



Introduction: the four ages of SOD1 discovery
There are roughly 500 new scientific publications relating to superoxide dismutase-1 (SOD1) every year. The deluge of information over the last few years is however relatively small in comparison with totality of knowledge accrued over the 80 years in which SOD1 has been a subject of scientific investigation. This level of interest reflects its prevalence and importance in our physiological processes, the relative ease with which one can work with SOD1 and its limitless capacity to retain enigma. The story of SOD1 recapitulates that of the golden age in Life Sciences which occurred over the last century. As new technologies have become available, SOD1 has proved an interesting and amenable test-case. Biophysics has been fruitful in providing answers to questions in SOD1 biology. In this section, we give a history of the discovery of SOD1, which can be divided into four distinct periods, and then describe how biophysics has shaped our view of SOD1 in relation to the neurodegenerative disease amyotrophic lateral sclerosis (ALS).
A source of copper in blood
The first age of SOD1 discovery began two centuries ago with the work of chemists, such as Christian Friedrich Bucholz, who found copper to be a constituent of plant and animal tissue. Subsequently, in the latter half of the 19th century, came the discovery of zinc in biological materials. The copper and zinc under investigation here comprised several different sources but a portion of it was present as co-factors for what would become known as haemocuprein and later SOD1.
In the late 1920s and 30s, a copper source was identified in the blood that could be freed from its organic component with the addition of hydrochloric or trichloroacetic acids but could not be removed by dialysis except at low pH (Warburg and Krebs, Reference Warburg and Krebs1927; Locke et al., Reference Locke, Main and Rosbash1932). Eisler et al. (Reference Eisler, Rosdahl and Theorell1936) found that all of the copper in blood serum was bound to protein. In seminal papers, Thaddeus Mann and David Keilin described the purification of a ‘distinctly blue’ protein, which they named haemocuprein, from cow, sheep and horse blood together with the first of many methods for its crystallisation (Mann and Keilin, Reference Mann and Keilin1938a, Reference Mann and Keilin1938b). Calculation of its copper content indicated that one 35 kDa SOD1 molecule contained two copper atoms. Subsequent experiments on erythrocuprein purified from human blood refined the molecular mass to 33 kDa and indicated it possessed high thermal stability (Markowitz et al., Reference Markowitz, Cartwright and Wintrobe1959). Cerebrocuprein and hepatocuprein isolated from brain and liver, respectively, were found to be identical to the protein isolated from blood but no function could be ascribed (Mann and Keilin, Reference Mann and Keilin1938b; Porter et al., Reference Porter, Folch, Beggrovs and Ainsworth1957; Carrico and Deutsch, Reference Carrico and Deutsch1969). Variable copper metalation, dependent on source, and the ability to reversibly bind copper led to the hypothesis that erythrocuprein acts as copper storage sink rather than an enzyme (Mohamed and Greenberg, Reference Mohamed and Greenberg1954).
Concurrently with the initial characterisation of erythrocuprein, Linus Pauling's investigation of the implications of quantum mechanics on covalent bonding had predicted the existence of three-electron bonds (Pauling, Reference Pauling1931). This led Edward Neuman to the discovery of the potassium superoxide state of the substance then known as potassium tetroxide. Superoxide was found to be paramagnetic with one unpaired electron (Neuman, Reference Neuman1934). Superoxide anion is the product of univalent reduction of dioxygen. Its primary reaction in aqueous solvents is the spontaneous, second-order dismutation to hydrogen peroxide and water (McClune and Fee, Reference McClune and Fee1976). Understanding of the biological implications of superoxide generation began with biochemical characterisation of xanthine oxidase. Cytochrome c was found to be reduced by xanthine oxidase in the presence of its substrates. The rate of this reaction was increased under aerobic conditions and hypothesised to be mediated by superoxide (Horecker and Heppel, Reference Horecker and Heppel1949; Rabani and Stein, Reference Rabani and Stein1962). Conversely, the reaction of xanthine oxidase with its substrate was shown to catalyse a chain reaction of aerobic auto-oxidation of sulphite via superoxide free radicals (Fridovich and Handler, Reference Fridovich and Handler1958, Reference Fridovich and Handler1961). Their observations of superoxide led Joe McCord and Irwin Fridovich to propose a scheme for the dismutation reaction (Fig. 1a) and write, ‘once generated, this radical could be very damaging to the components of the cell by virtue of its high reactivity’ (McCord and Fridovich, Reference McCord and Fridovich1968).
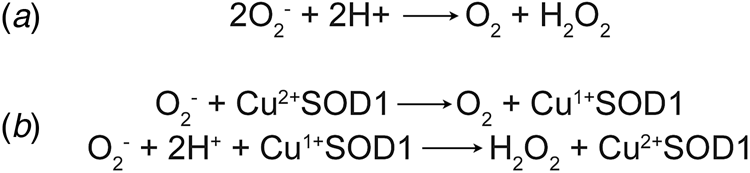
Fig. 1. Dismutation of superoxide anion. (a) The spontaneous dismutation of superoxide to oxygen and hydrogen peroxide (McCord and Fridovich, Reference McCord and Fridovich1968). (b) Two steps in the SOD1 catalysed dismutation of superoxide which cyclically reduces then oxidises the copper centre (Klug et al., Reference Klug, Rabani and Fridovich1972).
A cupro-enzyme with dismutase activity
Discovery of erythrocuprein's activity, and with it the beginning of the second age of SOD1 discovery, was a combination of many years of the rigorous biochemistry described above, and luck. McCord and Fridovich (Reference McCord and Fridovich1968) found that cytochrome c reduction by xanthene oxidase activity could be inhibited by carbonic anhydrase and myoglobin. On the verge of publication, the authors found that inhibition was due to an unknown impurity in both carbonic anhydrase and myoglobin preparations. From that position, the SOD activity of the impurity, erythrocuprein, was proven and the centrality of its copper cofactor in the dismutation reaction (McCord and Fridovich, Reference McCord and Fridovich1969; Klug et al., Reference Klug, Rabani and Fridovich1972) (Fig. 1b). In parallel, SOD1 was discovered to bind zinc in stoichiometric amounts to copper (Carrico and Deutsch, Reference Carrico and Deutsch1970).
Why does nature use superoxide dismutases?
‘There is a bizarre enzymatic activity universally present in respiring cells. The substrate is an unstable free radical that can be present only in minuscule amounts at any instant, and the reaction catalyzed proceeds at a rapid rate even in the absence of the enzyme’ (Fridovich, Reference Fridovich1975).
When a SOD was first isolated from Escherichia coli, it was found to be pink and contained manganese rather than copper and zinc (Keele et al., Reference Keele, McCord and Fridovich1970). Iron SODs are related to these manganese SODs and another evolutionarily distinct form are the nickel SODs (Smith and Doolittle, Reference Smith and Doolittle1992; Youn et al., Reference Youn, Kim, Roe, Hah and Kang1996). Copper/zinc superoxide dismutases (CuZnSOD) are found in the cytoplasm, intermembrane space of mitochondria, peroxisomes, nucleus, and the extracellular space of eukaryotes, and the periplasm of some prokaryotes. Manganese SODs are found in eukaryotic mitochondrial matrices as well as the prokaryotic cytoplasm, as are iron SODs. Nickel SODs are also largely prokaryotic. In almost every biological space, there is a SOD enzyme that operates within it. When oxygen is used in metabolism, SOD activity tends to be high and when organisms are exposed to oxygen, but it is not used in metabolism, SOD activity is low or inducible (Tally et al., Reference Tally, Goldin, Jacobus and Gorbach1977). The exception which proves this rule is the obligate anaerobes which have very low or non-existent SOD activity (McCord et al., Reference McCord, Keele and Fridovich1971). Thus, enzymatic SOD activity is primarily a response to the presence or use of oxygen (Gerschman et al., Reference Gerschman, Gilbert, Nye, Dwyer and Fenn1954) but may have evolved prior to Earth's great oxidation event, concomitantly with oxygenic photosynthesis. Please see excellent reviews of SOD evolution (Case, Reference Case2017) and structure (Perry et al., Reference Perry, Shin, Getzoff and Tainer2010).
Superoxide is produced by many eukaryotic cellular processes and in different locations. The respiratory electron transport chain (ETC) found in the mitochondrial inner membrane provides the proton-motive force for ATP generation but contains at least 11 sites where electrons can be lost to the surrounding oxygen-rich environment (Wong et al., Reference Wong, Dighe, Mezera, Monternier and Brand2017). Here superoxide is a Janus-faced component of the oxidative stress-redox signalling equilibrium; it is potentially damaging to any biological molecule, including mitochondrial DNA, while it also acts as a signal-transducer modulating respiration in a negative freed-back loop. Electrons can be shed by the mitochondrial ETC into the matrix where it forms superoxide and is detoxified by manganese SODs. Conversely, the ETC complex III (cytochrome bc1 complex) can shed electrons from its Qo site into the intermembrane space (Han et al., Reference Han, Williams and Cadenas2001; Bleier and Dröse, Reference Bleier and Dröse2013) where it is likely to undergo dismutation catalysed by SOD1. Xanthene oxidase, inducible nitric oxide synthase and NADPH-oxidase enzymes all produce superoxide in various cellular compartments, and as a result, SOD1 is usually found in their environs. An example is peroxisomes, which are voracious oxygen consumers and prolific reactive oxygen species producers, and as a result, they too house SOD1 (Fransen et al., Reference Fransen, Nordgren, Wang and Apanasets2012).
SOD1 structure discovery
Metal binding and disulphide bonding
With a function attributed to the previously mysterious erythrocuprein (McCord and Fridovich, Reference McCord and Fridovich1969), means of purification and enzymatic assay well described, the third age of SOD1 discovery began in earnest with biochemical and biophysical characterisation. Human and bovine SOD1 were each found to be composed of two sub-units of 16 kDa with strong interactions. This indicated that SOD1 is dimeric with high dimer affinity (Keele et al., Reference Keele, McCord and Fridovich1971; Hartz and Deutsch, Reference Hartz and Deutsch1972). Keele et al. (Reference Keele, McCord and Fridovich1971) found that disulphide reduction along with dodecyl sulphate denaturation was necessary to form monomeric SOD1. They concluded that a disulphide bond linked the SOD1 subunits. This was subsequently disproved by Hartz and Deutsch (Reference Hartz and Deutsch1972), but these experiments form the first understanding of the role of metalation and disulphide state in the stability of SOD1. A SOD1 intra-subunit disulphide was later confirmed in the wheat CuZnSOD orthologue and a similar bond was shown to link bovine SOD1 Cys55 and Cys144 (Cys57 and Cys146 human numbering) (Beauchamp and Fridovich, Reference Beauchamp and Fridovich1973; Abernethy et al., Reference Abernethy, Steinman and Hill1974). Analysing circular dichroism (CD) spectra from metalated, metal-apo and disulphide-reduced bovine SOD1, Wood et al. (Reference Wood, Dalgleish and Bannister1971) predicted the proximity of copper and zinc in the SOD1 structure and indicated the presence of one tryptophan per subunit. From electron paramagnetic resonance studies of SOD1 with the spectroscopically silent zinc replaced by cobalt, Rotilio et al. (Reference Rotilio, Calabrese, Mondovì and Blumberg1974) again demonstrated the proximity of the metal sites but also showed the zinc ion is not solvent exposed.
Wood et al. (Reference Wood, Dalgleish and Bannister1971) showed SOD1 has a β-sheet rich fold that is not significantly perturbed by metal removal. However, metal-free SOD1 is more susceptible to chaotrope-induced unfolding than the metalated form. They also ascertained that the disulphide bond has an impact on SOD1 tertiary structure. Forman and Fridovich (Reference Forman and Fridovich1973) found similar characteristics using enzyme activity and resistance to proteolysis as a measure of stability. With this approach, they gleaned that the zinc ion has a greater impact on stability than activity. The paradigm established here of metal loss and disulphide reduction contributing to SOD1 monomerisation and instability would become increasingly important following linkage with ALS two decades later.
The first crystallographic bovine SOD1 structure
The pursuit of a crystallographic structure of SOD1 ensued shortly after SOD catalytic activity was demonstrated. For the first decade, this work was dominated by Jane and David Richardson at Duke University which yielded the structure of bovine SOD1 and invaluable insight on protein structure as a whole.
X-ray diffraction from SOD1 was first observed from crystals grown in phosphate buffer and 2-methyl-2,4-pentanediol which diffracted to 2 Å resolution using the precession method (Richardson et al., Reference Richardson, Bier and Richardson1972). With the revolution in synchrotron radiation crystallography still a decade away, data from multiple crystals were collected using a Picker 4-circle x-ray diffractometer based on a sealed tube. The structure was solved at 5.5 Å resolution by multiple isomorphous replacements using a combination of heavy atom derivatives soaked for up to 3 months with mercury substituted in the SOD1 zinc site (Thomas et al., Reference Thomas, Rubin, Bier, Richardson and Richardson1974). The authors noted a 15 Å diameter hollow passing through the centre of each monomer which they attributed to adoption of an antiparallel β-cylinder structure (Fig. 2a). A year later brought a refinement of this structure to 3 Å resolution and with it tracing the back-bone, assignment of Cα positions and positioning of some side chains (PDB: 1SOD) (Richardson et al., Reference Richardson, Thomas, Rubin and Richardson1975a, Reference Richardson, Thomas and Richardson1975b). This joined the list of a dozen or so high-resolution structures at the time that included myoglobin, haemoglobin, lysozyme, ribonucleases A and S, carboxypeptidase, cytochrome c, chymotrypsin and immunoglobins.
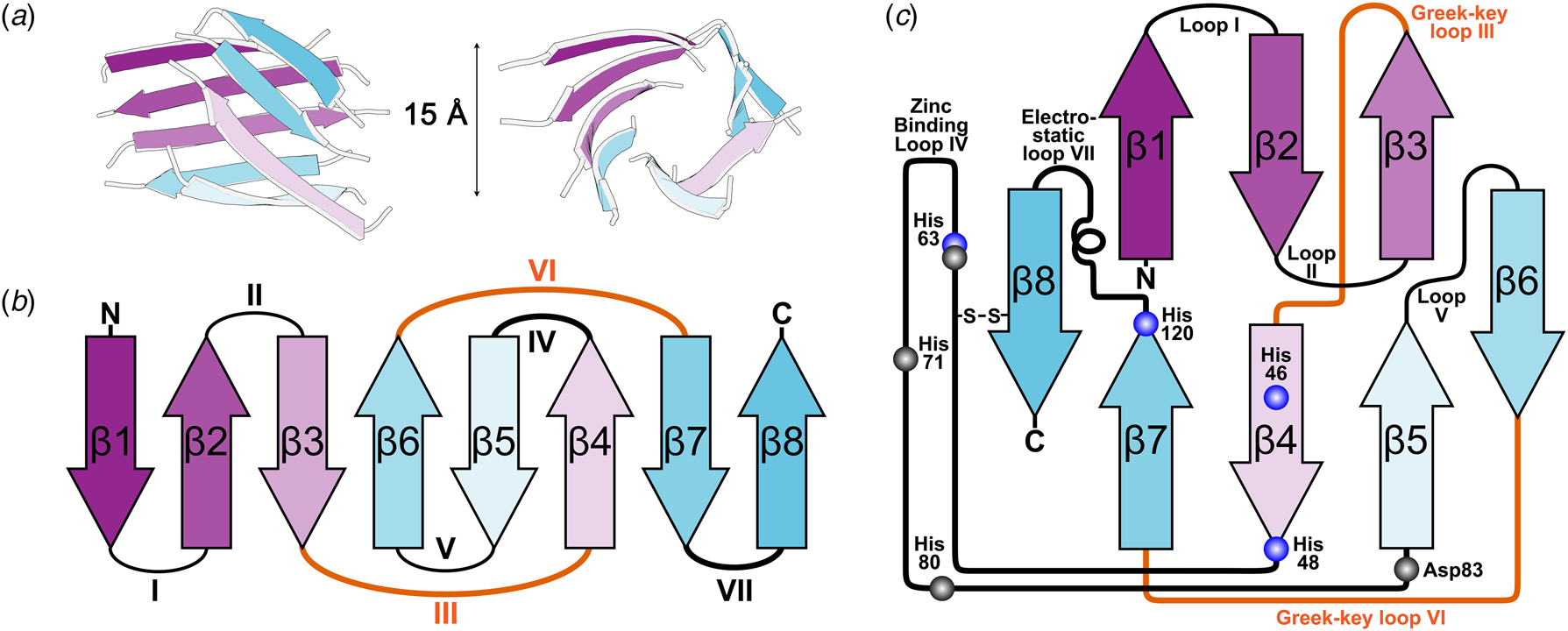
Fig. 2. The β-strand, loop and barrel structure of human SOD1. (a) β-barrel component of human SOD1 with approximately 15 Å diameter as described for bovine SOD1 (Thomas et al., Reference Thomas, Rubin, Bier, Richardson and Richardson1974). (b) Greek-key assembly of SOD1 β-strands showing loops which facilitate position swap of β-strands 4 and 6 in orange. (c) Arrangement of human SOD1 secondary structures within the β-barrel with residues involved in copper and zinc binding highlighted blue and grey respectively.
The above research indicated that the SOD1 monomer possesses an eight stranded anti-parallel β-barrel (Fig. 2b), with the hydrophobic inner core, which constitutes roughly half of the protein backbone. The remainder of the structure is found in two loops (Fig. 2c). The ‘external loop’ (loop VII, amino acids 122–144 in the human enzyme and now commonly called the electrostatic loop), which was visible in the 5.5 Å resolution map, had unknown functional significance. A second loop (loop IV, amino acids 49–83, named the zinc loop) provides three coordinating ligands to the zinc ion and is held against the β-barrel by a disulphide bond but also forms part of the dimer interface along with the C-terminus and part of the β-barrel surface. Copper and zinc coordinating amino acids could be defined and the copper site geometry was categorised as distorted square planar with solvent accessibility to the copper ion (Fig. 3). The proposition that both subunits in the SOD1 dimer are conformationally identical was raised along with interface hydrogen bonding (Fig. 3).
