


Introduction
Systemic and tissue imbalances of metal levels are often found in neurodegenerative disorders such as Parkinson's disease (PD) (Fink, Reference Fink2006), implying that metal ions may play roles in disease progression. A unifying molecular event in all neurodegenerative disorders is misfolding and aberrant self-assembly of proteins into amyloid fibers with a hallmark cross-β-sheet arrangement. Conformational changes resulting in aggregation of the neuronal protein α-synuclein (αS) into amyloid fibers is directly related to PD (Galvin et al. Reference Galvin1999; Winner et al. Reference Winner2011). Many synthetic as well as naturally occurring molecules can modulate αS amyloid formation in vitro, for example ring-fused two-pyridones (Horvath et al. Reference Horvath2012), other amyloidogenic proteins (i.e., cross-reactivity) (Horvath & Wittung-Stafshede, Reference Horvath and Wittung-Stafshede2016), bacterial proteins (Chorell et al. Reference Chorell2015) and metal ions (Davies et al. Reference Davies2016; Montes et al. Reference Montes2014).
Copper (Cu) in both redox states (oxidized = Cu(II); reduced = Cu(I)) can bind to αS (Camponeschi et al. Reference Camponeschi2013; De Ricco et al. Reference De Ricco2015a) and structural features, binding sites, and affinities for the interaction between αS and Cu(II), as well as Cu(I), have been the focus of in vitro spectroscopic investigations (Binolfi et al. Reference Binolfi2006, Reference Binolfi2011; Camponeschi et al. Reference Camponeschi2013; De Ricco et al. Reference De Ricco2015a), Fig. 1a. Although most divalent cations can accelerate αS amyloid formation, Cu(II) has the largest such effect in vitro (Davies et al. Reference Davies2016; Montes et al. Reference Montes2014). Notably, the effect of Cu(I) binding on αS amyloid formation in vitro has not been reported. It was recently suggested that αS was able to act as a ferri-reductase, reducing iron using Cu(I)/Cu(II) as a catalytic redox center (Brown, Reference Brown2013) but it awaits to be revealed if Cu-mediated redox reactions are related to αS function or merely part of a cascade of deleterious processes (De Ricco et al. Reference De Ricco2015b). It remains unknown how, and in what redox state, Cu would reach αS in vivo. The cytoplasm is a reducing environment so most Cu ions are in the Cu(I) form (Hatori and Lutsenko, Reference Hatori and Lutsenko2013); in addition, there is no free Cu in cells (Ohrvik & Thiele, Reference Ohrvik and Thiele2014; Tottey et al. Reference Tottey, Harvie and Robinson2005; Waldron et al. Reference Waldron2009).
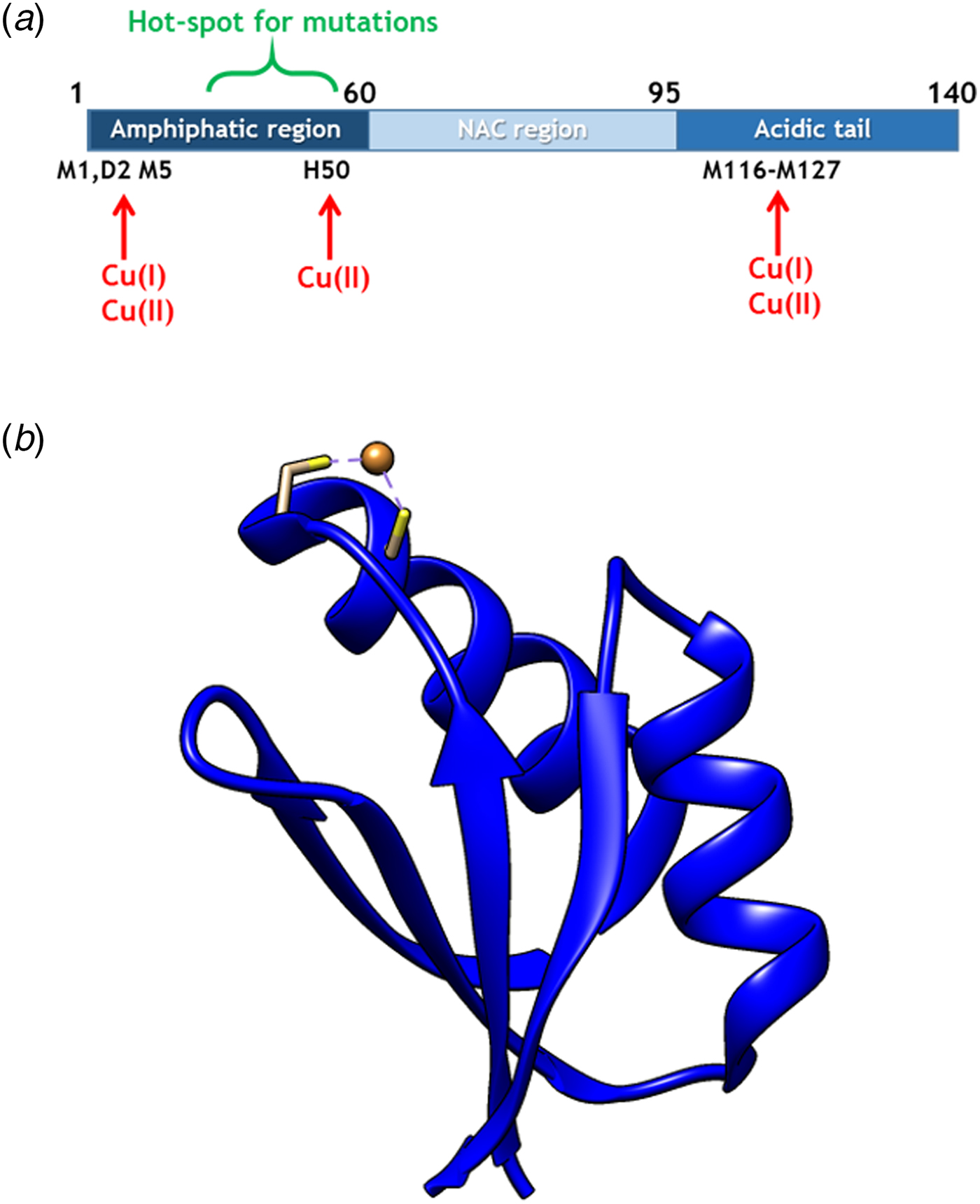
Fig. 1. (a) Scheme of αS sequence (key parts noted) with Cu-binding sites indicated. (b) Structure of Atox1 with Cu ion and Cu-coordinating Cys highlighted (1TL4).
In cells, Cu(I) is transported by copper transport proteins to specific targets that require Cu for function (De Feo et al. Reference De Feo2009). Such an elaborate system is necessary for tight control of metal distribution in time and space, and in order to keep Cu(I) soluble as free Cu(I) is chemically instable. In the human cytoplasm, the Cu chaperone Atox1 (Fig. 1b), a small single-domain protein with a CXXC (C, cysteine; X, any residue) Cu-binding motif (Boal & Rosenzweig, Reference Boal and Rosenzweig2009), brings Cu from the Cu importer Ctr1 in the plasma membrane to two large, multi-domain P1B type ATPases in the Golgi membrane, ATP7A and ATP7B, for loading of Cu-dependent enzymes (Lutsenkoet al. Reference Lutsenko, LeShane and Shinde2007). Movement of Cu(I) between these transport proteins is facilitated by direct metal-bridged protein–protein complexes (Niemiec et al. Reference Niemiec, Dingeldein and Wittung-Stafshede2015). Recently, new functions and partner proteins for Atox1 have been reported, implying an extended functional repertoire (Blockhuys & Wittung-Stafshede, Reference Blockhuys and Wittung-Stafshede2017; Blockhuys et al. Reference Blockhuys2017).
