeating helix–loop–helix–loop motifs, in which the backbone and sequence is identical for each repeat. This procedure generates well-packed superhelical repeat proteins; the desired superstructure can be specified by adding a pseudo-energy term that penalizes for geometries that do not match the desired superhelical curvature and rise. Using this method, Brunette et al. explored the structure space for helical repeat proteins containing a range of helix–loop–helix geometries (Brunette et al., Reference Brunette, Parmeggiani, Huang, Bhabha, Ekiert, Tsutakawa, Hura, Tainer and Baker2015). The resulting structures have been verified at atomic resolution, and a number of geometries not yet seen in crystal structures of natural proteins were designed and experimentally demonstrated. These methods have been extended to the design of filamentous arrays formed from previously characterized de novo-designed helical bundles (Shen et al., Reference Shen, Fallas, Lynch, Sheffler, Parry, Jannetty, Decarreau, Wagenbach, Vicente, Chen, Wang, Dowling, Oberdorfer, Stewart, Wordeman, De Yoreo, Jacobs-Wagner, Kollman and Baker2018). Using Rosetta, a variety of well packed motifs are sampled and replicated to create a range of superhelical geometries. Of 124 designs tested, 34 formed filaments – six of which were structurally characterized and found to agree with the underlying design to varying degree of accuracy.
Elongation in two dimensions: planar lattice-like structures
The design of lattice-like structures can be realized by de novo designed protein into a unit cell, and arranging its orientation and sequence to create a stable assembly. The first structurally verified de novo design of a two-dimensional (2D) assembly focused on P321 and P6 arrays of three-helix bundles (Lanci et al., Reference Lanci, MacDermaid, Kang, Acharya, North, Yang, Qiu, DeGrado and Saven2012). This work has been expanded to design arrays based on tetrameric bundles (Zhang et al., Reference Zhang, Polzer, Haider, Tian, Villegas, Kiick, Pochan and Saven2016). Both examples employed the SCAD sequence design algorithm to generate the sequence. In each case, the predicted models were in outstanding agreement with the experimental structures. More recently, similar methods have been used by Baker et al., to design a number of different lattices, in this case using natural proteins with cyclic symmetry as the basic building blocks (Gonen et al., Reference Gonen, DiMaio, Gonen and Baker2015). Together, these studies demonstrate the ability to design with Ångstrom-level accuracy over length scales on the order of tens to hundreds of nanometers.
Assembly of cages by combining multiple symmetry elements
The predictable nature and robustness of coiled-coil assemblies was expanded to form large cages (Fletcher et al., Reference Fletcher, Harniman, Barnes, Boyle, Collins, Mantell, Sharp, Antognozzi, Booth, Linden, Miles, Sessions, Verkade and Woolfson2013) as well as distinct supramolecular polyhedral nanostructures that can assemble both in vitro and in vivo (Gradišar et al., Reference Gradišar, Božič, Doles, Vengust, Hafner-Bratkovič, Mertelj, Webb, Šali, Klavžar and Jerala2013; Ljubetič et al., Reference Ljubetič, Lapenta, Gradišar, Drobnak, Aupič, Strmšek, Lainšček, Hafner-Bratkovič, Majerle, Krivec, Benčina, Pisanski, Veličković, Round, Carazo, Melero and Jerala2017; Park et al., Reference Park, Bedewy, Berggren and Keating2017). Marsh and coworkers developed a flexible, symmetry directed approach for creating protein cages by fusing coiled-coil forming peptides to a natural trimeric protein (Sciore et al., Reference Sciore, Su, Koldewey, Eschweiler, Diffley, Linhares, Ruotolo, Bardwell, Skiniotis and Marsh2016). It is also possible to build polyhedral using natural homo-oligomeric proteins as building blocks. Yates and coworkers (Padilla et al., Reference Padilla, Colovos and Yeates2001; Yeates, Reference Yeates2017) described general principles for the design of symmetrical virus-like assemblies that form large molecular cages by rigid fusions of two oligomeric proteins – for example, one that forms a C 2 dimer with one that forms a C 3 trimer – so that the symmetry axes match the symmetry axes of Euclidean solids. This method has been used to create novel cages with varying symmetries including icosahedral assemblies (King et al., Reference King, Sheffler, Sawaya, Vollmar, Sumida, Andre, Gonen, Yeates and Baker2012; Bale et al., Reference Bale, Gonen, Liu, Sheffler, Ellis, Thomas, Cascio, Yeates, Gonen, King and Baker2016; Hsia et al., Reference Hsia, Bale, Gonen, Shi, Sheffler, Fong, Nattermann, Xu, Huang, Ravichandran, Yi, Davis, Gonen, King and Baker2016). Baker and coworkers have implemented and extended this approach to allow design of large, well-defined, virus-like protein cages with atomic accuracy, including proteins capable of encapsulating their own DNA (Butterfield et al., Reference Butterfield, Lajoie, Gustafson, Sellers, Nattermann, Ellis, Bale, Ke, Lenz, Yehdego, Ravichandran, Pun, King and Baker2017). Hilvert and coworkers further modified these computationally designed protein cages to deliver oligonucleotides to efficiently regulate gene expression in mammalian cells (Edwardson and Hilvert, Reference Edwardson and Hilvert2019).