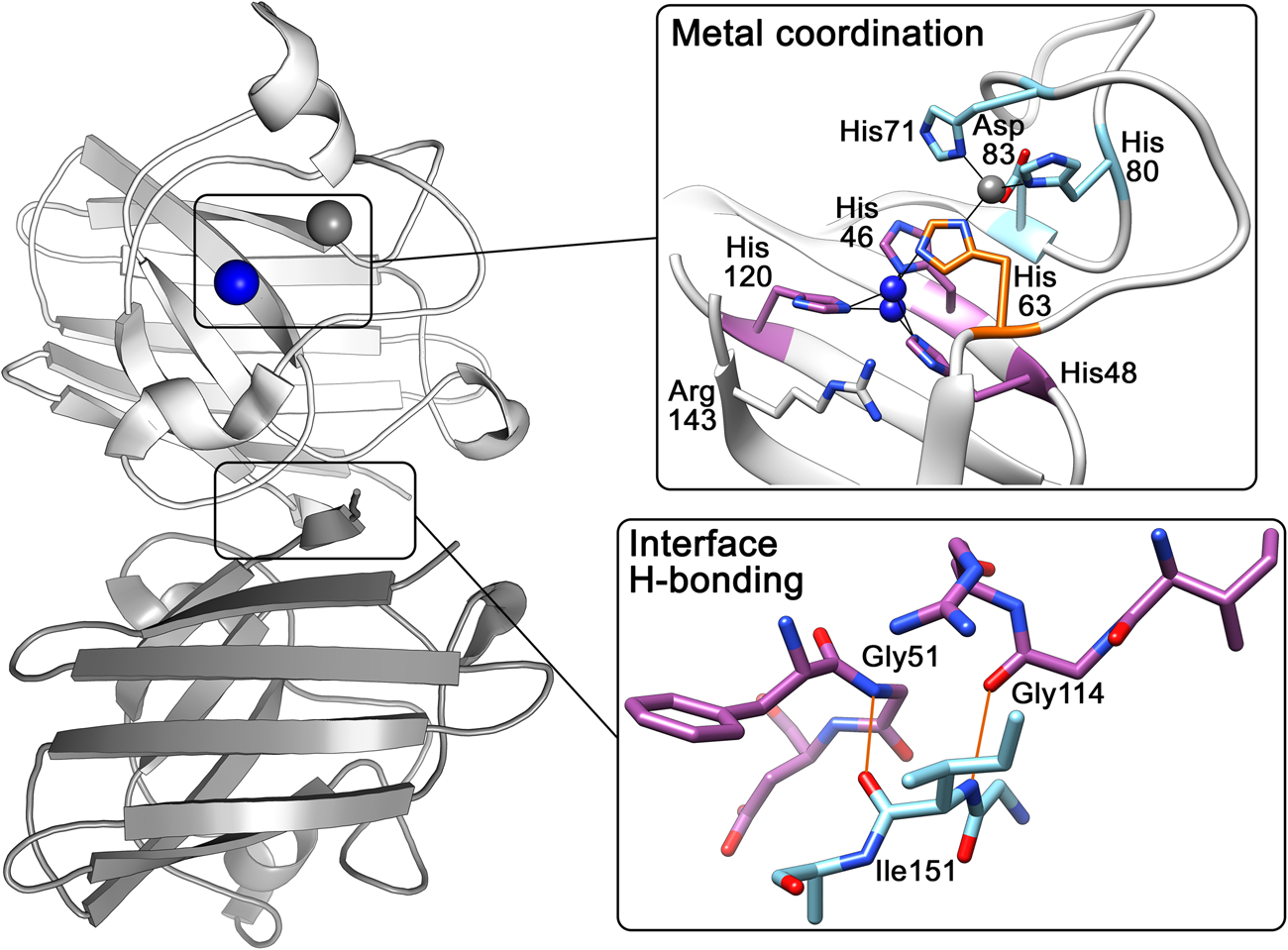
Fig. 3. Human SOD1 metal binding and dimerisation regions. Upper panel – Copper and zinc coordinating (purple and light blue respectively) sites are linked by His63 (orange). The copper ion (blue) can be found solvent exposed at two positions dependent on oxidation state while the zinc ion (grey) is internalised. Lower panel – Hydrogen bonding (orange) at the SOD1 dimer interface is mediated by backbone interactions of Ile151 with Gly51 and Gly114.
Immunoglobin-like fold
Of particular interest was the similarity between the SOD1 β-barrel structure and other immunoglobulin domains despite sequence and functional diversity. Where SOD1 had disulphide, zinc binding and external loops connecting β-strands, hyper-variable loops could be found in immunoglobulin structures (Richardson et al., Reference Richardson, Thomas, Rubin and Richardson1975a). This led to the deduction of a nomenclature for β-sheet structures, the ‘Greek key’ sobriquet for the β-barrel arrangement typical of SOD1-like proteins, and a series of protein domain structure rules. The latter included conservation of β-structure over sequence, conservation of disulphide connectivity and the potential for insertions and deletions to occur in loops but not within secondary structure elements thus engineering functionality without detriment to stability (Richardson et al., Reference Richardson, Richardson, Thomas, Silverton and Davies1976; Richardson, Reference Richardson1977). It was also noted that similar domains may use different interaction surfaces for oligomerisation, a fact now easily observed by comparison of prokaryotic and eukaryotic CuZnSOD dimerisation (Bourne et al., Reference Bourne, Redford, Steinman, Lepock, Tainer and Getzoff1996).
Structure refinement and a catalytic mechanism
Working in the Richardsons' laboratory, Elizabeth Getzoff and John Tainer were subsequently able to extend the limit of usable data to 2 Å and improve the bovine SOD1 model (PDB: 2SOD) with the stereochemistry and non-crystallographic symmetry restrained with Hendrickson–Konnert least-squares refinement (Tainer et al., Reference Tainer, Getzoff, Beem, Richardson and Richardson1982). This allowed for determination of all non-hydrogen atom positions, placement of a single water molecule 3.2 Å from the copper ion and detailed assessment of hydrogen bonding interactions that hold the β-barrel and loops in place. Following further refinement and charge analysis, it became clear that loops IV and VII create both the active site and the electrostatic forces in and beyond the active site channel that speed diffusion of superoxide towards the positively charged copper, zinc and Arg141 (human Arg143) in the correct orientation to maximise productive interactions (Getzoff et al., Reference Getzoff, Tainer, Weiner, Kollman, Richardson and Richardson1983). In the active site, superoxide displaces the copper bound water and hydrogen bonds with Arg141 side-chain guanidinium. The unpaired superoxide electron reduces copper (II) and oxygen is released. The bond between copper and His61 (human His63) is broken and the histidine is protonated after which it hydrogen bonds with a second superoxide anion. Arg141 again hydrogen bonds with the substrate. Addition of another proton and bond rearrangement creates hydrogen peroxide and oxidises copper (I) (Tainer et al., Reference Tainer, Getzoff, Richardson and Richardson1983). The structure of the fully reduced Cu(I) enzyme showing complete breakage of the Cu-His61 bond came two decades later at 1.15 Å resolution using a powerful synchrotron beam and cryogenically maintained crystals (Hough and Hasnain, Reference Hough and Hasnain2003).
Recombinant human SOD1 crystallography
Taking advantage of advances in recombinant DNA technology throughout the 1970s, Getzoff and Tainer embarked on the biophysical characterisation of human SOD1. Expressed in Saccharomyces cerevisiae and crystallised with ammonium sulphate and poly-ethylene glycol precipitants, recombinant wild-type human SOD1 was found to form a variety of crystals which diffracted to at least 2.8 Å resolution on a rotating anode x-ray generator setup. Seeding could again be used to stimulate growth and wild-type crystals were used to grow a free sulphydryl knock-out, thermostable mutant (Cys6Ala/Cys111Ser) following mutagenesis of the wild-type expression plasmid (Parge et al., Reference Parge, Getzoff, Scandella, Hallewell and Tainer1986). It was later shown that the removal of these free cysteines promotes refolding after thermal denaturation (Hallewell et al., Reference Hallewell, Imlay, Lee, Fong, Gallegos, Getzoff, Tainer, Cabelli, Tekamp-Olson, Mullenbach and Cousens1991). This work culminated in the crystal structures of recombinant wild-type and thermal-stable human SOD1 (Parge et al., Reference Parge, Hallewell and Tainer1992). Diffraction data were now collected using synchrotron radiation on film at the Stanford Synchrotron Radiation Laboratory or on a Xentronics multi-wire proportional counting area detector; two developments that would revolutionise protein crystallography. Structural work continued from this point with increasing crystallographic resolution allowing ever greater detail to be revealed (Strange et al., Reference Strange, Antonyuk, Hough, Doucette, Valentine and Hasnain2006) and nuclear magnetic resonance spectroscopy accessing solution states of SOD1 important in its journey from polypeptide to active protein (Banci et al., Reference Banci, Benedetto, Bertini, Del Conte, Piccioli and Viezzoli1998, Reference Banci, Bertini, Cramaro, Del Conte and Viezzoli2002, Reference Banci, Bertini, Cantini, D'Amelio and Gaggelli2006). In our laboratory, we routinely produce crystal structures of SOD1 in a high-throughput fashion. From initial exposure of a crystal to x-rays through to seeing a 3D model of SOD1 now takes only hours rather than years of painstaking data collection, data reduction and model building. The combination of all of this work has given us an exquisitely detailed picture of SOD1 structure.
Amyotrophic lateral sclerosis
ALS is a neurodegenerative disease historically stated as only affecting the upper and lower motor neurons. Neuron death leads to a progressive inability to control the musculature and ultimately death usually within 5 years. We now understand that ALS is a motor system disorder affecting neurons and their support cells and forms a spectrum of clinical characteristics with fronto-temporal lobar degeneration. Schizophrenia and cognitive impairment are also associated with ALS; however, this is rare for SOD1-related ALS. Following the first characterisation by Jean–Martin Charcot in 1869, conjecture on the aetiology of ALS varied between sporadic and familial (Kurland and Mulder, Reference Kurland and Mulder1955).
SOD1 and familial ALS
The existence of SOD1 polymorphisms, prevalent within a specific geographical location and inherited from one generation to the next, that could change the physico-chemical properties of the enzyme was understood long before linkage with ALS was proven (Brewer, Reference Brewer1967; Beckman, Reference Beckman1973; Lepock et al., Reference Lepock, Frey and Hallewell1990; Banci et al., Reference Banci, Bertini, Cabelli, Hallewell, Tung and Viezzoli1991). We now know that a portion of ALS incidence, roughly 20%, is attributed to inheritable genetic factors, termed familial ALS (fALS). A concerted effort to discover a chromosomal location associated with ALS came to fruition in 1991 with the discovery of the 21q22.1 locus (Siddique et al., Reference Siddique, Figlewigz, Pericak-Vance, Haines, Rouleau, Jeffers, Sapp, Hung, Bebout, McKenna-Yasek, Deng, Horvitz, Gusella, Brown and Roses1991). In 1993, the focus of SOD1 structure-function research shifted considerably with the knowledge that 11 amino acid substitutions in the SOD1 primary sequence were causative for ALS (Rosen et al., Reference Rosen, Siddique, Patterson, Figlewicz, Sapp, Hentati, Donaldson, Goto, O'Regan and Deng1993). In the following years, many new ALS-related mutations were discovered including the Asp90Ala identity of the polymorphism described by Beckman (Reference Beckman1973). In the last decade and a half, SOD1 has been joined by a disparate group of genes which have varying modes of inheritance and degrees of ALS penetrance. Thus, ALS is a genetically heterogeneous disease. The year 1993 marked the beginning of the fourth age of SOD1. Here biophysics would play a fundamental role in our understanding of the transformation which turns this normally cytoprotective enzyme into a neurotoxin.
Biophysical experiments establish the nature of mutant SOD1
Prior to linkage with ALS, Parge et al. (Reference Parge, Getzoff, Scandella, Hallewell and Tainer1986) conjectured on the possibility that β-barrel and dimer interface mutations would affect SOD1 stability. Months after linkage with SOD1, a working hypothesis for toxicity was devised by mapping ALS mutations on the crystal structure of wild-type SOD1. Deng et al. (Reference Deng, Hentati, Tainer, Iqbal, Cayabyab, Hung, Getzoff, Hu, Herzfeldt, Roos, Warner, Deng, Soriano, Smyth, Parge, Ahmed, Roses, Hallewell, Pericak-Vance and Siddique1993) then proposed that mutations to amino acids and conformations conserved in human, yeast and bovine SOD1 structures would destabilise β-barrel, dimer interface and loops. This hypothesis later became known as frame-work destabilisation (DiDonato et al., Reference DiDonato, Craig, Huff, Thayer, Cardoso, Kassmann, Lo, Bruns, Powers, Kelly, Getzoff and Tainer2003). Over the coming years, SOD1 cytoplasmic inclusions were found in the motor neurons and associated cells of post-mortem mutant SOD1 transgenic mice and human ALS patient tissues (Kato et al., Reference Kato, Shimoda, Watanabe, Nakashima, Takahashi and Ohama1996; Shibata et al., Reference Shibata, Hirano, Kobayashi, Siddique, Deng, Hung, Kato and Asayama1996a; Bruijn et al., Reference Bruijn, Becher, Lee, Anderson, Jenkins, Copeland, Sisodia, Rothstein, Borchelt, Price and Cleveland1997, Reference Bruijn, Houseweart, Kato, Anderson, Anderson, Ohama, Reaume, Scott and Cleveland1998). These observations were linked to framework destabilisation by in vitro biophysics experimentation which characterised SOD1 thermal instability (Rodriguez et al., Reference Rodriguez, Valentine, Eggers, Roe, Tiwari, Brown and Hayward2002; Tiwari and Hayward, Reference Tiwari and Hayward2005), unfolding propensity (Cardoso et al., Reference Cardoso, Thayer, DiDonato, Lo, Bruns, Getzoff and Tainer2002; Lindberg et al., Reference Lindberg, Tibell and Oliveberg2002; DiDonato et al., Reference DiDonato, Craig, Huff, Thayer, Cardoso, Kassmann, Lo, Bruns, Powers, Kelly, Getzoff and Tainer2003), dimer destabilisation (Hough et al., Reference Hough, Grossmann, Antonyuk, Strange, Doucette, Rodriguez, Whitson, Hart, Hayward, Valentine and Hasnain2004) and formation of high molecular mass soluble and insoluble structures (DiDonato et al., Reference DiDonato, Craig, Huff, Thayer, Cardoso, Kassmann, Lo, Bruns, Powers, Kelly, Getzoff and Tainer2003; Elam et al., Reference Elam, Taylor, Strange, Antonyuk, Doucette, Rodriguez, Hasnain, Hayward, Valentine, Yeates and Hart2003a; Ray et al., Reference Ray, Nowak, Strokovich, Brown, Walz and Lansbury2004). A consistent theme is the effect of SOD1 disulphide reduction and the inability to correctly bind copper and zinc on toxic properties. The structure of metal-deficient SOD1 shows loop disorder and exposure of the β-barrel (Strange et al., Reference Strange, Antonyuk, Hough, Doucette, Rodriguez, Hart, Hayward, Valentine and Hasnain2003). In vivo, metal-free, disulphide-reduced SOD1 is over-represented within SOD1 aggregates indicating a defective post-transitional modification (PTM) pathway (Jonsson et al., Reference Jonsson, Graffmo, Andersen, Brännström, Lindberg, Oliveberg and Marklund2006a; Karch et al., Reference Karch, Prudencio, Winkler, Hart and Borchelt2009; Bourassa et al., Reference Bourassa, Brown, Borchelt, Vogt and Miller2014). This brings the story of the pathogenesis of SOD1-reated ALS back to the first experiments on the newly named SOD1 in the 1970s which showed that the presence of metal cofactors and the intra-subunit disulphide bond were prerequisites for stability and activity.
The pathogenesis of SOD1-related ALS
SOD1 mutations
There are more than 180 SOD1 polymorphisms associated with ALS, the majority of which are collated in the ALSoD database (Abel et al., Reference Abel, Powell, Andersen and Al-Chalabi2012). Most are single amino acid substitutions, but truncations and frame-shift mutations that lead to truncations can also be found (Fig. 4a). The most common SOD1 polymorphisms are Ala4Val (Deng et al., Reference Deng, Hentati, Tainer, Iqbal, Cayabyab, Hung, Getzoff, Hu, Herzfeldt, Roos, Warner, Deng, Soriano, Smyth, Parge, Ahmed, Roses, Hallewell, Pericak-Vance and Siddique1993), predominantly found in North America, and Asp90Ala (Andersen et al., Reference Andersen, Nilsson, Ala-Hurula, Keränen, Tarvainen, Haltia, Nilsson, Binzer, Forsgren and Marklund1995) which is found in 5% of the Scandinavian population (Andersen et al., Reference Andersen, Forsgren, Binzer, Nilsson, Ala-Hurula, Keränen, Bergmark, Saarinen, Haltia, Tarvainen, Kinnunen, Udd and Marklund1996). These two polymorphisms reflect the diversity of SOD1 ALS mutations. They are found in different exons in the SOD1 gene (1 and 4, respectively) which code for amino acids with different properties in different parts of the SOD1 protein with different secondary structure. Ala4Val causes ALS with early onset and very short duration (Rosen et al., Reference Rosen, Bowling, Patterson, Usdin, Sapp, Mezey, McKenna-Yasek, O'Regan, Rahmani and Ferrante1994) whereas Asp90Ala leads to long survival times post presentation (Andersen et al., Reference Andersen, Forsgren, Binzer, Nilsson, Ala-Hurula, Keränen, Bergmark, Saarinen, Haltia, Tarvainen, Kinnunen, Udd and Marklund1996).