If alterations in Cu levels are a cause or a consequence in amyloidogenic diseases is unclear. Perhaps Cu transport proteins deliver Cu(I) to αS such that, possibly requiring Cu(I) to Cu(II) oxidation, aggregation is modulated? Or Cu transport proteins may instead be protective and remove Cu from αS before aggregation is triggered? We note that Atox1 and other Cu transporters are expressed in most neuronal cells (Davies et al. Reference Davies2013; Montes et al. Reference Montes2014); thus, interactions with αS are feasible. To begin to address this knowledge gap, we here tested if Atox1 can deliver Cu(I) to αS, and if so, what are consequences on amyloid formation.
Materials and methods
Proteins
A construct with the wild-type αS gene was transformed into BL21(DE3) (Novagen) cells. Cells were first grown to OD600 of 0.6 in LB containing 100 µg ml−1 carbenicillin at 37 °C and then induced with 1 mM IPTG (isopropyl b-D-1-thiogalactopyranoside) and grown overnight at 25 °C. The cells were lysed by sonication in an ice bath through a sonicator probe in pulse mode (20 mM Tris–HCl buffer, pH 8.0) in the presence of a protease inhibitor cocktail (Roche). After sonication, the lysate was treated with a universal nuclease (Pierce) for 15 min at room temperature. The lysate was then heated at 90 °C for 10 min followed by centrifugation for 30 min at 15 000 g. After filtration through a 0.2 µm filter, the sample was loaded on to pre-equilibrated 5 mL HiTrap Q FF anion exchange column (GE Healthcare) and eluted by a linear gradient of 1 M NaCl in 20 mM Tris–HCl buffer pH 8.0. Fractions containing αS were combined and concentrated with Ultra-15 Ultracel 10 K centrifugal filter devices (Millipore). The concentrate was loaded on to Hiload 16/600 Superdex 75 pg column (GE Healthcare) and retrieved in 20 mM Tris–sulfate buffer pH 7.4.
A pET3a construct (including a tag with a repressor protein and a His-stretch followed by a Caspase 7 cleavage site) of the variant αS A107C was transformed into BL21(DE3) (Novagen) cells, which were then grown in similar conditions as for wild-type αS. The cells were harvested by centrifugation and re-suspended in Buffer A (20 mM Tris–HCl pH 8, 20 mM imidazole and 8 M urea), then sonicated and centrifuged for 30 min at 15 000 g. The supernatant was filtered through a 0.2 µm filter and applied to pre-equilibrated 5 ml Hi Trap Ni-NTA column (GE healthcare) with buffer A. After washing the column with 5 column volumes of buffer B (20 mM Tris–sulfate pH 7.4, 20 mM imidazole, 50 mM NaCl), the protein was eluted with buffer B with 250 mM imidazole. The tag was cleaved by adding Caspase 7 in the ratio of 1:100 (caspase 7: αS A107C w/w) in the presence of 5 mM TCEP (tris (2-carboxyethyl) phosphine) and incubated overnight at 4 °C. The cleaved protein was loaded onto a 5 mL HiTrap Q FF anion exchange column (GE Healthcare) and eluted by a linear gradient of 1 M NaCl in 20 mM Tris-sulfate buffer, pH 7.4. The eluted fractions were pooled, concentrated and loaded onto a Hiload 16/600 Superdex 75 column (GE Healthcare). The purified protein was eluted in 20 mM Tris–HCl buffer pH 7.4 with 1 mM TCEP. Truncated αS (containing residues 1–97) was expressed using the same vector (pET3a) as the A107C αS mutant and the purification followed the same procedure. The final gel filtration step was performed in 20 mM Tris–HCl buffer pH 7.4. Purified CCS was obtained from the Umeå University Protein Expertise Platform (http://www.kbc.umu.se/english/pep/services/).
Until use, αS protein samples were frozen in liquid nitrogen and stored at −80 °C. Purity for αS variants was confirmed by single bands on SDS–PAGE and single elution peaks in SEC. The concentrations of wild-type and A107C αS were determined using ε280 = 5960 M−1 cm−1 (four Tyr; no Trp); ε280 = 1490 M−1 cm−1 was used for the truncated αS (one Tyr).