Elongation in three dimensions: crystal engineering
Progressing from the design of 2D arrays to macroscopic 3D crystals represents the highest level of complexity. Conceptually, this can be achieved by engineering the assembly of a 2D lattice (e.g. as discussed above in the section ‘Elongation in two dimensions: planar lattice-like structures’) into a third dimension. However, designing predetermined crystal structures is subtle, given the size and complexity of proteins and the myriad noncovalent interactions that govern protein crystallization. Saven, DeGrado, and coworkers developed a computational approach to design a helical bundle that assembles in P6, a polar, layered crystallographic space group with both C 2 and C 3 symmetry axes (Lanci et al., Reference Lanci, MacDermaid, Kang, Acharya, North, Yang, Qiu, DeGrado and Saven2012). A C 3-symmetric helical bundle was placed along the three-fold axis, and its orientation and unit cell parameters were systematically varied to create a sequence-structure energy landscape using the SCADS program for computational protein design. A hierarchy of interactions of graded stability was used in the design. Strongly stabilizing hydrophobic and packing interactions were engineered to stabilize the core of the three-helix bundle, while weaker packing interactions between surface-exposed Gly and Ala interactions were used to stabilize lateral interactions between the helices. Finally, end-to-end hydrogen bonds between helical ends stabilized the stacking of columns of helical bundles. A 2.1 Å resolution X-ray crystal structure of one such designed protein exhibits sub-Ångstrom agreement with the computational model in the spacing and parallel ordering of neighboring proteins in the crystal. The crystals have large hexagonal channels, which should be able to accommodate a variety of small- to meso-sized molecular cargos. For example, similar crystals of designed coiled coils have been found to organize C60 derivatives into arrays with interesting electronic properties (Kim et al., Reference Kim, Ko, Kim, Kim, Paul, Zhang, Murray, Acharya, DeGrado, Kim and Grigoryan2016).
Summary and outlook
In the past several decades, the design of de novo proteins with predetermined structures and functions has progressed from an outrageous concept to a routine accomplishment, with far-reaching implications for the fields of chemistry, nanoscience, and biotechnology. De novo design is a compellingly critical test of our understanding of protein structure and function. If we understand proteins we should be able to design them from scratch. This approach translates our passive understanding of proteins to an active understanding that is already enabling the design of proteins and biomimetic polymers with properties not available in nature.
The first grand challenge our field encountered was the protein folding problem – how does an amino acid sequence code for the 3D structure of a protein? Today, we understand the principles of protein folding sufficiently well to design proteins with a large range of sizes, sizes, dynamic properties and with thermodynamic stabilities far exceeding those seen in nature. Given this ability to control tertiary structure, protein designers are also tackling the problem of designing function. Initial work in this area has been primarily fundamental, as we have progressed from a passive understanding to the active understanding needed to design functional proteins from scratch. Nevertheless, practical applications have already emerged and clearly will expand. In this review, we focused on three functions: binding, catalysis, and vectoral transport through membranes. Sufficient progress has been made in each to reasonably extrapolate what we might reasonably expect to achieve in the next decade.
The first clearly defined achievements in the area of binding focused on selective and geometrically specific recognition of transition metal ions. The initial designs focused on binding of metal ions in relatively stable, common geometries, as in structural metal sites in proteins. With time, de novo design proteins were produced with more interesting metal sites capable of catalyzing a variety of oxidative, reductive, and hydrolytic processes. Thus, de novo design is now increasingly used to understand how proteins influence the reactivity and catalytic properties of their metal ion cofactors. Furthermore, a large number of proteins have been designed to bind non-biological metal ions and metal-organic complexes in precisely predetermined structures and environments. These accomplishments raise the possibility of designing cofactor-containing proteins for diverse applications ranging from optical devices to catalysts that combine the advantages of traditional transition metal catalysts with the versatility, programmability, and water solubility of proteins.