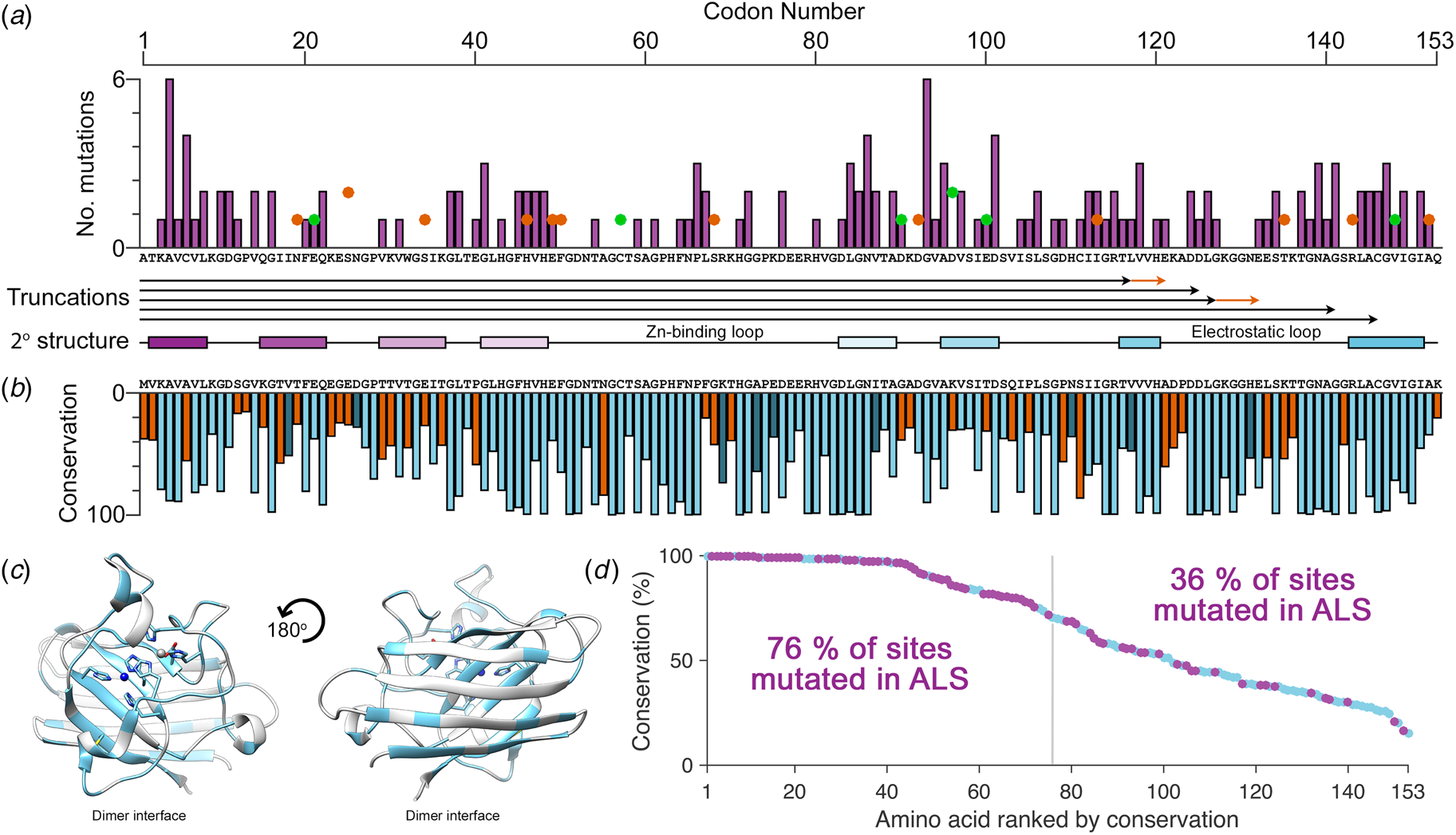
Fig. 4. The relationship between eukaryotic Cu/Zn superoxide dismutase codon conservation and human SOD1 ALS mutations. (a) The frequency of ALS mutations (purple bars), non-ALS polymorphisms (orange spots) and mutations with a disputed role in ALS (green spots) in the human SOD1 primary structure. Known truncation mutants are shown with black arrows and areas that differ from the wild-type sequence following a frame-shift mutation are shown in orange. The SOD1 secondary structure is also shown with β-strands coloured as in Fig. 2. (b) Consensus sequence of eukaryotic CuZnSODs. Light blue indicates identity between human SOD1 and the eukaryotic consensus, dark blue indicates conservative substitutions and orange indicates non-conservative substitutions. (c) Human SOD1 structure showing amino acids with >70% sequence conservation across eukaryotes highlighted in light blue. (d) Human SOD1 amino acids ranked by eukaryotic conservation (light blue). Sites that have at least one ALS-causing mutation are highlighted in purple. Sites that are highly conserved are more likely to have at least one ALS mutation.
Much effort relating genotype, molecular characteristics and disease phenotype has yielded proposed relationships between SOD1 thermal stability, half-life, aggregation, K m for hydrogen peroxide, hydrogen bond stability and free energy of wild-type/mutant heterodimerisation with disease course (Yim et al., Reference Yim, Kang, Chock, Stadtman and Yim1997; Sato et al., Reference Sato, Nakanishi, Yamamoto, Andersen, Ogawa, Fukada, Zhou, Aoike, Sugai, Nagano, Hirata, Ogawa, Nakano, Ohi, Kato, Nakagawa, Hamasaki, Shimizu and Sakoda2005, Reference Sato, Nakanishi, Yamamoto, Andersen, Ogawa, Fukada, Zhou, Aoike, Sugai, Nagano, Hirata, Ogawa, Nakano, Ohi, Kato, Nakagawa, Hamasaki, Shimizu and Sakoda2005; Wang et al., Reference Wang, Johnson, Agar and Agar2008; Prudencio et al., Reference Prudencio, Hart, Borchelt and Andersen2009; Shi et al., Reference Shi, Acerson, Abdolvahabi, Mowery and Shaw2016). However, the small numbers of people which are known to have carried most SOD1 polymorphisms, which in some cases may be as low as one individual, and the wide distribution of longevities following the onset of symptoms significantly hamper the power of these investigations. Other predisposition genes, epigenetic and environmental factors are additionally all likely to contribute to the phenotype which eventually leads to symptom onset further complicating this task of unpicking causality in SOD1 ALS.
SOD1-ALS inheritance and penetrance
The majority of SOD1 mutations cause ALS in an autosomal dominantly inherited fashion. In the heterozygous state, Ala4Val SOD1 phenotypic penetrance is high. At age 70, 91% of Ala4Val allele-positive individuals will have developed ALS (Cudkowicz et al., Reference Cudkowicz, McKenna-Yasek, Sapp, Chin, Geller, Hayden, Schoenfeld, Hosler, Horvitz and Brown1997) but examples do exist of Ala4Val carriers who remain symptom free (Weiss et al., Reference Weiss, Ravits, Schuman and Carter2006). By contrast, Asp90Ala SOD1 generally results in recessively inherited ALS but has also been described as dominantly inherited with reduced penetrance in genetically distinct families (Andersen et al., Reference Andersen, Nilsson, Ala-Hurula, Keränen, Tarvainen, Haltia, Nilsson, Binzer, Forsgren and Marklund1995; Robberecht et al., Reference Robberecht, Aguirre, Van den Bosch, Tilkin, Cassiman and Matthijs1996). There are several other examples of ALS mutant SOD1 homozygosity including a 13-year-old who had two copies of the Asn86Ser allele and died only 3.5 months after presentation. Heterozygotes with the same mutation have been noted to be asymptomatic or present with typical mutant SOD1 phenotype (Hayward et al., Reference Hayward, Brock, Minns and Swingler1998; Millecamps et al., Reference Millecamps, Salachas, Cazeneuve, Gordon, Bricka, Camuzat, Guillot-Noël, Russaouen, Bruneteau, Pradat, Le Forestier, Vandenberghe, Danel-Brunaud, Guy, Thauvin-Robinet, Lacomblez, Couratier, Hannequin, Seilhean, Le Ber, Corcia, Camu, Brice, Rouleau, LeGuern and Meininger2010). Individuals have also been found homozygous for a Leu126Ser substitution and a ΔGly27/Pro28 exon 2 splicing-site variant (Kato et al., Reference Kato, Aoki, Ohta, Nagai, Ishizaki, Nakamura and Itoyama2001a; Zinman et al., Reference Zinman, Liu, Sato, Wakutani, Marvelle, Moreno, Morrison, Mohlke, Bilbao, Robertson and Rogaeva2009). A further example describes three compound heterozygous Asp90Ala/Asp96Asn individuals who presented with early-onset ALS but very long disease duration (Hand et al., Reference Hand, Mayeux-Portas, Khoris, Briolotti, Clavelou, Camu and Rouleau2001). These cases are indicative that a necessary load of toxic SOD1 species must be met before cell death ensues. As ever though SOD1 provides an enigmatic outlier; a homozygous Leu84Phe 48-year-old individual remained asymptomatic while an unrelated heterozygous individual presented with ALS at age 45 (Boukaftane et al., Reference Boukaftane, Khoris, Moulard, Salachas, Meininger, Malafosse, Camu and Rouleau1998).
The Ile113Thr substitution is one of the most common ALS SOD1 mutations but the resulting phenotype can appear as sporadic ALS due to reduced penetrance (Rosen et al., Reference Rosen, Siddique, Patterson, Figlewicz, Sapp, Hentati, Donaldson, Goto, O'Regan and Deng1993; Suthers et al., Reference Suthers, Laing, Wilton, Dorosz and Waddy1994). This high phenotypic variability also includes variable disease onset and duration (Orrell et al., Reference Orrell, Habgood, Malaspina, Mitchell, Greenwood, Lane and deBelleroche1999; Lopate et al., Reference Lopate, Baloh, Al-Lozi, Miller, Fernandes Filho, Ni, Leston, Florence, Schierbecker and Allred2010). These characteristics are in stark contrast to Ala4Val SOD-related ALS despite the proximity of the two mutations; Ala4 and Ile113 side-chains are separated by only 3.7 Å when the protein is folded and mature. Other mutations have been noted to yield ALS with incomplete penetrance including Gly61Arg (Conforti et al., Reference Conforti, Barone, Fermo, Giliberto, Patti, Gambardella, Quattrone and Zappia2011), and Leu67Pro (del Grande et al., Reference del Grande, Luigetti, Conte, Mancuso, Lattante, Marangi, Stipa, Zollino and Sabatelli2011) however, in the absence of detailed immunohistochemistry, it is difficult to ascertain if these are truly ALS-causing SOD1 mutations or harmless polymorphisms co-incident with sporadic or low penetrance non-SOD1 fALS as has been noted for Asp90Ala and Glu100Lys families (Felbecker et al., Reference Felbecker, Camu, Valdmanis, Sperfeld, Waibel, Steinbach, Rouleau, Ludolph and Andersen2010). Indeed, SOD1 polymorphisms Ser25Thr, Ser25Asn, Ser34Ile, His46Tyr, Glu49Val, Phe50Cys, Ser68Phe, Asp92Gly, Ile113Met, Thr135Ile, Arg143Gly and Ala152Thr are known to exist in the wider population but, to date, are not associated with ALS in any way (Lek et al., Reference Lek, Karczewski, Minikel, Samocha, Banks, Fennell, O'Donnell-Luria, Ware, Hill, Cummings, Tukiainen, Birnbaum, Kosmicki, Duncan, Estrada, Zhao, Zou, Pierce-Hoffman, Berghout, Cooper, Deflaux, DePristo, Do, Flannick, Fromer, Gauthier, Goldstein, Gupta, Howrigan, Kiezun, Kurki, Moonshine, Natarajan, Orozco, Peloso, Poplin, Rivas, Ruano-Rubio, Rose, Ruderfer, Shakir, Stenson, Stevens, Thomas, Tiao, Tusie-Luna, Weisburd, Won, Yu, Altshuler, Ardissino, Boehnke, Danesh, Donnelly, Elosua, Florez, Gabriel, Getz, Glatt, Hultman, Kathiresan, Laakso, McCarroll, McCarthy, McGovern, McPherson, Neale, Palotie, Purcell, Saleheen, Scharf, Sklar, Sullivan, Tuomilehto, Tsuang, Watkins, Wilson, Daly and MacArthur2016). This is despite several occupying positions where other substitutions do cause ALS (His46Arg and Ile113Thr, e.g.) and several non-conservative substitutions at functionally important sites (Arg143 in the copper active site (Getzoff et al., Reference Getzoff, Tainer, Weiner, Kollman, Richardson and Richardson1983) and Phe50 in the dimerisation region (Banci et al., Reference Banci, Bertini, Chiu, Mullenbach and Viezzoli1995)). Furthermore, several SOD1 polymorphisms linked to ALS have a disputed role in disease pathogenesis including Asn19Ser, Glu21Lys, Thr54Arg, Leu67Arg, Val148Ile, Asp90Ala, Glu100Lys, Asp96Asn and Asp96Val (Mayeux et al., Reference Mayeux, Corcia, Besson, Jafari-Schluep, Briolotti and Camu2003; Andersen et al., Reference Andersen, Restagno, Stewart and Chiò2004; Felbecker et al., Reference Felbecker, Camu, Valdmanis, Sperfeld, Waibel, Steinbach, Rouleau, Ludolph and Andersen2010; Fujisawa et al., Reference Fujisawa, Yamaguchi, Kadowaki, Tsukamoto, Tsuburaya, Tsubota, Takahashi, Naguro, Takahashi, Goto, Tsuji, Nishitoh, Homma and Ichijo2015). Thus, not all SOD1 mutants cause ALS and those that do may do so irregularly. Felbecker et al. (Reference Felbecker, Camu, Valdmanis, Sperfeld, Waibel, Steinbach, Rouleau, Ludolph and Andersen2010) quantified this effect and estimated that only one-third of known SOD1 polymorphisms are actually pathogenic.
SOD1 mutations within the primary and tertiary structure
Eukaryotic CuZnSOD sequence conservation is high in functional parts of the molecule including interface, metal binding, β-barrel core and disulphide regions, while β-strands 2, 3 and 6 on the opposing face of the β-barrel are divergent (Fig. 4b, c). Highly conserved SOD1 residues are more likely to have associated ALS mutations while poorly conserved residues are much less likely to harbour ALS mutations (Fig. 4d). Fifty-seven human SOD1 amino acids are not associated with any polymorphism, and while they are found sparsely through-out the primary sequence, they cluster in the β-strand 2–3 region and the zinc-binding loop. The zinc loop and disulphide sub-loop substructure within, overlaps with a region of very high sequence conservation extending through amino acids 43–86. This indicates that substitutions in the zinc loop have been sufficiently disadvantageous to be selected against over the whole course of eukaryotic evolution and recent human history. Conversely, β-strand 2–3 along with structurally adjacent β-strand 6 is an area of high sequence divergence. This non-functional side of the SOD1 β-barrel underwent change during early primate evolution that has been conserved in the Hominidae. The absence of ALS mutations in this region may reflect strong recent selection to inhibit SOD1 aggregation as is the case for other characteristics such as charge and thermostability (Dasmeh and Kepp, Reference Dasmeh and Kepp2017) or indicate that substitutions are tolerated. The lack of sequence conservation and the existence of three non-ALS polymorphisms seems to support the latter. We predict that non-ALS allelic variation will be found to be high in this non-functional area of the SOD1 β-barrel as more sequence data become available.
Some SOD1 amino acids are more frequently mutated than others. For example, mutation of 4/4 cysteines but only 1/9 lysine sites is known to be causative for ALS. Glycine is the most common amino acid within the SOD1 sequence (16.2%). Of the 25 glycine residues within SOD1, 15 of those sites (60%) can harbour ALS mutations with a total of 29 different mutations. Gly93, for example, can be found mutated to serine, valine, alanine, cysteine, arginine and aspartic acid. Life expectancy following the onset of disease symptoms occupies a spectrum from 2 to 10 years which appears specific to each Gly93 substitution and also dictates the biophysical properties of the protein (Cudkowicz et al., Reference Cudkowicz, McKenna-Yasek, Sapp, Chin, Geller, Hayden, Schoenfeld, Hosler, Horvitz and Brown1997; Pratt et al., Reference Pratt, Shin, Merz, Rambo, Lancaster, Dyer, Borbat, Poole, Adams, Freed, Crane, Tainer and Getzoff2014).
Loss or gain of function?
SOD1 is abundant within motor neurons (Pardo et al., Reference Pardo, Xu, Borchelt, Price, Sisodia and Cleveland1995); however, reduced SOD activity of roughly 50% was quickly discovered following linkage between ALS and SOD1 (Bowling et al., Reference Bowling, Schulz, Brown and Beal1993; Deng et al., Reference Deng, Hentati, Tainer, Iqbal, Cayabyab, Hung, Getzoff, Hu, Herzfeldt, Roos, Warner, Deng, Soriano, Smyth, Parge, Ahmed, Roses, Hallewell, Pericak-Vance and Siddique1993). This led investigators to suspect that SOD1-ALS was a loss of function disease. The resulting accumulation of superoxide radical, oxidative damage and mitochondrial pathology provided a logical route from SOD1 mutations to neuronal death (Bowling et al., Reference Bowling, Schulz, Brown and Beal1993). Progressing from this work, inhibition of SOD1 by metal chelation or antisense knock-down reduced the abundance of SOD1 protein along with SOD and choline acetyltransferase activities. The resulting oxidative stress, leading to apoptotic cell death, could be repressed with antioxidants (Rothstein et al., Reference Rothstein, Bristol, Hosler, Brown and Kuncl1994; Troy and Shelanski, Reference Troy and Shelanski1994). However, the location of ALS mutations on the SOD1 structure ruled out a direct effect on catalysis (Deng et al., Reference Deng, Hentati, Tainer, Iqbal, Cayabyab, Hung, Getzoff, Hu, Herzfeldt, Roos, Warner, Deng, Soriano, Smyth, Parge, Ahmed, Roses, Hallewell, Pericak-Vance and Siddique1993). Investigating how ALS mutations reduce SOD1 activity, Borchelt et al. (Reference Borchelt, Lee, Slunt, Guarnieri, Xu, Wong, Brown, Price, Sisodia and Cleveland1994) concluded that a range of specific activities could be found for a variety of SOD1 mutants from zero up to 150% of wild-type for Gly85Arg and Gly37Arg, respectively. However, overall Gly37Arg SOD1 activity and stability were reduced in lymphoblasts from an fALS individual. This correlated with the reduction of SOD1 polypeptide half-life in a cell model with Ala4Val and Gly85Arg having the shortest at 7.5 h as opposed to wild-type at roughly 30 h. The first biophysical characterisation of disease-relevant mutations on purified protein was performed by mapping human ALS substitutions onto S. Cerevisiae Cu/ZnSOD (Nishida et al., Reference Nishida, Gralla and Valentine1994). On addition of copper and zinc to the metal-free protein, Gly85Arg was found to have 40% of wild-type activity while Gly93Ala had 80%. The latter could restore insensitivity to oxidative stress in SOD1 knock-out yeast. These results were the first indications that SOD1-ALS does not proceed by a loss of function mechanism.
A small proportion of dominantly inherited diseases are caused by haploinsufficiency; however, diseases with protein loss of function are usually inherited recessively. This led to questioning of the validity of the loss of function hypothesis. A dominant negative mechanism, where the mutant protein effects the functionality of the wild-type, formed the basis of a second hypothesis and, to this end, Gly37Arg SOD1 was shown to heterodimerise with wild-type (Borchelt et al., Reference Borchelt, Lee, Slunt, Guarnieri, Xu, Wong, Brown, Price, Sisodia and Cleveland1994). However, Borchelt et al. (Reference Borchelt, Guarnieri, Wong, Lee, Slunt, Xu, Sisodia, Price and Cleveland1995) found that co-expression of Gly41Asp and Gly85Arg SOD1 mutants with wild-type did not affect the activity or half-life of the wild-type form. Furthermore, high-level transgenic expression of Gly93Ala human SOD1 in mice led to an ALS-like phenotype at 3–4 months old whereas lower expression did not (Gurney et al., Reference Gurney, Pu, Chiu, Dal Canto, Polchow, Alexander, Caliendo, Hentati, Kwon and Deng1994). This forms a parallel with the notion that high expression of SOD1 in spinal motor neurons predisposes them to mutant SOD1-mediated cell death (Pardo et al., Reference Pardo, Xu, Borchelt, Price, Sisodia and Cleveland1995), and SOD1 mutants induce apoptosis in neuronal culture where as the wild-type protein does not (Rabizadeh et al., Reference Rabizadeh, Gralla, Borchelt, Gwinn, Valentine, Sisodia, Wong, Lee, Hahn and Bredesen1995). Borchelt et al. (Reference Borchelt, Guarnieri, Wong, Lee, Slunt, Xu, Sisodia, Price and Cleveland1995) concluded ‘that it is unlikely that a partial loss of free radical scavenging activity underlies motor neuron disease in fALS’ and ‘the dominant character of SOD1-linked fALS is due to a gain of some injurious property of mutant SOD1 subunits’. Indeed, complete knock-out of SOD1 does not cause ALS-like symptoms (Reaume et al., Reference Reaume, Elliott, Hoffman, Kowall, Ferrante, Siwek, Wilcox, Flood, Beal, Brown, Scott and Snider1996). More recently, reduced expression resulting from a 50 bp deletion in the SOD1 promotor region was shown not to cause ALS in humans but also does not change the ALS phenotype in people harbouring mutant SOD1 (Ingre et al., Reference Ingre, Pinto, Birve, Press, Danielsson, de Carvalho, Guđmundsson and Andersen2013). The possible implications of SOD1 loss of enzymatic activity in ALS have been excellently reviewed by Saccon et al. (Reference Saccon, Bunton-Stasyshyn, Fisher and Fratta2013) and will not be described further.