Atox1 in a pET21(b) vector was transformed into BL21(DE3) plysS (Novagen) cells. Transformants were first grown to OD600 of 0.6 at 37 °C, and then induced with 1 mM IPTG and grown for overnight at 25 °C. The cells were harvested by centrifugation and re-suspended in Buffer A (20 mM MES, 1 mM EDTA, 1 mM TCEP, pH 5.7) containing protease inhibitor cocktail (Roche), followed by sonication. The lysate was treated with a universal nuclease (Pierce) for 30 min at 4 °C and then centrifuged for 30 min at 15 000 g. After filtration through a 0.2 µm filter, the supernatant was loaded onto a pre-equilibrated 5 mL HiTrap SP HP cation exchange column (GE Healthcare). The protein was eluted by a linear gradient of buffer B (20 mM MES, 1 mM TCEP, 1 M NaCl, pH 5.7). Fractions containing protein of the correct molecular weight were pooled and concentrated with Ultra-15 Ultracel 3 K centrifugal filter devices (Millipore). The concentrated protein sample was loaded on to Hiload 16/600 Superdex 75 pg column (GE Healthcare) and retrieved in Chelex-treated storage buffer C (20 mM Tris-sulfate pH 7.4, 150 mM NaCl, 1 mM TCEP). The concentration of Atox1 was determined using ε280 = 2980 M−1 cm−1. Upon purification, Atox1 is eluted in the apo-form. Cu(I)-loading of Atox1 is achieved by the addition of equimolar Cu(II) in the presence of 5-fold excess 1,4-dithiothreitol (DTT), as previously reported (Niemiec et al. Reference Niemiec, Weise and Wittung-Stafshede2012, Reference Niemiec, Dingeldein and Wittung-Stafshede2015).
Amyloid formation assay
αS amyloid formation reactions were conducted in 96-well half-area transparent bottom plates with a non-binding (non-ionic hydrophilic) surface (Corning, CLS3881) with one 2-mm glass bead in each well using a plate reader-incubator instrument (BMG Labtech, Fluostar Optima). Time-resolved measurements were performed in TBS (0.05 M Tris–HCl buffer, pH 7.4 with 0.15 M NaCl; 93 318 Sigma-Aldrich) buffer in the presence of 20 µM Thioflavin T (ThT; T3516 Sigma-Aldrich) at 37 °C using 200 rpm agitation for 5 min during the 20 min measurement cycles. Samples were typically incubated for 60 h and fluorescence measured every 20 min. ThT was excited at 440 nm and emission was recorded at 480 nm. All ThT experiments were performed in triplicate at each time and repeated at least three independent times.
Atomic force microscopy (AFM)
Aggregated samples from ThT experiments were diluted into MilliQ water (10–20 times) and deposited on freshly cleaved mica. After 10 min, the mica was rinsed with filtered Milli-Q water and dried under a gentle nitrogen stream. Images were recorded on an NTEGRA Prima setup (NT-MDT) using a gold-coated single crystal silicon cantilever (NT-MDT, NSG01, spring constant of ~5.1 N/m) and a resonance frequency of ~180 kHz. 512-pixel images were acquired with 0.5 Hz scan rate. Images were analyzed using the WSxM 5.0 software(Horcas et al. Reference Horcas2007).
Size exclusion chromatography (SEC)
500 µl of samples containing 70 µM αS and/or 70 µM apo- or holo-Atox1 were injected on a Superdex 75 10/300 column (GE Healthcare) using an Äkta purifier (GE healthcare) system. Running buffer was 20 mM TRIS pH 7.6 with 150 mM NaCl and 350 µM DTT. Pre-mixing of Cu(II), DTT and apo-Atox1, results in Cu(I) loading of the protein, i.e., Cu-Atox1 (Niemiec et al. Reference Niemiec, Weise and Wittung-Stafshede2012, Reference Niemiec, Dingeldein and Wittung-Stafshede2015). Elution profiles were probed by absorbance at two wavelengths in parallel: 280 nm (protein) and 254 nm (sulfur-to-Cu charge transfer). 1 ml fractions were collected and sent for metal analysis by ICP-MS (Chalmers ICP-MS facility). SEC was also used to quantify the amount of soluble αS after aggregation reactions. For such analysis, the aggregated samples were spun down (12 000 g, 15 min) and the supernatant loaded on the Superdex 75 10/300 column using the same conditions as for the fresh protein mixtures. The amount of αS was determined by peak intensity at 280 nm at the 9.5 ml elution volume corresponding to αS. The samples were also analyzed for protein content by SDS–PAGE electrophoresis.