A second binding functionality that has been achieved involves the design of peptides and proteins that bind to protein interfaces. De novo design methods have enabled the design of proteins that are smaller and much more stable toward chemical, enzymatic, and thermal denaturation than natural proteins such as antibodies. De novo design is also providing increasingly good starting points for experimental optimization of binding affinity and specificity. De novo designed proteins have considerable potential as therapeutics for pharmaceutical intervention of unmet medical needs.
The design of proteins that bind complex, highly functionalized small molecules remains a larger challenge that has only now being addressed . The design of small molecule binders requires mastery of some of the most difficult problems in protein design. First, a binding cavity must be constructed to encompass the molecule of interest. In early studies where this was accomplished (Di Costanzo et al., Reference Di Costanzo, Wade, Geremia, Randaccio, Pavone, DeGrado and Lombardi2001; Lombardi et al., Reference Lombardi, Nastri and Pavone2001; Geremia et al., Reference Geremia, Di Costanzo, Randaccio, Engel, Lombardi, Nastri and DeGrado2005; Lombardi et al., Reference Lombardi, Pirro, Maglio, Chino and DeGrado2019), building a small-molecule binding site was very destabilizing to the protein conformation and required careful optimization of other regions of the tertiary structure (Faiella et al., Reference Faiella, Andreozzi, de Rosales, Pavone, Maglio, Nastri, DeGrado and Lombardi2009). Once, a cavity has been constructed, the designed protein must also position polar sidechains appropriately to form highly directional hydrogen-bonded interactions to the ligand (in cases where the binding of densely functionalized polar ligands is desired). Finally, when the target small molecule contains a number of rotatable bonds, the ligand–protein interactions need to be highly favorable to compensate for the unfavorable entropy associated with binding the small molecule in a single conformation. While challenging, we expect that advances in sampling and scoring ligand–protein poses will enable successful design of small molecule-binding proteins without the need for repeated cycles of experimental optimization. The attainment of this ability will be an important step in the design of proteins that catalyze kinetically challenging reactions with efficiencies approaching those of natural enzymes.
We have also seen significant progress in the design of proteins that assemble in membranes and other non-aqueous or heterogeneous environments. It is now possible to design membrane proteins and assemblies with very high stabilities and predictable structures. We have also seen the first examples of proteins that facilitate transport of electrons and polar solutes across phospholipid bilayers. Applications of such systems to single-molecule sensing are likely to follow. For example, highly engineered variants of natural proteins are now used for sequencing RNA and DNA using the nanopore technology (Branton et al., Reference Branton, Deamer, Marziali, Bayley, Benner, Butler, Di Ventra, Garaj, Hibbs, Huang, Jovanovich, Krstic, Lindsay, Ling, Mastrangelo, Meller, Oliver, Pershin, Ramsey, Riehn, Soni, Tabard-Cossa, Wanunu, Wiggin and Schloss2008). It will be exciting to construct proteins from scratch for such demanding applications.
Finally, methods for protein design are developing very rapidly. In this review, we saw that the earliest proteins were designed using simple physical principles and molecular mechanics force fields. More recently developed methods increasingly rely on backbone fragments and statistical quantities derived from structural bioinformatics to sample foldable protein structures and sequences. Nevertheless, the same physical principles are involved and incorporated into modern force fields for protein design. In the coming years, there will doubtlessly be improvements in both approaches. Advances in computing will allow all-atom molecular dynamics calculations using both implicit and explicit solvents at various steps within the design workflow. Such methods will allow one to better model non-canonical structures and to evaluate the potential success of designs. In parallel, the power of bioinformatics will increase dramatically with the inclusion of machine learning (Mackenzie et al., Reference Mackenzie, Zhou and Grigoryan2016; Mackenzie and Grigoryan, Reference Mackenzie and Grigoryan2017; Eguchi and Huang, Reference Eguchi and Huang2019). Advanced non-supervised approaches will enable one to discover highly favorable atomic arrangements that are difficult to sample with high precision and quantify with current methods. Machine-learning methods will contribute to the identification of stable ‘designable’ tertiary structures that can be designed using the 20 commonly occurring amino acids. Generative adversarial networks will be used to generate both tertiary structures and sequences starting with only a rough draft of the desired structure.
In summary, de novo protein design has evolved into a vibrant approach for testing hypotheses concerning the fundamental aspects of protein folding and function, and it is now brimming with potential for applications in sensing, catalysis, pharmaceuticals, and nanotechnology. Given recent improvements in computing, including advanced methods for machine learning, one can expect advances to accelerate dramatically in the coming years.
Acknowledgements
The authors thank he NIH (Grants GM122603 to W.F.D. and GM119634 to I.V.K. ), the NSF (Grant 1709506 to W.F.D.) and the CRDF (Grant No. OISE-18-63891-0 to I.V.K.) for support.