Aggregation, a toxic gain of function
While this work was ongoing, SOD1 was found to be a component of cytoplasmic hyaline inclusions and Lewy bodies in the spinal cord of SOD1 fALS patients (Shibata et al., Reference Shibata, Hirano, Kobayashi, Asayama, Umahara and Ikemoto1993, Reference Shibata, Hirano, Kobayashi, Siddique, Deng, Hung, Kato and Asayama1996a). Bruijn et al. (Reference Bruijn, Becher, Lee, Anderson, Jenkins, Copeland, Sisodia, Rothstein, Borchelt, Price and Cleveland1997) were then able to demonstrate the formation of these aggregates in Gly85Arg SOD1 transgenic mice that express the mutant protein at a level comparable with native mouse SOD1 but which contributes little to cellular SOD activity. The human mutant SOD1 increased in abundance as the mice aged as opposed to the native mouse form which remained static. They found astrocytic SOD1 Lewy-like bodies prior to ALS-like symptom onset and they increased in number as the disease progressed. Within motor neurons, very few indistinct SOD1 aggregates were observed before symptoms which progressed to Lewy-like bodies and irregular inclusions concomitantly with the disease. This combined evidence from mice and humans of abnormal mutant SOD1 aggregation provided a putative gain of toxic function to complement dominant SOD1-related fALS inheritance. A detailed description of SOD1 aggregates will follow but first we look at how ALS mutations affect the SOD1 structure.
Mutant SOD1 structure and phenotype
Of the 11 ALS SOD1 substitution mutations initially discovered by Rosen et al. (Reference Rosen, Siddique, Patterson, Figlewicz, Sapp, Hentati, Donaldson, Goto, O'Regan and Deng1993), six have been described by crystal structures. The complete library of crystallographic and NMR structures of SOD1 now comprises wild-type in various maturation states and mutants from all broad groups including dimer interface, β-barrel, metal-binding and loop regions. Mutants are found in metalation states dependent on mutant characteristics, expression platform, post-purification processing and crystallisation conditions. In some cases, mutant structures are very similar to the wild-type but often the destabilising nature of the resulting structural change is underestimated. The singular finding from this painstaking biophysical characterisation is that all ALS mutations frustrate molecular packing and reduce the likelihood that SOD1 will populate the canonical mature state. There is no single mechanism by which this destabilisation occurs, indeed every conceivable destabilisation strategy is represented; hydrogen bonds are weakened or broken, hydrophobic interactions are negated and salt-bridges removed. Most frequently however, the attractive and repulsive Van der Waals interactions which support the SOD1 structure are disturbed and new repulsive clashes are introduced. What follows is a dissection of the destabilising effects found in SOD1 ALS amino acid substitution mutants available in the Protein Data Bank (PDB) with correlation drawn to phenotypic data (Fig. 5). Where noted, mutations have been introduced into the thermostable SOD1 background (Cys6Ala/Cys111Ser) described by Parge et al. (Reference Parge, Getzoff, Scandella, Hallewell and Tainer1986).
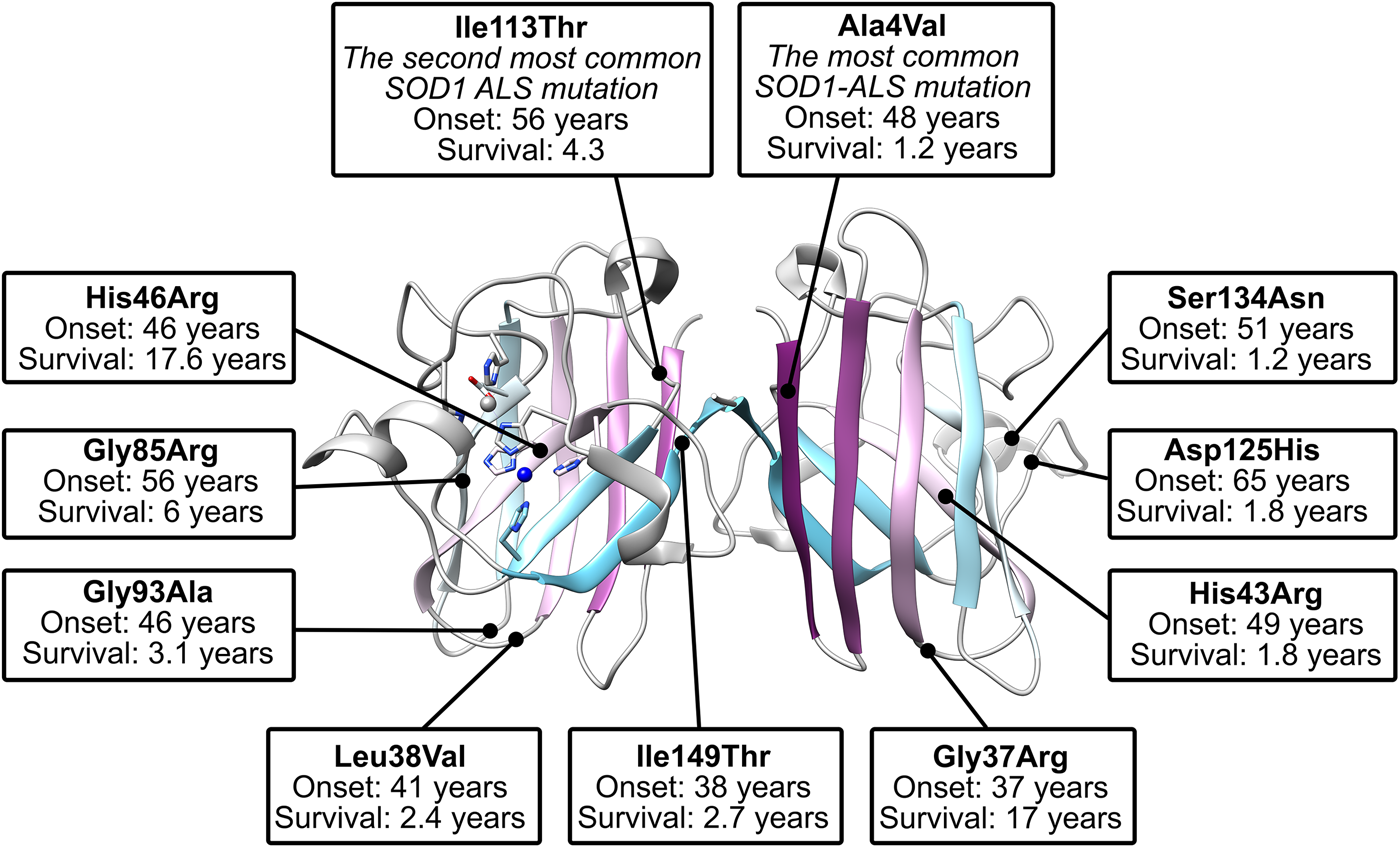
Fig. 5. The location of commonly studied ALS mutants within the SOD1 dimer structure. Shown are those mutants that have been described in crystal structures and have phenotypic data available from more than two cases (Wang et al., Reference Wang, Johnson, Agar and Agar2008).
Gly37Arg
The first structure of a mutant protein related to ALS as a whole was the crystallographic structure of Gly37Arg SOD1 (PDB: 1AZV) (Hart et al., Reference Hart, Liu, Pellegrini, Nersissian, Gralla, Valentine and Eisenberg1998). This mutation is found in loop III, adjacent to the Leu38 β-barrel plug and is conserved in 96% of eukaryotic CuZnSODs. The structure represents mature SOD1 with both metal ions and intra-subunit disulphide. The authors observed asymmetry in copper site geometry and subunit B-factors. As with many ALS-related SOD1 mutant structures, the structural effect of the mutation is subtle. Comparison of wild-type and Gly37Arg SOD1 reveals a steric clash between the Arg37 and loop I Pro13 (Fig. 6). In the wild-type form, the closest distance between these two residues, Gly37 Cα and Pro13 carbonyl oxygen, is 3.8 Å. However, substitution for arginine would reduce this nearest contact between Cβ and Cγ side-chain atoms to 2.3 Å. This is well within the 3.2 Å sum of Van der Waals radii for carbon and oxygen atoms and would be energetically unfavourable. As a result, the backbone of loops I and III distort to accommodate the 3.3 Å separation found in the crystal structure. The Arg37 side-chain has comparatively high B-factors with the exception of the Cβ atom. While the side-chain is conformationally dynamic, the Cβ position is dictated by the position of loop III. Therefore, any mutation of Gly37, which by necessity incorporates an additional Cβ atom, will lead to SOD1 structural frustration and ALS. The reported Gly37Val cases led to a 14-month and 4.6-year disease progression (Kobayashi et al., Reference Kobayashi, Kuroda, Kawata, Mochizuki, Mizutani, Komori, Ikeuchi and Koide2012; Bali et al., Reference Bali, Self, Liu, Siddique, Wang, Bird, Ratti, Atassi, Boylan, Glass, Maragakis, Caress, McCluskey, Appel, Wymer, Gibson, Zinman, Mozaffar, Callaghan, McVey, Jockel-Balsarotti, Allred, Fisher, Lopate, Pestronk, Cudkowicz and Miller2017) in contrast to 16.5 years mean for Gly37Arg (Cudkowicz et al., Reference Cudkowicz, McKenna-Yasek, Sapp, Chin, Geller, Hayden, Schoenfeld, Hosler, Horvitz and Brown1997; Bali et al., Reference Bali, Self, Liu, Siddique, Wang, Bird, Ratti, Atassi, Boylan, Glass, Maragakis, Caress, McCluskey, Appel, Wymer, Gibson, Zinman, Mozaffar, Callaghan, McVey, Jockel-Balsarotti, Allred, Fisher, Lopate, Pestronk, Cudkowicz and Miller2017), possibly due to the increased repulsion presented by two conformationally restricted carbon atoms positioned between loops II and III rather than one.
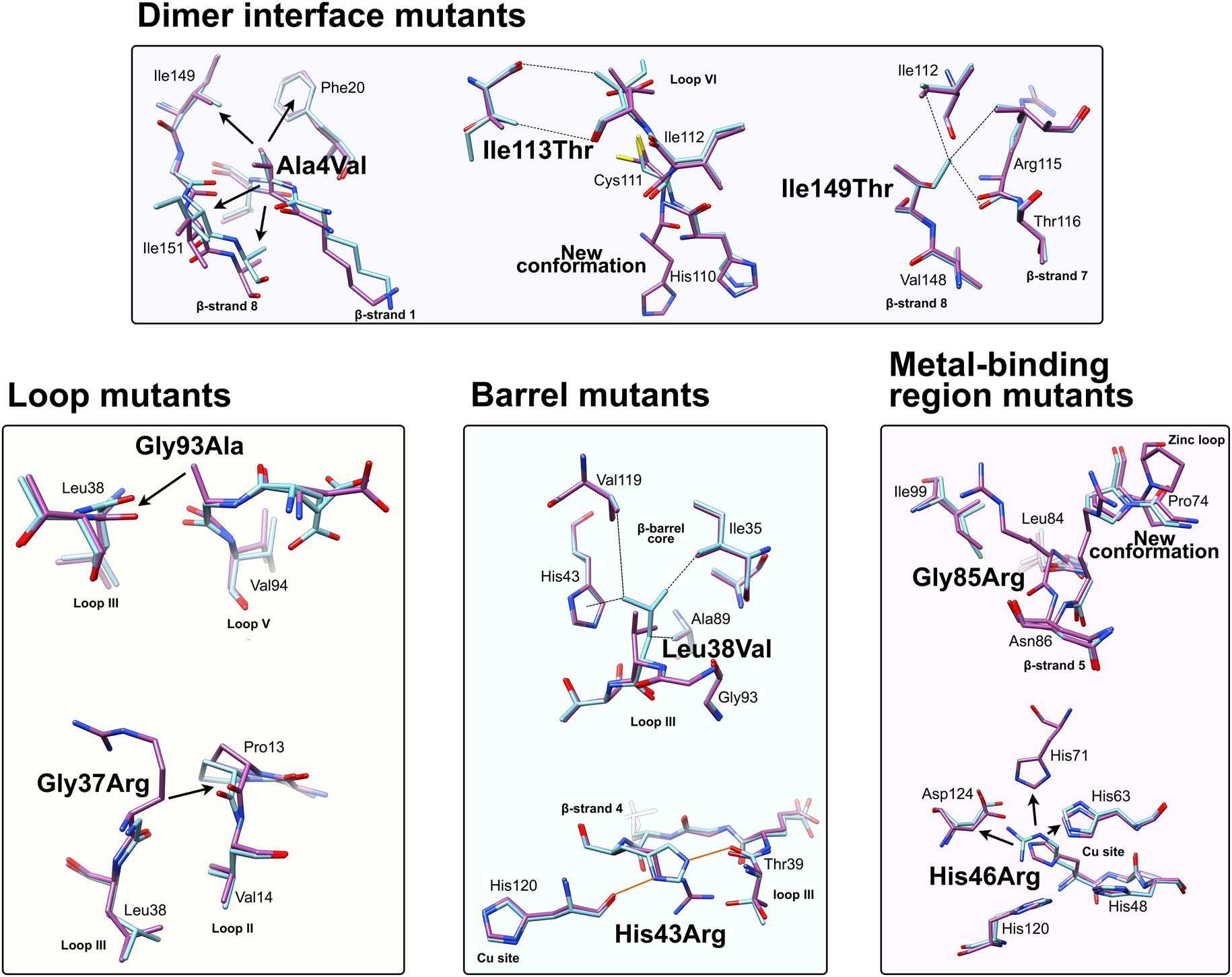
Fig. 6. Local structural destabilisation of SOD1 by ALS mutations in crystallography. Metal binding mutants; Ala4Val shows repulsion around the mutation site while Ile113Thr and Ile149Thr disrupt Van der Waals interactions at the dimer interface and the SOD1 core, respectively. Loop mutants; Gly93Ala causes repulsion between loop III and loop V and Gly37Arg causes repulsion between Loop III and Loop II. β-barrel mutants; Leu38Val disrupts Van der Waals interactions in the SOD1 core and His43Arg breaks a network of stabilising hydrogen bonds. Metal binding-region mutants; Gly85Arg causes repulsion and zinc loop disruption whereas His46Arg prevents copper binding and destabilises the electrostatic loop. Hydrogen bonds are shown in orange lines, repulsive interactions are shown as arrows, Van der Waals interactions are shown as dotted lines.
Ala4Val
Crystallographic characterisation of Ala4Val mutant SOD1, the most frequently occurring ALS SOD1 mutation (Cudkowicz et al., Reference Cudkowicz, McKenna-Yasek, Sapp, Chin, Geller, Hayden, Schoenfeld, Hosler, Horvitz and Brown1997; Wang et al., Reference Wang, Johnson, Agar and Agar2008; Bali et al., Reference Bali, Self, Liu, Siddique, Wang, Bird, Ratti, Atassi, Boylan, Glass, Maragakis, Caress, McCluskey, Appel, Wymer, Gibson, Zinman, Mozaffar, Callaghan, McVey, Jockel-Balsarotti, Allred, Fisher, Lopate, Pestronk, Cudkowicz and Miller2017), also indicated conservation of fold, metalation and disulphide bond but rearrangement around the mutation site situated close to the dimer interface. The valine side-chain is clearly defined by electron density in thermostable (PDB: 1N19) (Cardoso et al., Reference Cardoso, Thayer, DiDonato, Lo, Bruns, Getzoff and Tainer2002) and wild-type (PDB: 1UXM) (Hough et al., Reference Hough, Grossmann, Antonyuk, Strange, Doucette, Rodriguez, Whitson, Hart, Hayward, Valentine and Hasnain2004) backgrounds as it is in the metal-free (PDB: 3GZQ) (Galaleldeen et al., Reference Galaleldeen, Strange, Whitson, Antonyuk, Narayana, Taylor, Schuermann, Holloway, Hasnain and Hart2009) structure. This indicates it is conformationally restricted. Introducing two additional carbon atoms inside the tightly packed hydrophobic core of the β-barrel pushes surrounding Phe20, Leu106, Ile113, Ile149 and Ile151 away from the mutation site (Cardoso et al., Reference Cardoso, Thayer, DiDonato, Lo, Bruns, Getzoff and Tainer2002) (Fig. 6). These permutations change inter-monomer orientation (Hough et al., Reference Hough, Grossmann, Antonyuk, Strange, Doucette, Rodriguez, Whitson, Hart, Hayward, Valentine and Hasnain2004). Mean interface Gly51-Ile151 and Ile151-Gly114 hydrogen bonding distances are decreased 2.04 to 1.99 and 1.93 to 1.87 Å (calculated as acceptor–hydrogen distance or 2.78 to 2.75 and 2.87 to 2.83 Å calculated as acceptor–donor distance) with concomitant changes to bond angles +1.7 and −1.4°. Thus, on average, the Ile151-Gly114 hydrogen bond appears strengthened by the mutation. However, the variability of Ile151-Gly115 hydrogen bond distances and angles observed in Ala4Val SOD1 structures is far higher than those in wild-type structures; standard deviation 0.02–0.14 Å and 1.9–4.5°. This describes a frustrated molecule which is not able to settle into the tightly packed and stable structure occupied by wild-type SOD1. Alanine is found at this position in 88% of eukaryotic CuZnSODs; however, five other ALS-related mutations are found at codon 4, Ala4Ser, Ala4Thr, Ala4Pro, Ala4Asp (Naruse et al., Reference Naruse, Iwata, Takahashi, Ichihara, Kamei, Yamatoku, Hirayama, Suzuki, Aoki, Miyagawa, Shimizu, Tsuji and Goto2013) and Ala4Phe (Baek et al., Reference Baek, Koh, Park, Kim, Kim, Kwon, Ki and Kim2011). Each increases the number of side-chain atoms within the core of the SOD1 β-barrel. While these mutations cause variable onset of ALS, the disease course is invariably short, 7 months to 2 years, as a result.