Surface plasmon resonance (Biacore)
The αS Ala107Cys variant was biotinylated with Ez-Link BMCC-Biotin (Cat. No.: 21900, Thermo Fisher Scientific) following the manufacturer's instructions. After the reaction, the mixture was loaded on an Enrich70 24 ml SEC (BioRad) using an NGC chromatography system (BioRad) to remove excess biotin. Biotinylated αS was immobilized on one channel of a streptavidin-coated sensor chip SA (GE Healthcare) to a level of ~1000 response units. Samples were injected for 180 s pulses in 10 mM HEPES pH 7.4 buffer containing 150 mM NaCl. Measurements were performed on a Biacore X100 instrument. The measured response units were background corrected by subtracting signal of the non-modified flow channel.
Results and discussion
Purified αS aggregates into amyloid fibers in vitro upon incubation at pH 7, 37 °C and, with agitation, αS (70 µM) aggregates with a lag time of about 10–15 h, as determined using the ThT assay (Fig. 2a). Addition of 1:1 molar ratio of Cu(II) speeds up aggregation (Fig. 2a), as expected. The presence of apo-Atox1 has only a small delaying effect on the lag time for aggregation, whereas Cu(I)-loaded Atox1 distinctly reduced αS amyloid formation in a concentration-dependent manner (Fig. 2a,b). Atomic force microscopy (AFM) revealed the presence of typical αS amyloids for samples with only αS, αS in the presence of Cu(II), and αS with apo-Atox1. In contrast, but in accord with the ThT data, in the presence of Cu(I)-Atox1, no αS amyloids were detected by AFM (Fig. 2c). Notably, aggregation experiments in the presence of Cu(I) (without Atox1, e.g. as a Cu(I)-DTT complex) are technically challenging, as Cu(I) will oxidize and/or precipitate during these typically 1–2 days long experiments. Instead, we used silver ions (Ag(I)) as a redox-inert model for Cu(I) ions that is stable in solution. Because the presence of equimolar AgNO3 did not affect αS amyloid formation (Fig. S1A), it appears that the inhibitory effect of Cu-Atox1 arises due to the ternary complex formation, and not Cu(I) delivery to αS.

Fig. 2. (a) Representative ThT fluorescence curves probing amyloid formation of 70 µM αS alone (black), and in the presence of equimolar Cu(II) (blue), apo-Atox1 (green) and Cu-Atox1 (red). (b) Analysis of ThT emission at 60 h for different αS/Atox1 mixtures. Average values and standard deviations were calculated based on at least four repetitions. (c) AFM images of aggregation end-products in A.
As a complement to the AFM (lack of observable amyloids) and ThT emission (low emission implying no amyloids) data for αS mixed with Cu-Atox1, quantification of soluble αS after aggregation experiments showed that there was much more monomeric αS when it was incubated with Cu-Atox1 as compared with when incubated alone or with apo-Atox1 (Fig. S2A). As a test of specificity, we probed the effect of another cytoplasmic Cu-chaperone, the copper chaperone for superoxide dismutase (CCS), on αS amyloid formation. However, the presence of Cu-loaded CCS had no effect on αS amyloid formation (Fig. S2B), implying that the Cu(I)-Atox1-mediated inhibition of αS aggregation is protein specific.
To test for direct interaction of Cu-Atox1 with αS, we turned to surface plasmon resonance (Biacore). Using αS attached to the surface of a Biacore chip via cysteine-modification, Atox1 binding was detected as an increase in the response signal which corresponds to the mass of bound material. Whereas apo-Atox1 did not interact with αS, we found a concentration-dependent interaction between αS and Cu(I)-Atox1 in the Biacore experiments. By fitting concentration-dependent endpoint response values, a dissociation constant for the Cu-Atox1/αS complex of around 5 µM was determined (Fig. 3a, Fig. S1B).

Fig. 3. (a) Binding data from Biacore experiments for the interaction of Cu-Atox1 with an αS-coated surface. (b) Cu content (shown as % of total protein, assuming one Cu per protein) determined by ICP-MS for the αS peak eluted on a Superdex 75 column of αS alone and the mixture αS + Cu-Atox1.
To probe the protein–protein pair further, we analyzed Cu-Atox1/αS mixtures with SEC. Initial mixtures contained concentrations of proteins similar to in the ThT aggregation experiments, which are then diluted during chromatography. SEC traces probed at 280 nm (protein) and 254 nm (254/280 nm ratio probes Cu-loading of Atox1 ( Niemiec et al. Reference Niemiec, Weise and Wittung-Stafshede2012, Reference Niemiec, Dingeldein and Wittung-Stafshede2015); ligand-to-metal charge transfer) showed that samples that had been subjected to agitation and incubation, i.e., ‘amyloid inhibitory condition’ contained significant fractions of monomeric αS (cf. Fig. S2A) and Atox1, in accord with no amyloid formation.
SEC analysis of freshly mixed samples, mimicking the Biacore experiments, showed the absence of a detectable Cu-Atox1/αS complex but according to the 254/280 nm ratio of the resulting Atox1 peak, Cu had been removed from Atox1 (Fig. S3). Inductively coupled plasma mass spectrometry (ICP-MS) elemental analysis of Cu in the αS fractions (cannot be determined via 254/280 nm absorption ratio) from Cu-Atox1/αS mixtures revealed increased amounts of Cu in αS; thus, some Cu had moved from Atox1 to αS (Fig. 3b). Because the eluted protein fractions contain protein in concentrations near the determined dissociation constant for the Cu-Atox1/αS complex, it is reasonable that the Cu-Atox1/αS complex detected in Biacore dissociates when running through SEC. If Cu is bridging the ternary complex, it is indeed reasonable that some Cu is found in αS upon dissociation.
Our findings suggest that Cu(I)-loaded Atox1 can form a Cu(I)-dependent complex with αS monomers that results in blockage of αS amyloid formation (Fig. 4). The apo-form of Atox1 may interact weakly with αS, and this appears to delay aggregation to some extent. The Atox1–Cu–αS complex appears to involve bridging of Cu(I), as, if the complex is dissociated (eg. during SEC), some of the Cu(I) is then found in αS. Cu(I) can bind to sites in both N- and C-termini in αS (Binolfi et al. Reference Binolfi2006, Reference Binolfi2011; Camponeschi et al. Reference Camponeschi2013; De Ricco et al. Reference De Ricco2015a), see Fig. 1a. In aggregation experiments with a truncated variant of αS (αS residues 1–97; i.e., lacking the C-terminal 43 residues), we found Cu(I)-Atox1 to inhibit the amyloid formation of the αS variant but apo-Atox1 had no effect (Fig. S4). Because these results are similar to those found for wild-type αS, we propose that Cu(I)-Atox1 interacts with the N-terminal Cu(I) site of αS. This interaction may sterically block assembly of core regions of αS into amyloid fibers, but further mechanistic studies are desired.