Gly93Ala
Gly93 is found in loop V where it is distant from metal binding, disulphide and dimer interface regions but is conserved in 90% of eukaryotic CuZnSODs. Six different SOD1 codon 93 mutations are known to cause ALS: Gly93Ala, Gly93Cys, Gly93Ser, Gly93Val, Gly93Asp and Gly93Arg with very similar mean age at onset (47.8 years) but high variance (±14.8 years) and mutant-specific duration variability (Gly93Ala 2.2 ± 1.5 years, Gly93Cys 10.1 ± 6.1 years and Gly93Asp 10.5 ± 5.5 years) (Cudkowicz et al., Reference Cudkowicz, McKenna-Yasek, Sapp, Chin, Geller, Hayden, Schoenfeld, Hosler, Horvitz and Brown1997). Comparison of metalated Gly93Ala SOD1 (PDB: 3GZP, 3GZO, 2WKO) with wild-type SOD1 shows that introduction of the alanine Cβ would cause a steric clash with the loop III Leu38 carbonyl (2.7 ± 0.1 Å). To mitigate this effect, the Leu38 peptide bond rotates towards the SOD1 core increasing this distance to 3.1 ± 0.1 Å. In addition, the peptide bonds of residues 92 and 93 rotate moving their carbonyls away from the core and shifting the overall position of loop V away from Leu38 in the loop III (Galaleldeen et al., Reference Galaleldeen, Strange, Whitson, Antonyuk, Narayana, Taylor, Schuermann, Holloway, Hasnain and Hart2009) (Fig. 6).
Leu38Val
Leu38 caps one end the β-barrel with its side-chain inserted into the hydrophobic core. This arrangement creates stabilising Van der Walls interactions with Val14, Ile35, His43, Ala89, Ala95, Val119 and Leu144. It is conserved in 84% of eukaryotic CuZnSODs. The atomic resolution crystal structure of Leu38Val SOD1 (PDB: 2WYT) shows interactions with Ile35, Ala95, Val119 are lengthened by substitution for valine, and without rearrangement to accommodate the substitution, clashes would be created between Val38, His43, Ala89 side chains and Gly93 carbonyl (Antonyuk et al., Reference Antonyuk, Strange and Hasnain2010). A favourable rotamer is selected to minimise clashes with His43; however, the backbone must distort slightly due to repulsion between the Val38 and Ala89 side-chains. In turn, this pushes Val14 away from the site of mutation (Fig. 6). The overall effect of Leu38Val is therefore to reduce structural cohesion within the core of the SOD1 β-barrel. In 10% of eukaryotic CuZnSODs, an aspartic acid substitutes for leucine underlining the importance of maintaining packing through Van der Waals interactions in the core of the molecule.
The Leu38Arg mutation is also associated with ALS. Here, the extra side-chain length must create highly destabilising clashes with the amino acids which normally form stabilising interactions with Leu38. Phenotypic data for Leu38Arg are sparse but show typical disease onset and progression (Millecamps et al., Reference Millecamps, Salachas, Cazeneuve, Gordon, Bricka, Camuzat, Guillot-Noël, Russaouen, Bruneteau, Pradat, Le Forestier, Vandenberghe, Danel-Brunaud, Guy, Thauvin-Robinet, Lacomblez, Couratier, Hannequin, Seilhean, Le Ber, Corcia, Camu, Brice, Rouleau, LeGuern and Meininger2010) while Leu38Val yields early-onset ALS with a very short disease course. These effects are comparable with mutation of Leu106 which caps the opposite end of the SOD1 β-barrel and forms hydrophobic Van der Waals interactions with Ala4, Phe20, Glu22, Ile112 and Ile113. Many of these interactions would be weakened or become repulsive on mutation to valine. Like Leu38Val, Leu104Val gives rise to ALS with early onset and short duration (Cudkowicz et al., Reference Cudkowicz, McKenna-Yasek, Sapp, Chin, Geller, Hayden, Schoenfeld, Hosler, Horvitz and Brown1997). Leu106Phe increases the side-chain mass inside the SOD1 core like Leu38Arg, likely creating clashes with Phe20 and Val29. This causes a disease with typical age at onset but faster than usual progression (Battistini et al., Reference Battistini, Ricci, Lotti, Benigni, Gagliardi, Zucco, Bondavalli, Marcello, Ceroni and Cereda2010).
Gly85Arg
Structural perturbations arising from substituting arginine for Gly85 are more pronounced in comparison with those described above. Within the SOD1 monomers found in seven available crystal structures (PDB: 2VR6, 2VR7, 2VR8, 2ZKW, 2ZKX, 3CQP, 3CQQ), the mutation site adopts two different backbone conformations. Eleven of 18 are found in a wild-type conformation with the arginine side-chain extending perpendicularly from β-strand 5 and clashing with zinc loop Pro74. Alternatively, seven of 18 monomers show a 180° rotation around the Leu84 Cα-C ψ bond, which is accommodated by a further rotation around the Asn86 N-Cα ϕ bond, and the arginine side-chain extends towards β-strand 6. In the latter case, zinc loop Pro74 is pushed away from the mutation site slightly changing the position of the upper zinc loop. In the former case, the steric clash between arginine and Pro74 is too severe to mitigate with a similar small shift and Pro74 rotates almost 90° (Fig. 6). This disrupts hydrogen bonding between Pro74 and Arg79, and destabilises or reorients the upper zinc-binding and electrostatic loops with knock-on effects on metal binding (Cao et al., Reference Cao, Antonyuk, Seetharaman, Whitson, Taylor, Holloway, Strange, Doucette, Valentine, Tiwari, Hayward, Padua, Cohlberg, Hasnain and Hart2008). A Gly85Ser substitution has also been observed with early onset and fast disease progression (Takazawa et al., Reference Takazawa, Ikeda, Hirayama, Kawabe, Nakamura, Ito, Kano, Yoshii, Tanaka, Sobue and Iwasaki2010).
Ile113Thr
Ile113Thr is the second most common SOD1 ALS mutation (Bali et al., Reference Bali, Self, Liu, Siddique, Wang, Bird, Ratti, Atassi, Boylan, Glass, Maragakis, Caress, McCluskey, Appel, Wymer, Gibson, Zinman, Mozaffar, Callaghan, McVey, Jockel-Balsarotti, Allred, Fisher, Lopate, Pestronk, Cudkowicz and Miller2017). Ile113 residues interact across the SOD1 dimer interface; Van der Waals interactions are present between the Ile113 side chain, the opposing Ile113 carbonyl oxygen and also Gly114 Cα. These inter-molecular interactions are lengthened by the Ile113Thr substitution (PDB: 1UXL) (Fig. 6). In addition, new repulsive interactions are created with Ile151 side-chain and Cys111 sulphydryl. While there are no changes to interface hydrogen bonding in the crystal structure of Ile113Thr, the noted effects on interactions with Ile151 and Gly114 may affect the strength of these bonds in solution when higher energy states are accessible. As a result of the substitution, Cys111 adopts two side-chain rotamers and a new loop VI conformation, which extends up to Leu106, is stabilised in 30% of monomers. As noted by Hough et al. (Reference Hough, Grossmann, Antonyuk, Strange, Doucette, Rodriguez, Whitson, Hart, Hayward, Valentine and Hasnain2004), a hydrophobic contact with Ala4 is also lost leading to destabilisation compared to wild-type. These structural changes resulting from the mutation are subtle, and while Ile113 is conserved in 58% of eukaryotic CuZnSODs, it is often replaced by valine or leucine indicating a tolerance of these conservative substitutions. This is reflected in late disease onset, longer than average disease course and incomplete penetrance (Suthers et al., Reference Suthers, Laing, Wilton, Dorosz and Waddy1994; Cudkowicz et al., Reference Cudkowicz, McKenna-Yasek, Sapp, Chin, Geller, Hayden, Schoenfeld, Hosler, Horvitz and Brown1997).
Ile149Thr
The Ile149 side-chain Cδ1 has Van der Waals interactions with Val47, Ile112, Val148 and Leu117. The structure of Ile149Thr in a thermostable background (PDB: 4OH2) shows space is created in the core of the molecule on mutation to threonine and these hydrophobic interactions are lost. Ile112 relaxes into the newly available space as does the Arg115 peptide bond carbonyl which rotates, possibly weakening the hydrogen bond with Arg115 which links β-strands 7 and 8 (Fig. 6). Ile113 and Ile149 are foundation residues, they serve only structural purposes, there are no serious repulsive clashes generated by the conservative mutations described but a vacuum is created in the molecule which undermines structural cohesion.
His43Arg
His43 is found on β-strand 4 and forms strong hydrogen bonds with Thr39 in loop III and the copper coordinating ligand His120 in loop VII, the electrostatic loop. Held in this position, His43 provides support to Leu38 through Van der Waals interactions. The crystal structure of His43Arg, in a thermostable background (PDB: 1PTZ), shows the arginine maintaining hydrogen bonding with Thr39 and making a further hydrogen bond with Glu40 through its side-chain guanidinium group but loses the interaction with His120. To avoid a major steric clash with loop III, the Arg43 side-chain adopts a rotamer shifted away from Leu38 (Fig. 6). This reduces buried packing surface between the two residues fivefold and removes many of the Van der Waals interactions provided by His43 (DiDonato et al., Reference DiDonato, Craig, Huff, Thayer, Cardoso, Kassmann, Lo, Bruns, Powers, Kelly, Getzoff and Tainer2003).
His46Arg
His46 coordinates copper in the SOD1 active site and as a result is ubiquitously conserved across eukaryotic CuZnSODs. Large tertiary structure defects arising from His46Arg mutation are clear in crystallo (PDB: 1OZT and 1OEZ); in both metal-free and zinc-bound states, the electrostatic and zinc-binding loops are destabilised which enables non-native loop and β-barrel interactions between neighbouring molecules (Elam et al., Reference Elam, Taylor, Strange, Antonyuk, Doucette, Rodriguez, Hasnain, Hayward, Valentine, Yeates and Hart2003a). This results from: (1) Lost His71-zinc coordination, changing both geometry, position and very likely affinity for zinc. (2) Clashes with copper and zinc coordinating His63 pushing it away from the site of mutation with similar effects. (3) Clashes with electrostatic loop Asp124, Thr137 and Ala140 (Fig. 6). His46Arg SOD1-related ALS presents at the typical age for SOD1 ALS but has a very long disease course which can last more than 20 years (Aoki et al., Reference Aoki, Ogasawara, Matsubara, Narisawa, Nakamura, Itoyama and Abe1993; Bali et al., Reference Bali, Self, Liu, Siddique, Wang, Bird, Ratti, Atassi, Boylan, Glass, Maragakis, Caress, McCluskey, Appel, Wymer, Gibson, Zinman, Mozaffar, Callaghan, McVey, Jockel-Balsarotti, Allred, Fisher, Lopate, Pestronk, Cudkowicz and Miller2017). Please see ‘Structural instability’ for a discussion of the relationship between His46Arg stability and phenotype.
Ser134Asn, Asp125His, Asp124Val and His80Arg
The electrostatic loop mutation Ser134Asn has much the same effect on SOD1 as His46Arg despite the very different location (Elam et al., Reference Elam, Taylor, Strange, Antonyuk, Doucette, Rodriguez, Hasnain, Hayward, Valentine, Yeates and Hart2003a). While the Asn134 residue itself cannot be seen in the crystal structure (1OZU) due to disorder in the electrostatic loop, the reasons are clear; severe clashes with loop residues 125, 127–131 which prevent adoption of the canonical helical structure. Ser134 mutation to asparagine prevents adequate metal binding leading to hydrophobicity, abnormal intermolecular contacts and aggregation (Hayward et al., Reference Hayward, Rodriguez, Kim, Tiwari, Goto, Cabelli, Valentine and Brown2002; Elam et al., Reference Elam, Taylor, Strange, Antonyuk, Doucette, Rodriguez, Hasnain, Hayward, Valentine, Yeates and Hart2003a; Banci et al., Reference Banci, Bertini, D'Amelio, Gaggelli, Libralesso, Matecko, Turano and Valentine2005; Marucci et al., Reference Marucci, Morandi, Bartolomei, Salvi, Pession, Righi, Lauria and Foschini2007). Counterintuitively however, the Ser134Asn phenotype is not aggressive despite the above molecular characteristics. Two heterozygous individuals developed late-onset ALS (63 and 52 years old) with variable progression (9 months and at least 2 years) while another heterozygous individual had no symptoms at 70 years old (Watanabe et al., Reference Watanabe, Aoki, Abe, Shoji, Iizuka, Ikeda, Hirai, Kurokawa, Kato, Sasaki and Itoyama1997; Marucci et al., Reference Marucci, Morandi, Bartolomei, Salvi, Pession, Righi, Lauria and Foschini2007). A fourth individual, who had trisomy 21 Down's syndrome and two copies of the Ser134Asn SOD1 allele, developed ALS at 34 years of age which progressed over 3 years (Marucci et al., Reference Marucci, Morandi, Bartolomei, Salvi, Pession, Righi, Lauria and Foschini2007) and is reminiscent of the gene dosage effect observed in homozygous Asp90Ala ALS patients.
The crystal structure of Asp125His (PDB: 1P1V) (Elam et al., Reference Elam, Malek, Rodriguez, Doucette, Taylor, Hayward, Cabelli, Valentine and Hart2003b) shows very similar destabilisation of the electrostatic loop between amino acids 129 and 135 as described for Ser134Asn. This is due to steric clashes with Gly126, Ser134 and Asn139 that prevent folding of the electrostatic loop helix. The clash with Ser134 causes the mutated His125 to flip rotamer and project into the solvent in two of three cases. In addition, a hydrogen bond which links Gly127 and the Asp125 side-chain is broken. Individuals with this mutation died of ALS at age 57 and 71, the latter following a 20-month disease duration (Enayat et al., Reference Enayat, Orrell, Claus, Ludolph, Bachus, Brockmüller, Ray-Chaudhuri, Radunovic, Shaw and Wilkinson1995).
Asp124Val also causes destabilisation of electrostatic loop residues 124–139 and upper zinc loop amino acids 66–79. The crystal structure of this mutant (PDB: 3H2P) shows the bonding network which links copper coordinating His46 through Asp124 to zinc coordinating His71 is lost on mutation to valine (Seetharaman et al., Reference Seetharaman, Winkler, Taylor, Cao, Whitson, Doucette, Valentine, Schirf, Demeler, Carroll, Culotta and Hart2010). This removes a vital stabilising link between the β-barrel core, disulphide sub-loop and electrostatic loop. Metal binding is consequently compromised.
His80Arg is the only verified de novo SOD1 mutation known to have caused ALS. Crystal structures of His80Arg SOD1 (PDB: 3QQD and 3H2Q) show destabilisation of the upper zinc and electrostatic loops due to complete loss of the zinc site and severe steric clashes with both His71 and Lys136 (Seetharaman et al., Reference Seetharaman, Winkler, Taylor, Cao, Whitson, Doucette, Valentine, Schirf, Demeler, Carroll, Culotta and Hart2010). His80Arg has been found in only one person who presented with ALS symptoms at 24 years of age and died 18 months later (Alexander et al., Reference Alexander, Traynor, Miller, Corr, Frost, McQuaid, Brett, Green and Hardiman2002). This early disease onset would reduce the propagation of this mutation through future generations and illustrates that the majority of SOD1 mutations have been able to persist because they generally cause disease after the reproductive period of human life.
His46, His80, Ser134, Asp125 and Asp124 are ubiquitous across eukaryotic CuZnSODs. With the exception of His80, mutation of these residues gives rise to forms of ALS where the disease presents late in life or with a protracted disease course. These mutations, to very highly conserved amino acids, often scramble or completely prevent important metal binding and manifest as phenotypically less severe than Leu38 and Leu106 mutant ALS. Mutations outside of the β-barrel core and dimerisation regions appear better tolerated by some molecular or cellular mechanism.
SOD1 maturation and ALS mutants
Following ribosomal translation, the progression from nascent and unfolded protein to the active and mature enzyme is crucially important in the production of SOD1. This post-translational maturation pathway includes folding, metal binding, intra-subunit disulphide formation and dimerisation. Appraisal of the mutant SOD1 crystal structures described above in conjunction with a wealth of biophysical, biochemical and cell biology research reveals common themes of structural instability, incomplete or aberrant metalation, disulphide reduction, dimerisation defects and propensity to exist in an unfolded state. Below we review the post-translational processes in the sequence they are likely to occur within cells and highlight how the different steps can be affected by ALS mutations paying particular attention to those SOD1 mutants which do not fit overall trends.
Folding
SOD1 folding can happen spontaneously or aided by the human copper chaperone for SOD1 (hCCS), but does not require ATP-dependent chaperones (Bruns and Kopito, Reference Bruns and Kopito2007; Luchinat et al., Reference Luchinat, Barbieri and Banci2017). By analysing protease K-catalysed degradation following SOD1 translation, Bruns and Kopito (Reference Bruns and Kopito2007) showed that copper binding has no effect on folding. Conversely, removal of zinc from the folding environment prevents transformation into a highly protease-resistant form but not a moderately resistant intermediate. This is indicative of a two-step folding mechanism that comprises sequential zinc-independent β-barrel formation and zinc-dependent loop ordering. Analogously, using chaotropic unfolding, Lindberg et al. (Reference Lindberg, Normark, Holmgren and Oliveberg2004, Reference Lindberg, Byström, Boknäs, Andersen and Oliveberg2005) described a two-step folding mechanism for metal-free SOD1 with the denatured monomer folding independently through a transition state prior to a dimerisation event. Monomer folding is slow when compared to other two state proteins but in as expected for highly β-structure-rich proteins (Kayatekin et al., Reference Kayatekin, Cohen and Matthews2012). Folding is the rate-limiting step and subsequent dimerisation is limited only by diffusion. Different SOD1 mutations perturb either one or both of the steps in this process and drive the protein towards immature states (Lindberg et al., Reference Lindberg, Byström, Boknäs, Andersen and Oliveberg2005).
Folding in the presence of zinc is faster than for the metal-free SOD1; however, the low affinity of the nascent protein for zinc may mean folding occurs slowly through metal-free species and zinc is bound later (Kayatekin et al., Reference Kayatekin, Zitzewitz and Matthews2008). Metal-free SOD1 monomer folding is nucleated by hydrophobic side-chain interactions within the core of the β-barrel on β-strands 1, 2, 3, 4 and 7 (Nordlund and Oliveberg, Reference Nordlund and Oliveberg2006) (Fig. 7). This folding nucleus operates independent of loop structures and metal binding with the enthalpic barrier to monomer folding related to the dehydration of these hydrophobic core residues (Kayatekin et al., Reference Kayatekin, Cohen and Matthews2012; Yang et al., Reference Yang, Wang, Logan, Mu, Danielsson and Oliveberg2018). The primary sequence distance between β-strands 4 and 7 (70 residues) offers a rationale for the relatively slow folding of β-rich SOD1 and the susceptibility for aggregation. The retention of N-terminal β-strands in the core of SOD1 aggregate structures could also be a facet of their swift assembly following translation while other strands involved in the transition state structure leave the ribosome much later.