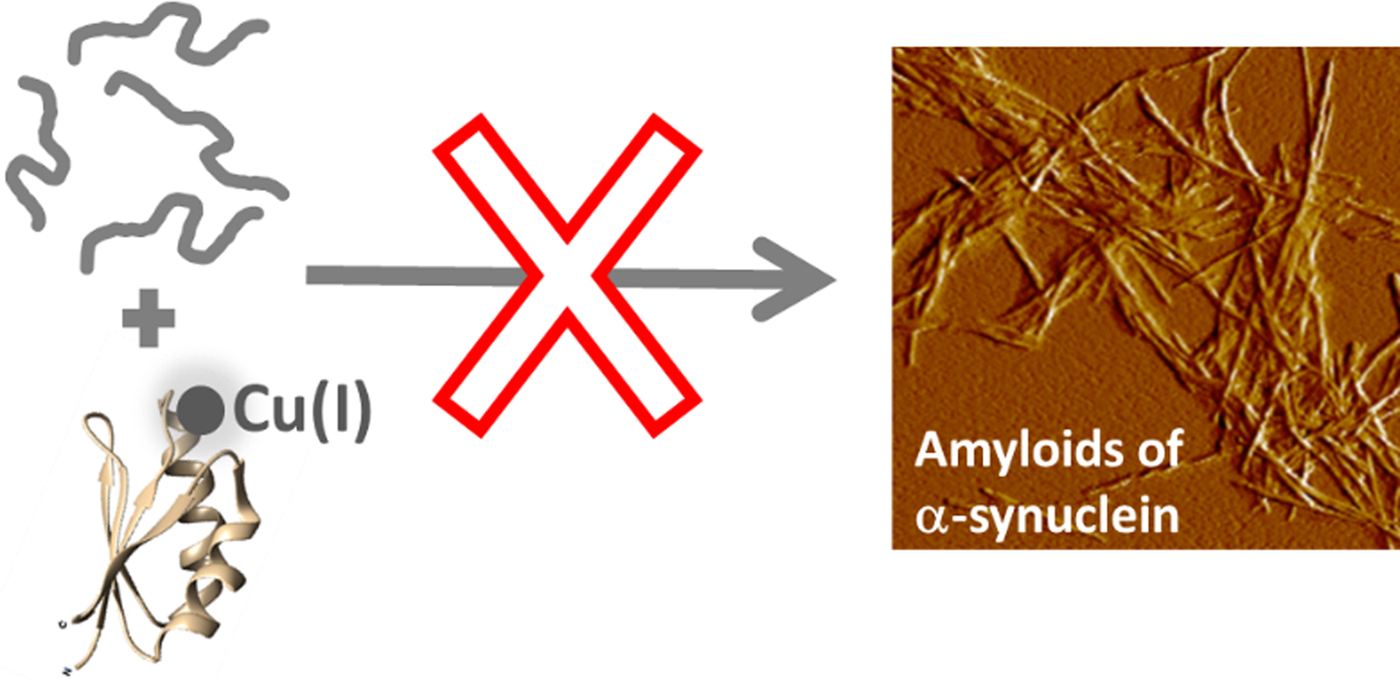
Fig. 4. Cartoon illustration of the main conclusion of this study, i.e., that Cu-loaded Atox1 blocks monomeric αS from aggregation into amyloid fibers in vitro.
In conclusion, we propose that metal-dependent chaperoning, exemplified by Cu(I)-Atox1, may be another cellular mechanism, in addition to the protein chaperone network, that controls the proteome. In similarity with respect to the outcome but opposite in terms of the role of the metal, metallothioneins can scavenge Cu(II) and Zn(II) from amyloidogenic peptides and protect against aggregation (Atrian-Blasco et al. Reference Atrian-Blasco2017; Okita et al. Reference Okita2017). For the specific case of PD, a significant decrease in total tissue Cu in substantia nigra has been reported (Davies et al. Reference Davies2014; Okita et al. Reference Okita2017). In addition to perturbing the antioxidant activity of Cu-dependent SOD1, our results imply that this reduction in tissue Cu may also abolish a Cu(I)-dependent chaperoning ability of Atox1; both being alterations that would further promote PD.
Box: speculation
In addition to the myriad of well-established chaperone and chaperonin proteins that aid in protein folding and quality control, we propose that metal-dependent chaperoning, here shown for a cytoplasmic copper chaperone, is an additional cellular process that modulates and controls the proteome. We speculate that interactions between the copper chaperone Atox1 and αS normally hold back αS amyloid formation and this interaction may also deliver copper ions to αS for (yet, unknown) metal-dependent αS functions. In contrast, when cellular metal homeostasis and/or redox status is disrupted, metal-dependent chaperone interactions may be abolished and deleterious aggregation of amyloidogenic proteins (such as αS) can flourish.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0033583518000045
Acknowledgement
We thank Stellan Holgersson (Chalmers) for ICP-MS analysis.
Financial support
Funding is acknowledged from the Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Olle Engkvist foundation and the Chalmers Foundation.
Conflict of interest
None.






 Open access
Open access