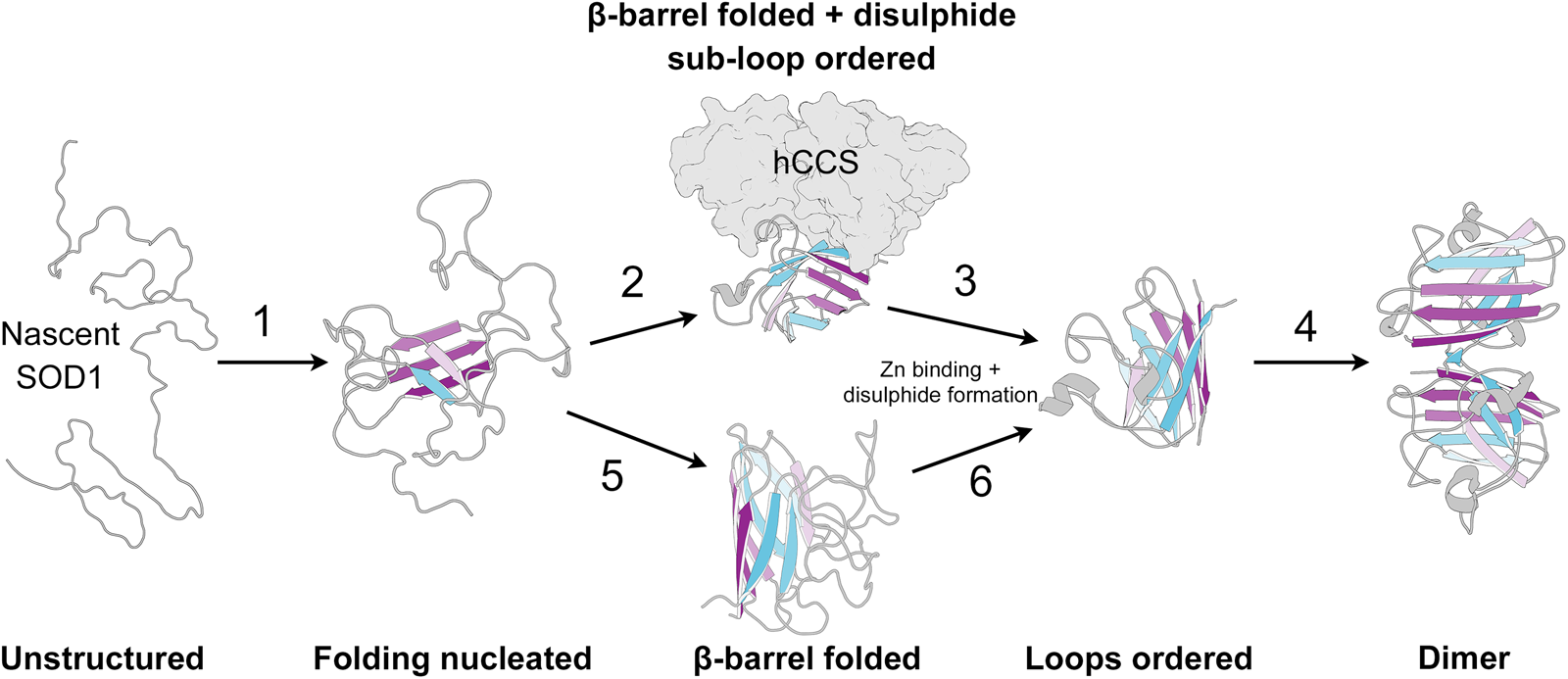
Fig. 7. The SOD1 folding pathway. Step 1: Nascent and completely unstructured SOD1 folding is nucleated by residues in β-strands 1, 2, 3, 4 and 7. Step 2: The molecular chaperone activity of hCCS then promotes folding of the remaining β-barrel structure and disulphide subloops. Step 3: SOD1 binds zinc and disulphide formation imparts stability on the disulphide sub-loop. This weakens hCCS-SOD1 heterodimer affinity. Step 4: Strong SOD1 interface Gly51-Ile151 hydrogen bonding and a stable dimer interface both promote SOD1 homodimerisation. Steps 5 and 6: hCCS mediated folding can be circumvented through spontaneous β-barrel organisation and zinc binding however the mechanism of disulphide formation, and therefore the formation of a stable dimer interface, through this hCCS-independent route is not entirely clear. SOD1 β-strands are coloured as in Fig. 2.
Metal binding
SOD1 is thought to acquire free zinc from its surroundings while copper is acquired from hCCS or an hCCS-independent route (Subramaniam et al., Reference Subramaniam, Lyons, Liu, Bartnikas, Rothstein, Price, Cleveland, Gitlin and Wong2002; Carroll et al., Reference Carroll, Girouard, Ulloa, Subramaniam, Wong, Valentine and Culotta2004). Zinc binding may occur in the free monomer state, or following chaperone-assisted folding when complexed with hCCS (Potter et al., Reference Potter, Zhu, Shaw, Rodriguez, Doucette, Sohn, Durazo, Faull, Gralla, Nersissian and Valentine2007; Kayatekin et al., Reference Kayatekin, Zitzewitz and Matthews2008; Leinartaite et al., Reference Leinartaite, Saraboji, Nordlund, Logan and Oliveberg2010; Banci et al., Reference Banci, Barbieri, Bertini, Luchinat, Secci, Zhao and Aricescu2013; Boyd et al., Reference Boyd, Liu, Bulla and Winkler2018). The canonical β-barrel fold of mature wild-type SOD1 is retained on the removal of metal cofactors under test-tube conditions (Wood et al., Reference Wood, Dalgleish and Bannister1971). In the metal-free state, the electrostatic and zinc loops have a high degree of disorder; however, lack of copper metalation does not significantly change the loop structure (Strange et al., Reference Strange, Antonyuk, Hough, Doucette, Rodriguez, Hart, Hayward, Valentine and Hasnain2003; Sekhar et al., Reference Sekhar, Rumfeldt, Broom, Doyle, Bouvignies, Meiering and Kay2015). The stabilising effect of zinc was discovered by Forman and Fridovich (Reference Forman and Fridovich1973), and our understanding of loop ordering on zinc binding reinforces that notion. However, zinc also positions the redox potential of the copper ion to enable catalytic cycling between Cu(I) and Cu(II) states (Nedd et al., Reference Nedd, Redler, Proctor, Dokholyan and Alexandrova2014). Furthermore, characterisation of Candida albicans SOD5 superoxide dismutase showed this enzyme has an intact copper site but ALS-like mutations to zinc site His71 and His80 (human numbering) which prevent zinc binding (Gleason et al., Reference Gleason, Galaleldeen, Peterson, Taylor, Holloway, Waninger-Saroni, Cormack, Cabelli, Hart and Culotta2014). As a result, C. albicans SOD5 has reduced copper affinity compared with other CuZnSODs (Robinett et al., Reference Robinett, Culbertson, Peterson, Sanchez, Andes, Nett and Culotta2018). SOD5 is an extracellular enzyme which likely gains its metal cofactor through release of copper during host oxidative burst. Conversely, intracellular CuZnSODs, including human SOD1, must compete for copper with the high chelation capacity of the cellular interior making zinc binding a necessity. Thus, zinc binding serves roles in folding, stability, copper acquisition and catalysis.
The absence of free copper salts in biological systems was posited by Mann and Keilin (Reference Mann and Keilin1938a) at the outset of SOD1 research. The intracellular chelation capacity of yeast, for example, means that of the estimated 390 000 copper atoms present in the cell, less than one ion exists free in solution (Rae et al., Reference Rae, Schmidt, Pufahl, Culotta and O'Halloran1999). To service demand for the SOD1 copper cofactor, a relay system is necessary in order to prevent intra-cellular metal scavengers from limiting supply. Proteins responsible for the cellular trafficking of metals are termed metallochaperones and act primarily as sequestration and transport devices. Metallochaperones assist in the prevention of incorrect metal ion coordination resulting from the preference of metal binding proteins to coordinating metals at the high end of the Irving–Williams series. Sequestration also mitigates the hazard of reactive metals in solution, the case in point being free radical production by copper through redox chemistry.
Identification, structure and function of hCCS
The discovery of HAH1 and COX17 copper chaperones for the copper transporting P-type ATPases and cytochrome c oxidase, respectively, spurred the search for a protein that activated SOD1 with copper. Culotta et al. (Reference Culotta, Klomp, Strain, Casareno, Krems and Gitlin1997) discovered the copper chaperone for SOD1 (CCS) through the similarity of SOD1 and LYS7 yeast knock-out strains' oxygen-dependent inability to synthesise lysine and methionine. hCCS was discovered by EST sequence comparison with LYS7 and found to be expressed in all human cell types. Within the central nervous system, hCCS was found to be relatively abundant within motor neurons, deep cerebellar neurons and cortical neurons overlapping the distribution of SOD1 (Rothstein et al., Reference Rothstein, Dykes-Hoberg, Corson, Becker, Cleveland, Price, Culotta and Wong1999).
hCCS comprises two distinct domains which are connected by flexible linkers that allow a high degree of conformational freedom. In addition, an extended, largely unstructured region is found at the C-terminus (Wright et al., Reference Wright, Hasnain and Grossmann2011; Sala et al., Reference Sala, Wright, Antonyuk, Garratt and Hasnain2019). hCCS domain II (amino acids 86–234) is 47% identical to human SOD1 (Culotta et al., Reference Culotta, Klomp, Strain, Casareno, Krems and Gitlin1997; Casareno et al., Reference Casareno, Waggoner and Gitlin1998). Crystal structures of hCCS show that domain II has an anti-parallel Greek-key β-barrel and harbours a dimerisation interface very similar to that of SOD1 (Lamb et al., Reference Lamb, Wernimont, Pufahl, Culotta, O'Halloran and Rosenzweig1999; Sala et al., Reference Sala, Wright, Antonyuk, Garratt and Hasnain2019). This SOD1-like domain mediates a direct interaction with SOD1 (Casareno et al., Reference Casareno, Waggoner and Gitlin1998). While zinc binding is conserved between SOD1 and hCCS domain II, copper binding is not (Schmidt et al., Reference Schmidt, Ramos-Gomez and Culotta1999). Furthermore, loss of zinc prevents hCCS homodimerisation and heterodimerisation with SOD1 thereby reducing its ability to activate SOD1 (Endo et al., Reference Endo, Fujii, Sato, Taniguchi and Fujii2000; Allen and Dennison, Reference Allen and Dennison2014; Wright et al., Reference Wright, Antonyuk and Hasnain2016).
The first 85 amino acids, domain I, of hCCS and yCCS are homologous to the Atx1-like copper-binding proteins and contain the consensus MHCXXC copper (I)-binding motif which is present in HAH1, ATOX1, and both copper-transporting ATPases. Domain I binds copper (I) in a bis-cysteine arrangement with 1015 to 1018 M affinity (Brown et al., Reference Brown, Torres, Doan and O'Halloran2004; Banci et al., Reference Banci, Bertini, Ciofi-Baffoni, Kozyreva, Zovo and Palumaa2010, Reference Banci, Bertini, Cantini, Kozyreva, Massagni, Palumaa, Rubino and Zovo2012a; Allen et al., Reference Allen, Badarau and Dennison2012b) without significant main chain or side chain perturbations by comparison of NMR structures PDB: 2CRL and 2RSQ produced by the Yokoyama and Banci groups, respectively. The affinity of domain I for copper is less than that of the tetra/tri-histidine SOD1 site, therefore copper flow along the hCCS trafficking route is uni-directional (Banci et al., Reference Banci, Bertini, Ciofi-Baffoni, Kozyreva, Zovo and Palumaa2010). Heterodimer formation between SOD1 and hCCS along with the concerted action of domain I and a CSC motif in the C-terminal tail facilitates the delivery of both copper and the disulphide bond to the immature SOD1 substrate (Culotta et al., Reference Culotta, Klomp, Strain, Casareno, Krems and Gitlin1997; Furukawa et al., Reference Furukawa, Torres and O'Halloran2004).
The role of hCCS in SOD1-ALS
Mouse CCS knock-out does not change disease onset or life span in Gly37Arg, Gly93Ala and Gly85Arg SOD1 transgenic mice (Subramaniam et al., Reference Subramaniam, Lyons, Liu, Bartnikas, Rothstein, Price, Cleveland, Gitlin and Wong2002). Conversely, overexpression of hCCS in a mutant SOD1 background has a profound negative effect on disease; Gly93Ala SOD1/hCCS double transgenic mice have a mean survival of 36 days in comparison with 242 for single transgenic Gly93Ala SOD1 mice (Son et al., Reference Son, Puttaparthi, Kawamata, Rajendran, Boyer, Manfredi and Elliott2007; Proescher et al., Reference Proescher, Son, Elliott and Culotta2008). Reduced life span is associated with quickly appearing vacuolar mitochondrial pathology. This is of note because CCS can direct SOD1 to the mitochondria where aggregation on membranes or within the intermembrane space is observed in ALS mutant cell and animal models (Deng et al., Reference Deng, Shi, Furukawa, Zhai, Fu, Liu, Gorrie, Khan, Hung, Bigio, Lukas, Dal Canto, O'Halloran and Siddique2006; Kawamata and Manfredi, Reference Kawamata and Manfredi2008; Vande Velde et al., Reference Vande Velde, Miller, Cashman and Cleveland2008; Oladzad Abbasabadi et al., Reference Oladzad Abbasabadi, Javanian, Nikkhah, Meratan, Ghiasi and Nemat-Gorgani2013). In contrast, transgenic mice over expressing hCCS or wild-type SOD1/hCCS have normal life-span with no mitochondrial swelling (Son et al., Reference Son, Puttaparthi, Kawamata, Rajendran, Boyer, Manfredi and Elliott2007). Supplementation of copper effectively confers wild-type-like survival of Gly93Ala SOD1/hCCS overexpressing mice (Williams et al., Reference Williams, Trias, Beilby, Lopez, Labut, Samuel Bradford, Roberts, McAllum, Crouch, Rhoads, Pereira, Son, Elliott, Franco, Estévez, Barbeito and Beckman2016).
Incomplete metalation
Incomplete SOD1 metalation as a result of metal-binding ligand substitution was first observed by Banci et al. (Reference Banci, Bertini, Cabelli, Hallewell, Tung and Viezzoli1991), prior to the establishment of a link between SOD1 and ALS. One of the mutations described, Asp124Gly, was later shown to be associated with ALS (Andersen et al., Reference Andersen, Sims, Xin, Kiely, O'Neill, Ravits, Pioro, Harati, Brower, Levine, Heinicke, Seltzer, Boss and Brown2003). Following linkage with ALS, Deng et al. (Reference Deng, Hentati, Tainer, Iqbal, Cayabyab, Hung, Getzoff, Hu, Herzfeldt, Roos, Warner, Deng, Soriano, Smyth, Parge, Ahmed, Roses, Hallewell, Pericak-Vance and Siddique1993) predicted that the Gly85Arg SOD1 mutation would disturb His46 and Asp83 metal site co-ordinating ligands. This was proven shortly after by incorporating the mutation into yeast CuZnSOD (Nishida et al., Reference Nishida, Gralla and Valentine1994). Gly85Arg SOD1 activity was lost on addition of stoichiometric amounts of ethylenediaminetetraacetic acid, whereas wild-type and Gly93Ala retained their activity after the addition of more than 2000-fold excess. Visible light electronic absorption spectroscopy confirmed that zinc site geometry had indeed been compromised by the Gly85Arg substitution.
With the exception of the bridging ligand His63, every metal coordinating ligand (His63, His48, His71, His80, Asp83 and His120) has been found mutated in cases of ALS. On examination of the crystal structures of SOD1 where mutations are found in metal coordinating residues or close to metal sites, there is clear and rationalisable loss of metal cofactors. However, many non-metal-binding region SOD1 mutations including Ala4Val, Leu39Val, Gly41Ser, GLy85Arg, Ile113Thr, Gly37Arg and Gly72Ser also have reduced metalation when expressed in heterologous systems (Hayward et al., Reference Hayward, Rodriguez, Kim, Tiwari, Goto, Cabelli, Valentine and Brown2002; Luchinat et al., Reference Luchinat, Barbieri, Rubino, Kozyreva, Cantini and Banci2014). Furthermore, metal-free, disulphide-reduced Gly85Arg, His46Arg, Val148Gly and Glu100Gly SOD1 variants do not adopt a transient electrostatic loop conformational sub-population found in 2% of the metal-free, disulphide-reduced wild-type protein (Sekhar et al., Reference Sekhar, Rumfeldt, Broom, Doyle, Bouvignies, Meiering and Kay2015) that resembles the helix found in the mature protein. In the case of Ala4Val, Gly93Ala and Gly37Arg access to this conformation is partially restricted (Sekhar et al., Reference Sekhar, Rumfeldt, Broom, Doyle, Sobering, Meiering and Kay2016). While this helix does not contain any of the zinc coordinating residues, it folds adjacent to the zinc site and may aid acquisition of zinc. On addition of zinc, the electrostatic loop loses its conformational plasticity coalescing into the conformation found in the mature form (Strange et al., Reference Strange, Antonyuk, Hough, Doucette, Rodriguez, Hart, Hayward, Valentine and Hasnain2003; Culik et al., Reference Culik, Sekhar, Nagesh, Deol, Rumfeldt, Meiering and Kay2018).
The dissociation constants for zinc and copper binding to wild-type SOD1 were determined to be 10−14 and 10−18 M−1, respectively (Crow et al., Reference Crow, Sampson, Zhuang, Thompson and Beckman1997). Eighteen to 30-fold reduction in zinc affinity for Ala4Val, Ala4Thr, Ile113Thr and Leu38Val SOD1 was demonstrated while the reduction in copper affinity was much less severe. Within cells, and even with supplementation of growth media with zinc, many of these mutants fail to adequately zinc metalate and remain in an unstructured state (Luchinat et al., Reference Luchinat, Barbieri, Rubino, Kozyreva, Cantini and Banci2014). Absence of metals leads to the formation of high molecular mass SOD1 aggregates on incubation of purified protein at body temperature (Banci et al., Reference Banci, Bertini, Durazo, Girotto, Gralla, Martinelli, Valentine, Vieru and Whitelegge2007; Wright et al., Reference Wright, Antonyuk, Kershaw, Strange and Samar Hasnain2013). Mutant SOD1 found in the soluble fraction of transgenic mouse spinal cords is metalated, albeit with perturbed Cu:Zn ratio. However, analysis of aggregates from mutant SOD1 transgenic mice spinal cord indicated no increase of copper or zinc content despite increased SOD1 content. Thus, the SOD1 that accumulates in aggregates is metal free (Lelie et al., Reference Lelie, Liba, Bourassa, Chattopadhyay, Chan, Gralla, Miller, Borchelt, Valentine and Whitelegge2011). Additionally, several SOD1 conformation-specific antibodies, including the first described C4F6 that was raised against and specific for metal-free mutant SOD1, recognise their epitope in human ALS spinal tissue and lysates (Urushitani et al., Reference Urushitani, Ezzi and Julien2007; Brotherton et al., Reference Brotherton, Li, Cooper, Gearing, Julien, Rothstein, Boylan and Glass2012).
Disulphide bond
Wood et al. (Reference Wood, Dalgleish and Bannister1971) showed that removing metal cofactors and the disulphide bond from SOD1 led to the loss of secondary and tertiary structure. While SOD1 monomers will adopt their β-barrel fold in the absence of the intra-subunit disulphide, its presence stabilises the monomer and promotes dimerisation (Lindberg et al., Reference Lindberg, Normark, Holmgren and Oliveberg2004). Conversely, formation of the disulphide can promote acquisition of structure from an otherwise globally unfolded precursor through an oxidative folding mechanism (Capper et al., Reference Capper, Wright, Barbieri, Luchinat, Mercatelli, McAlary, Yerbury, O'Neill, Antonyuk, Banci and Hasnain2018). SOD1 must form and maintain its disulphide in the presence of reduced glutathione and the thioredoxin system which are purposed to reduce cytoplasmic thiols. When over-expressed in human cell culture, SOD1 does not naturally form the disulphide. Conversely, half of SOD1 monomers form the disulphide on co-expression with hCCS. Addition of copper to this system yields complete disulphide formation (Banci et al., Reference Banci, Barbieri, Bertini, Luchinat, Secci, Zhao and Aricescu2013).
The role the SOD1 intra-subunit disulphide plays in stabilisation and dimerisation is made clear in structures of the wild-type protein. Covalent bonding of Cys146, found in β-strand 8, to Cys57 anchors the zinc loop to the core of the protein. The disulphide sub-loop, a component of the zinc-binding loop IV, is defined by copper-coordinating His48 and metal-bridging ligand His63 at its N- and C-termini. The disulphide subloop forms 62% of the buried surface area of the SOD1 dimer interface (Fig. 8a) and also contains Gly51 which creates two of the four interface hydrogen bonds (Fig. 3). We have recently shown disulphide sub-loop plasticity to also be a necessary precursor of heterodimerisation with hCCS (Sala et al., Reference Sala, Wright, Antonyuk, Garratt and Hasnain2019).

Fig. 8. The SOD1 disulphide sub-loop dimer interface. (a) The disulphide subloop forms extensive contacts across the SOD1 dimer interface. (b) S–S bond lengths found in all human SOD1 structures in the Protein Data Bank. The 2.02–2.04 Å average for all protein structures (Thornton, Reference Thornton1981; Petersen et al., Reference Petersen, Jonson and Petersen1999) is shown in grey. By excluding bond lengths above 2.3 Å the mean SOD1 S–S bond length is found to be 2.08 Å, shown in blue.
Lyons et al. (Reference Lyons, Liu, Goto, Nersissian, Roe, Graden, Café, Ellerby, Bredesen, Gralla and Valentine1996) showed that mature Ala4Val, Gly85Arg and Gly93Ala SOD1 are less resistant to intra-subunit disulphide reduction on exposure to ascorbate than the wild-type form. Indeed, all SOD1 mutants are more susceptible to disulphide loss in a reducing environment akin to the cytoplasm, promoting monomerisation and loss of structural cohesion. This effect is more pronounced for metal-binding region mutants and metal-free wild-type SOD1 (Tiwari and Hayward, Reference Tiwari and Hayward2003). In addition, Alvarez-Zaldiernas et al. (Reference Alvarez-Zaldiernas, Lu, Zheng, Yang, Blasi, Solsona and Holmgren2016) showed that a reconstituted thioredoxin system was capable of reducing Ala4Val and Gly93Ala disulphides. This disulphide-reduced SOD1 is observed in SOD1 transgenic mice, particularly in the brain and spinal cord (Jonsson et al., Reference Jonsson, Graffmo, Andersen, Brännström, Lindberg, Oliveberg and Marklund2006a), and is a major component of intracellular aggregates (Karch et al., Reference Karch, Prudencio, Winkler, Hart and Borchelt2009).
The mean S–S length within a protein disulphide bond is 2.02–2.04 Å (Thornton, Reference Thornton1981; Petersen et al., Reference Petersen, Jonson and Petersen1999). The S–S distance variation across every SOD1 half cystine pair found in the PDB shows a mode at 2.04 Å with a long tail stretching out to 2.82 Å (Fig. 8b). SOD1 intra-subunit S–S bond lengths above 2.3 Å are found in His46Arg/His48Gln double mutant (PDB: 3GQF) (Winkler et al., Reference Winkler, Schuermann, Cao, Holloway, Borchelt, Carroll, Proescher, Culotta and Hart2009), Gly85Arg (PDB: 3CQP) (Cao et al., Reference Cao, Antonyuk, Seetharaman, Whitson, Taylor, Holloway, Strange, Doucette, Valentine, Tiwari, Hayward, Padua, Cohlberg, Hasnain and Hart2008), Gly93Ala (PDB: 3GZO) (Galaleldeen et al., Reference Galaleldeen, Strange, Whitson, Antonyuk, Narayana, Taylor, Schuermann, Holloway, Hasnain and Hart2009) and His80Arg SOD1 crystallographic structures (PDB: 3H2Q and 3QQD) (Seetharaman et al., Reference Seetharaman, Winkler, Taylor, Cao, Whitson, Doucette, Valentine, Schirf, Demeler, Carroll, Culotta and Hart2010). These structures are well refined with resolutions of ~1.95 Å. It is very likely that these S–S bonds have been broken through exposure to x-rays as is the case for some disulphides found in the Ile113Thr SOD1 structure (PDB: 1UXL) (Hough et al., Reference Hough, Grossmann, Antonyuk, Strange, Doucette, Rodriguez, Whitson, Hart, Hayward, Valentine and Hasnain2004). This underscores the fragility of the mutant SOD1 disulphide bond and the importance of accounting for radiation damage in published crystal structures. Omitting these bonds gives a mean SOD1 disulphide bond S–S distance of 2.08 Å, similar to the 2.05 Å distance observed in bovine SOD structures (Hough et al., Reference Hough, Strange and Hasnain2000).
Dimerisation
SOD1 dimerises through portions of β-strands 1, 8, loop VI and the disulphide subloop section of loop IV. Due to primary sequence separation of the N- and C-termini, SOD1 cannot dimerise before translation is complete. The interface is hydrophobic and excludes water. Ile151 residues create hydrogen bonds with Gly51 and Gly114 from the opposing monomer creating four interfacial hydrogen bonds. Overall, SOD1 dimer interface mutations disturb dimer interface packing but do not affect these bonds. Exceptions to this rule include hydrogen bond length variability for Ala4Val SOD1 which manifests as dimer reorientation, described in ‘Ala4Val’ section and Hough et al. (Reference Hough, Grossmann, Antonyuk, Strange, Doucette, Rodriguez, Whitson, Hart, Hayward, Valentine and Hasnain2004), and C-terminal truncations which remove Ile151 and therefore all interface hydrogen bonding.
As with the post-translational modifications described in the previous sections, homodimerisation increases SOD1 stability; obligate monomer SOD1 unfolds at lower chaotrope concentration than the dimer (Lindberg et al., Reference Lindberg, Byström, Boknäs, Andersen and Oliveberg2005). In vivo, dimerisation occurs after zinc binding and disulphide formation but in solution any state of wild-type SOD1 can be forced to dimerise by increasing its concentration (Arnesano et al., Reference Arnesano, Banci, Bertini, Martinelli, Furukawa and O'Halloran2004; Bruns and Kopito, Reference Bruns and Kopito2007; Hörnberg et al., Reference Hörnberg, Logan, Marklund and Oliveberg2007). When the SOD1 disulphide is reduced, the zinc-binding loop becomes dynamic and dimer affinity is reduced (Banci et al., Reference Banci, Benedetto, Bertini, Del Conte, Piccioli and Viezzoli1998). Conversely, SOD1 homodimer affinity is increased by progressively restricting zinc loop and disulphide subloop conformational sampling as SOD1 acquires post-translational modifications (Hörnberg et al., Reference Hörnberg, Logan, Marklund and Oliveberg2007). The reciprocity of dimerisation and disulphide bonding is illustrated by crystal structures where both disulphide bonding cysteines are mutated to alanine. In these cases, promoting dimerisation through high concentration forces the disulphide subloop to adopt the conformation common to disulphide intact structures (Hörnberg et al., Reference Hörnberg, Logan, Marklund and Oliveberg2007; Sala et al., Reference Sala, Wright, Antonyuk, Garratt and Hasnain2019).
The homodimer dissociation constant for mature wild-type SOD1 is in the low nanomolar to high picomolar range (10−8 to 10−10 M) (Khare et al., Reference Khare, Caplow and Dokholyan2004; McAlary et al., Reference McAlary, Yerbury and Aquilina2013). Many SOD1 ALS mutants, including Gly93Ala, have dimer affinity equal to that of wild-type but dimer interface mutants Ala4Val, Val148Gly and Ile149Thr reduce this affinity (McAlary et al., Reference McAlary, Yerbury and Aquilina2013; Capper et al., Reference Capper, Wright, Barbieri, Luchinat, Mercatelli, McAlary, Yerbury, O'Neill, Antonyuk, Banci and Hasnain2018). The metal-free, disulphide-intact, wild-type SOD1 dimer dissociation constant is high nanomolar and every mutant tested by Broom et al. (Reference Broom, Rumfeldt, Vassall and Meiering2015a) was destabilised by up to 3-orders of magnitude with homodimer affinity ranging up to 100 µM. As a result, reduced mutant homodimer formation, and indeed heterodimer formation with wild-type SOD1, is observed within cells for all SOD1 mutants with the exception of Asp90Ala (Kim et al., Reference Kim, Lee, Lee, Kwon, Genovesio, Fenistein, Ogier, Brondani and Grailhe2014). Additionally, an antibody raised against amino acids 143–151 in β-strand 8 that are only accessible when SOD1 is monomeric recognises its antigen in transgenic rodent and human ALS spinal cord samples. Immunoreactivity was highest for mitochondrial tissue fractions, particularly those from Gly85Arg mice, despite mitochondrial SOD1 being only a small component of total cellular SOD1. Tissues were stained increasingly through presymptomatic and initial symptom onset but declined at disease end stage (Rakhit et al., Reference Rakhit, Robertson, Vande Velde, Horne, Ruth, Griffin, Cleveland, Cashman and Chakrabartty2007).
Reducing the affinity of metal-free SOD1 homodimerisation does not necessarily mean increased populations of unfolded monomers. Ala4Val, His46Arg and Val148Gly can all exist as folded monomers (Broom et al., Reference Broom, Vassall, Rumfeldt, Doyle, Tong, Bonner and Meiering2015b). The existence of monomeric immature SOD1 with a conformationally flexible disulphide loop is also a prerequisite for the formation of a transient heterodimer with hCCS (Sala et al., Reference Sala, Wright, Antonyuk, Garratt and Hasnain2019). We do not know if the affinity of hCCS for its substrate is altered by ALS SOD1 mutations but there is evidence that some mutants are not able to interact with hCCS while others form stable complexes (Son et al., Reference Son, Fu, Puttaparthi, Matthews and Elliott2009). It is likely that mutation-dependent SOD1 homodimer affinity changes will also be reflected in the interaction with hCCS.
Structural instability
SOD1 zinc metalation, disulphide formation and dimerisation synergistically raise SOD1 thermal stability (Furukawa and O'Halloran, Reference Furukawa and O'Halloran2005). However, SOD1 nascent state stability is compromised by long, metal-binding loops. Shortening these functional structures increases metal-free, disulphide-reduced SOD1 stability (Yang et al., Reference Yang, Wang, Logan, Mu, Danielsson and Oliveberg2018). This trade-off between functionality and stability is represented throughout nature. For example, proteins from thermophiles tend to have trimmed loops. Closer to our subject, C. albicans SOD5 and the S. cerevisiae CCS SOD-like domain have substantially shorter electrostatic loops in comparison with human SOD1 and the hCCS SOD-like domain, respectively, because the former does not bind zinc (Culotta et al., Reference Culotta, Klomp, Strain, Casareno, Krems and Gitlin1997; Gleason et al., Reference Gleason, Galaleldeen, Peterson, Taylor, Holloway, Waninger-Saroni, Cormack, Cabelli, Hart and Culotta2014). Having shorter functional loops increases nascent-state stability but effective zinc binding has clearly outweighing functional benefits for SOD1.
The concept of reduced SOD1 metalation leading to structural instability was first described by Wood et al. (Reference Wood, Dalgleish and Bannister1971). ALS mutants are less resistant to chaotropic unfolding in both the metal-free and metalated states (Lindberg et al., Reference Lindberg, Tibell and Oliveberg2002; Stathopulos et al., Reference Stathopulos, Rumfeldt, Scholz, Irani, Frey, Hallewell, Lepock and Meiering2003). This work led Lindberg et al. (Reference Lindberg, Tibell and Oliveberg2002) to propose that destabilisation leads to aggregation, while Stathopulos et al. (Reference Stathopulos, Rumfeldt, Scholz, Irani, Frey, Hallewell, Lepock and Meiering2003) subsequently correlated decreased thermal stability, unfolding and aggregation.
Rodriguez et al. (Reference Rodriguez, Valentine, Eggers, Roe, Tiwari, Brown and Hayward2002) described reduced thermal stability of recombinant ALS SOD1 mutants in comparison with wild-type in metalation states as-isolated from insect cells. Furukawa and O'Halloran (Reference Furukawa and O'Halloran2005) noted that the 42.9 °C melting temperature of nascent wild-type SOD1, lacking both disulphide and metal ions, is above physiological temperature and sequential addition of zinc (58.4 °C) and the Cys57-Cys146 disulphide (74.6 °C) incrementally increases thermal stability. Thus, wild-type SOD1 thermal stability increases as it progresses along the maturation pathway from metal-free, disulphide-reduced, monomeric protein to the fully metalated, disulphide intact, dimeric, active enzyme. Increased dimer affinity, reduced populations of excited states and increased thermal stability are reflected in increased enzymatic activity and half-life (Furukawa and O'Halloran, Reference Furukawa and O'Halloran2005; Culik et al., Reference Culik, Sekhar, Nagesh, Deol, Rumfeldt, Meiering and Kay2018). Importantly, Gly93Ala and Ala4Val ALS substitutions reduced the melting temperature of the nascent protein below physiological temperature as ascertained by differential scanning calorimetry (DSC) experiments (Furukawa and O'Halloran, Reference Furukawa and O'Halloran2005). This is indicative that these species exist in a monomer unfolded state which was later observed by hydrogen/deuterium exchange and native mass spectrometry along with in-cell NMR for a range of mutants including Ala4Val and Gly93Ala (Shaw et al., Reference Shaw, Durazo, Nersissian, Whitelegge, Faull and Valentine2006; Luchinat et al., Reference Luchinat, Barbieri, Rubino, Kozyreva, Cantini and Banci2014; McAlary et al., Reference McAlary, Aquilina and Yerbury2016). Progression along the normal maturation pathway can stabilise SOD1 mutants in vitro, but frequently not to the same extent as wild-type, and this process is often ineffective within cells (Furukawa and O'Halloran, Reference Furukawa and O'Halloran2005; Luchinat et al., Reference Luchinat, Barbieri, Rubino, Kozyreva, Cantini and Banci2014).
The majority of ALS-related SOD1 mutations have been shown to be thermally destabilising in all states where testing was possible (Rodriguez et al., Reference Rodriguez, Valentine, Eggers, Roe, Tiwari, Brown and Hayward2002; Stathopulos et al., Reference Stathopulos, Rumfeldt, Scholz, Irani, Frey, Hallewell, Lepock and Meiering2003; Furukawa and O'Halloran, Reference Furukawa and O'Halloran2005; Münch et al., Reference Münch, O'Brien and Bertolotti2011; Vassall et al., Reference Vassall, Stubbs, Primmer, Tong, Sullivan, Sobering, Srinivasan, Briere, Dunn, Colón and Meiering2011; Doyle et al., Reference Doyle, Rumfeldt, Broom, Sekhar, Kay and Meiering2016). The immature state is where the biggest stability deficits are found (Furukawa and O'Halloran, Reference Furukawa and O'Halloran2005; Kayatekin et al., Reference Kayatekin, Zitzewitz and Matthews2010) but destabilisation of the apo state is not the whole story. Metal site mutants are equally, if not more stable than wild-type, when metal free (Rodriguez et al., Reference Rodriguez, Shaw, Durazo, Sohn, Doucette, Nersissian, Faull, Eggers, Tiwari, Hayward and Valentine2005).
While metal-free, disulphide intact His46Arg SOD1 has been shown to have similar thermal stability compared to wild-type (Münch et al., Reference Münch, O'Brien and Bertolotti2011; Abdolvahabi et al., Reference Abdolvahabi, Shi, Rasouli, Croom, Aliyan, Marti and Shaw2017). The metal-free, disulphide-reduced state is stabilised by 5 °C possibly affording it resistance to accumulation into large aggregates (Vassall et al., Reference Vassall, Stubbs, Primmer, Tong, Sullivan, Sobering, Srinivasan, Briere, Dunn, Colón and Meiering2011). His46Arg SOD1 is not unusual in this regard, Nordlund et al. (Reference Nordlund, Leinartaitė, Saraboji, Aisenbrey, Gröbner, Zetterström, Danielsson, Logan and Oliveberg2009) showed that mutation of all four zinc coordinating residues stabilises the immature protein. Unlike non-metal-binding site ALS mutants, however, His46Arg and His48Gln cannot progress along the normal maturation pathway to reach full stability and activity (Hayward et al., Reference Hayward, Rodriguez, Kim, Tiwari, Goto, Cabelli, Valentine and Brown2002; Bruns and Kopito, Reference Bruns and Kopito2007). This strands His46Arg in a pseudo-stable state with a nearly 30 °C thermal stability deficit in comparison with mature, wild-type SOD1 perhaps enabling degradation. His46Arg SOD1-related ALS has a long disease course, possibly due to these characteristics.
The Val148Ile dimer interface substitution has been shown to thermally stabilise the metal-free, disulphide-reduced state by 5 °C (Vassall et al., Reference Vassall, Stubbs, Primmer, Tong, Sullivan, Sobering, Srinivasan, Briere, Dunn, Colón and Meiering2011) but conversely its homodimerisation affinity is decreased sevenfold (Broom et al., Reference Broom, Rumfeldt, Vassall and Meiering2015a). DSC experiments performed on mature Val148Ile SOD1 show no difference in melting temperature over a range of protein concentrations but amide proton chemical shift temperature coefficients describe stabilisation across the protein sequence (Doyle et al., Reference Doyle, Rumfeldt, Broom, Sekhar, Kay and Meiering2016). We also note that, like wild-type, Val148Ile SOD1 is not recognised by a misfolding-specific antibody targeting the Derlin-1 binding site in the SOD1 N-terminus possibly because of the above stabilisation (Fujisawa et al., Reference Fujisawa, Yamaguchi, Kadowaki, Tsukamoto, Tsuburaya, Tsubota, Takahashi, Naguro, Takahashi, Goto, Tsuji, Nishitoh, Homma and Ichijo2015). Total erythrocyte Val148Ile SOD1 activity was found to be ~58% of controls, indicative of decreased half-life in common with other destabilised ALS mutants (described in the following section). While only one individual is known to have carried the Val148Ile mutation (Ikeda et al., Reference Ikeda, Abe, Aoki, Ogasawara, Kameya, Watanabe, Shoji, Hirai and Itoyama1995), inheritance in that individual's family was clearly dominant with disease onset very young with rapid progression (Sakuma et al., Reference Sakuma, Abe, Aoki, Ikeda, Okita, Hiwatari, Sakurai and Itoyama1995). Together, this is indicative of genuine SOD1-ALS. However, and as is often the case with SOD1-ALS mutations, drawing conclusions on causality from patients with very low-frequency alleles is challenging.
SOD1 like most proteins exists within a heterogenous soup of water, ions, small molecules and macromolecules within our cells. This molecular crowding can have varied effects on proteins under investigation (Ma et al., Reference Ma, Fan, Zhou, Zhou, Meng, Hu, Chen and Liang2012). However, the SOD1 β-barrel is generally destabilised by the cellular environment when compared with purified protein in aqueous buffer. In some cases, this can reduce its folding transition temperature by 40% (Danielsson et al., Reference Danielsson, Mu, Lang, Wang, Binolfi, Theillet, Bekei, Logan, Selenko, Wennerström and Oliveberg2015; Bille et al., Reference Bille, Jensen, Mohanty, Akke and Irbäck2019). The change in stability conferred by the environment occurs through interactions with β-strands 6 and 8 that stabilise the unfolded state (Bille et al., Reference Bille, Jensen, Mohanty, Akke and Irbäck2019). As such, environmental effects are strongly determined by the primary sequence. SOD1 ALS-like mutations largely exacerbate the overall destabilising trend on the β-barrel with the exception of His46Arg which has very slightly increased stability (Gnutt et al., Reference Gnutt, Timr, Ahlers, König, Manderfeld, Heyden, Sterpone and Ebbinghaus2019). This again highlights the cost-benefit relationship of introducing functionality. The conclusion of this work is that SOD1 species which appear folded under test-tube conditions may well be unfolded at physiological temperatures within cells. The adoption of a persistently unfolded state for many metal-free SOD1 ALS mutants in the cytoplasm is the result (Luchinat et al., Reference Luchinat, Barbieri, Rubino, Kozyreva, Cantini and Banci2014).
Activity and degradation
The final step in SOD1 maturation yields a stable and active enzyme. However, erythrocyte SOD1 activity is almost always reduced in ALS patients (Saccon et al., Reference Saccon, Bunton-Stasyshyn, Fisher and Fratta2013; Keskin et al., Reference Keskin, Birve, Berdynski, Hjertkvist, Rofougaran, Nilsson, Glass, Marklund and Andersen2017). Exclusion of Asp90Ala mutants leads to a mean activity of roughly 50% of that found in non-ALS individuals. Clearly an inability to correctly metalate will reduce overall SOD1 activity but a second, related factor may also play a role. Sato et al. (Reference Sato, Nakanishi, Yamamoto, Andersen, Ogawa, Fukada, Zhou, Aoike, Sugai, Nagano, Hirata, Ogawa, Nakano, Ohi, Kato, Nakagawa, Hamasaki, Shimizu and Sakoda2005) found erythrocytes taken from people heterozygous for an ALS SOD1 mutations had reduced abundance (0–58%) of the mutant SOD1 protein. Wild-type SOD1 is a long-lived protein with a 25-day half-life in cerebrospinal fluid. In transgenic rats, spinal cord wild-type SOD1 has a 15-day half-life whereas Gly93Ala SOD1 has a half-life of 9 days (Crisp et al., Reference Crisp, Mawuenyega, Patterson, Reddy, Chott, Self, Weihl, Jockel-Balsarotti, Varadhachary, Bucelli, Yarasheski, Bateman and Miller2015). Mutant SOD1 is also turned-over faster than wild-type in cell-based assays (Nakano et al., Reference Nakano, Inuzuka, Kikugawa, Takahashi, Sakimura, Fujii, Taniguchi and Tsuji1996; Kabuta et al., Reference Kabuta, Suzuki and Wada2006). Mature red blood cells do not contain a nucleus and therefore cannot transcribe new mRNAs encoding cellular proteins. Thus, it appears that no new SOD1 is created but mutant SOD1 is preferentially degraded. Over the life-time of a red blood cell, this reduces the abundance of mutant SOD1. In general, SOD1 degradation proceeds by proteasomal or macroautophagic routes (Kabuta et al., Reference Kabuta, Suzuki and Wada2006; Keskin et al., Reference Keskin, Forsgren, Lange, Weber, Birve, Synofzik, Gilthorpe, Andersen and Marklund2016). Ubiquitin E3 ligases dorphin, CHIP, mahogunin and cIAP have all been shown to ubiquitinate mutant SOD1 and signal its degradation (Choi et al., Reference Choi, Cho, Park, Park and Lee2004, Reference Choi, Kim, Lee, Cho, Ha, Park, Kim, Park and Kim2016; Sone et al., Reference Sone, Niwa, Kawai, Ishigaki, Yamada, Adachi, Katsuno, Tanaka, Doyu and Sobue2010; Chhangani et al., Reference Chhangani, Endo, Amanullah, Upadhyay, Watanabe, Mishra, Yamanaka and Mishra2016). However, SOD1 aggregation has been shown to sequester more than half of cellular ubiquitin thereby reducing the availability of free monomeric ubiquitin with downstream changes to the ubiquitination profile of more than 70 cellular proteins (Farrawell et al., Reference Farrawell, Lambert-Smith, Mitchell, McKenna, McAlary, Ciryam, Vine, Saunders and Yerbury2018). Thus, ALS SOD1 mutants that cause the greatest structural disturbance and instability lead to the greatest reductions in activity and abundance but are also more likely to upset proteostatic mechanisms through aggregation and proteasomal sequestration (Sato et al., Reference Sato, Nakanishi, Yamamoto, Andersen, Ogawa, Fukada, Zhou, Aoike, Sugai, Nagano, Hirata, Ogawa, Nakano, Ohi, Kato, Nakagawa, Hamasaki, Shimizu and Sakoda2005; McAlary et al., Reference McAlary, Aquilina and Yerbury2016; Keskin et al., Reference Keskin, Birve, Berdynski, Hjertkvist, Rofougaran, Nilsson, Glass, Marklund and Andersen2017; Farrawell et al., Reference Farrawell, Lambert-Smith, Mitchell, McKenna, McAlary, Ciryam, Vine, Saunders and Yerbury2018).
Cryptic mutations
The mutations described above induce structural changes to the SOD1 molecule that manifest as protein thermal instability, reduced homodimer affinity, inability to correctly metalate, reduced activity, reduced half-life in vivo and increased aggregation propensity. However, several mutations display stability and aggregation characteristics very similar to wild-type when observed in vitro. Shaw et al. (Reference Shaw, Moustakas, Whitelegge and Faull2010) termed these SOD1 variants ‘cryptic mutations’ because it is not clear where the source of their toxicity lies. These include Asp90Ala, Asn86Ser, Asp101Asn, Asn139Lys and Glu100Lys (Shaw and Valentine, Reference Shaw and Valentine2007). Cryptic mutations are usually surface exposed and involve the substitution of charged residues. While we do not have experimentally derived structures of these SOD1 mutants, analysis using the wild-type structure as a model provides us with an indication of their effect. In almost every case, a structural perturbation can be ascribed.
Surface mutations with mild phenotype
Asp90Ala
The Asp90Ala substitution does not affect dimer stability, metalation or activity (Andersen et al., Reference Andersen, Nilsson, Ala-Hurula, Keränen, Tarvainen, Haltia, Nilsson, Binzer, Forsgren and Marklund1995; Marklund et al., Reference Marklund, Andersen, Forsgren, Nilsson, Ohlsson, Wikander and Oberg1997; Rodriguez et al., Reference Rodriguez, Valentine, Eggers, Roe, Tiwari, Brown and Hayward2002; Lindberg et al., Reference Lindberg, Byström, Boknäs, Andersen and Oliveberg2005). The codon 90 position is not well conserved and the majority of eukaryotic CuZnSODs have a glycine residue at this site. Analysis of the wild-type SOD1 crystal structure shows the Asp90 side-chain making a series of hydrogen bonds within loop V with backbone amides of Asp92, Gly93 and Val94. Substitution with alanine would break these β-hairpin bonds but would not introduce steric repulsion within the loop, as is the case for Gly93 mutations. As a result, the Asp90Ala monomer is slightly destabilised (Marklund et al., Reference Marklund, Andersen, Forsgren, Nilsson, Ohlsson, Wikander and Oberg1997; Rodriguez et al., Reference Rodriguez, Valentine, Eggers, Roe, Tiwari, Brown and Hayward2002; Lindberg et al., Reference Lindberg, Byström, Boknäs, Andersen and Oliveberg2005; Byström et al., Reference Byström, Andersen, Gröbner and Oliveberg2010). An alternative Asp90Val mutation creates further monomer destabilisation and a more aggressive phenotype (Morita et al., Reference Morita, Abe, Takahashi, Onodera, Okumura, Niino, Tashiro, Nakano and Itoyama1998; Byström et al., Reference Byström, Andersen, Gröbner and Oliveberg2010).
Asn86Ser
The Asn86Ser mutation manifests ALS with a long disease course and incomplete penetrance but led to an extremely severe phenotype in a single homozygous individual (Hayward et al., Reference Hayward, Brock, Minns and Swingler1998; Sato et al., Reference Sato, Nakanishi, Yamamoto, Andersen, Ogawa, Fukada, Zhou, Aoike, Sugai, Nagano, Hirata, Ogawa, Nakano, Ohi, Kato, Nakagawa, Hamasaki, Shimizu and Sakoda2005). Asn86 stabilises the electrostatic loop through a hydrogen bond with Asp124 which itself hydrogen bonds with zinc- and copper-coordinating His71 and His46. Conversion to serine would destabilise these interactions but would not introduce any repulsive contacts. There are no metalation studies on this mutant and the monomer does not appear effected but the Asn86Ser dimer is destabilised (Byström et al., Reference Byström, Andersen, Gröbner and Oliveberg2010).
Other examples
Other charged, surface mutations which potentially fall into this category include Glu40Gly (Bertolin et al., Reference Bertolin, D'Ascenzo, Querin, Gaiani, Boaretto, Salvoro, Vazza, Angelini, Cagnin, Pegoraro, Sorarù and Mostacciuolo2014), Asp76Tyr (Andersen et al., Reference Andersen, Nilsson, Keränen, Forsgren, Hägglund, Karlsborg, Ronnevi, Gredal and Marklund1997), Asp76Val (Segovia-Silvestre et al., Reference Segovia-Silvestre, Andreu, Vives-Bauza, Garcia-Arumi, Cervera and Gamez2002), Asp11Tyr (Georgoulopoulou et al., Reference Georgoulopoulou, Gellera, Bragato, Sola, Chiari, Bernabei and Mandrioli2010), Glu21Lys (Jones et al., Reference Jones, Swingler and Brock1994a) and Glu121Gly (Canosa et al., Reference Canosa, Calvo, Moglia, Barberis, Brunetti, Cammarosano, Manera, Ilardi, Restagno and Chiò2015). In each case, no bonding or Van der Walls interactions are likely to be disturbed or the substitution could be mitigated without changing the overall structure.
Surface mutations with typical phenotype
Asp101Asn
The Asp101Asn substitution (Jones et al., Reference Jones, Shaw, Chari and Brock1994b) is also structurally conservative, removing only 1 Da of mass. However, it negates the electrostatic component of a salt-bridge formed by Asp101 and Arg79 which is conserved in more than 97% of eukaryotic CuZnSODs. This salt-bridge forms a strong connection between the C-terminal end of β-strand 6 and the upper zinc loop including spatially proximal zinc coordinating residues His80 and Asp38. An alternative substitution for glycine removes both electrostatic, hydrogen bonding and Van der Waals interactions. In agreement with proposed structural changes, Asp101Asn was shown to have stability equal to that of wild-type in the metal-free state in vitro while Asp101Gly destabilises both metal-free monomer and dimer states (Rodriguez et al., Reference Rodriguez, Shaw, Durazo, Sohn, Doucette, Nersissian, Faull, Eggers, Tiwari, Hayward and Valentine2005; Byström et al., Reference Byström, Andersen, Gröbner and Oliveberg2010). A detailed analysis of aggregation kinetics in HEK293 cells showed that Asp101Gly exhibits a short lag phase before the formation of insoluble aggregates. By contrast, Asp101Asn SOD1 initially expresses as soluble species, but following a long lag phase, this pool is transferred into degradation-resistant insoluble aggregates (Prudencio et al., Reference Prudencio, Hart, Borchelt and Andersen2009; Ayers et al., Reference Ayers, Lelie, Workman, Prudencio, Brown, Fromholt, Valentine, Whitelegge and Borchelt2014a). Given the structure of each mutation together with their biophysical and aggregation characteristics, it would be expected that Asp101Gly would yield a more severe phenotype relative to Asp101Asn. However, both mutations give rise to an early onset and rapidly progressing form of ALS (Wang et al., Reference Wang, Johnson, Agar and Agar2008). A possible explanation for Asp101 mutation toxicity lies in their inability to correctly metalate and potential weakening of the Asp101-Arg79 salt-bridge. Ayers et al. (Reference Ayers, Lelie, Workman, Prudencio, Brown, Fromholt, Valentine, Whitelegge and Borchelt2014a) found that both Asp101Asn and Asp101Gly SOD1 fail to acquire copper or zinc in a cell-based assay. This is unsurprising given that Asp101 anchors the zinc loop to β-strand 6. Thus, like His46Arg described in ‘Structural instability’ section, the absence of a stability deficit in the apo state is over-shadowed by the inability of the mutant to progress along the PTM pathway. Indeed, Asp101Asn has also been shown to have delayed folding kinetics (Bruns and Kopito, Reference Bruns and Kopito2007). A second contributory mechanism may also be the reduction of net negative charge, reviewed in ‘SOD1 net charge’ section.
ALS-associated mutations Asp101His and Asp101Try have also been observed and would create severe clashes with Arg79, Leu84, Val103 or Ile104 but result in a mild phenotype. Here the combination of severe structural instability together with an inability to metalate may create a protein that is easier to remove through proteostatic mechanisms or sequester thereby reducing the toxic protein load in comparison with Asp101Asn or Asp101Gly. It should be noted however that case numbers for the glycine, histidine and tyrosine substitutions are low making phenotype comparisons weak.
Other examples
Other examples of surface charge mutations which lead to a typical ALS phenotype include Arg110Gly (Kostrzewa et al., Reference Kostrzewa, Burck-Lehmann and Müller1994) which is likely to disturb the dimer interface and hydrogen bonding with loop VI helix, and Asn139Lys which is destabilised in the metal-free state (Rodriguez et al., Reference Rodriguez, Shaw, Durazo, Sohn, Doucette, Nersissian, Faull, Eggers, Tiwari, Hayward and Valentine2005; Münch and Bertolotti, Reference Münch and Bertolotti2010) because an internal electrostatic loop hydrogen bond is broken by the substitution. Little phenotype data are available for Asn139Lys but SOD1 activity is reduced 46.5% (Pramatarova et al., Reference Pramatarova, Figlewicz, Krizus, Han, Ceballos-Picot, Nicole, Dib, Meininger, Brown and Rouleau1995) and given its location is likely to reduce metalation. Like Asp101Asn described above, Asn139Lys has been shown to have delayed folding kinetics (Bruns and Kopito, Reference Bruns and Kopito2007).
The pathogenesis of ALS due to surface mutations
We can summarise the above sections by saying that, a substitution that perturbs the structure little, that does not affect the non-covalent bonding configuration or does not inhibit maturation processes, leads to incomplete disease penetrance often with later onset or longer disease course than mean. Conversely, large structural effects including breaking non-covalent interactions, introduction of repulsive interactions or inhibition of folding or PTMs lead to high penetrance and a natural history which is typical or comparatively severe for SOD1 ALS. As we have seen, however, there are exceptions to this dogma. The path from structure through stability and aggregation to disease phenotype cannot be unambiguously defined. Wang et al. (Reference Wang, Johnson, Agar and Agar2008) found that stability and aggregation account for only 69% of the variability in mutant SOD1 phenotypes. There must therefore be other factors that influence disease outcomes for particular substitution mutations.
SOD1 net charge
With a preponderance of aspartate and glutamate residues over arginines and lysines, the SOD1 monomer carries a hypothetical net charge of −6 at neutral pH. This is the maximum charge found on any vertebrate SOD1 (Sandelin et al., Reference Sandelin, Nordlund, Andersen, Marklund and Oliveberg2007). The structure surrounding ionisable side-chain groups together with the pH of the subcellular locale are important determinants of protein isoelectric point. Shi et al. (Reference Shi, Mowery and Shaw2013a) experimentally determined the net charge on metal-free, disulphide-reduced, monomeric, wild-type SOD1 at physiological pH to be 6.9. Formation of the SOD1 disulphide reduces the net charge to 6.05 per monomer. Binding of a zinc to disulphide intact SOD1 reduces the net charge to 4.3 and further addition of copper to create fully mature SOD1 decreases the net charge further to 3.7. Thus, the SOD1 PTM pathway increases stability but decreases monomer net charge.
ALS mutations and surface charge
ALS mutations have, on average, a propensity to reduce SOD1 net negative charge (Sandelin et al., Reference Sandelin, Nordlund, Andersen, Marklund and Oliveberg2007). While 76% of negatively charged sites have been found mutated in ALS cases, only 13% of positively charged amino acids are known to harbour ALS mutations, Lys3 and Arg115 only. This disparity is thought to reflect a role of net repulsive charge in maintaining free movement in the intracellular environment (Mu et al., Reference Mu, Choi, Lang, Mowray, Dokholyan, Danielsson and Oliveberg2017) and preventing aggregation; mutation of a positive residue increases net charge and therefore repulsion, whereas mutation of a negatively charged amino acid decreases net charge weakening repulsion and increasing aggregation propensity. In support of this, acetylation of SOD1 multiple lysine side-chains with aspirin (acetylsalicylic acid) increased fibrillation lag time and slowed growth (Abdolvahabi et al., Reference Abdolvahabi, Shi, Rhodes, Cook, Martí and Shaw2015). This could also be accomplished with a selection of charged anhydrides which modified only a single SOD1 lysine (Rasouli et al., Reference Rasouli, Abdolvahabi, Croom, Plewman, Shi, Ayers and Shaw2017). In both cases, inhibition of aggregation was not due to increased thermal stability as acylation ubiquitously decreased SOD1 melting temperature. However, inhibition of fibril growth was only observed in low ionic strength buffer indicating the charge effect may be limited under physiological conditions. Fibrillation inhibition also led to the accumulation of SOD1 into amorphous aggregates. This may be similar to the misfolding observed in the absence of large aggregates on cellular expression of SOD1 with poorly conserved lysines mutagenically removed (Crosby et al., Reference Crosby, Crown, Roberts, Brown, Ayers and Borchelt2018).
It is possible to fit a simple linear model of instability and survival time following disease onset by omitting outlier mutants that change net charge. On inspection of these outliers, Byström et al. (Reference Byström, Andersen, Gröbner and Oliveberg2010) found that net charge is a modifier of disease phenotype. For example, Gly41Asp has a very similar relative stability and disease onset compared with Leu106Val, but increased net charge, and therefore repulsion, raises disease duration from 2 to 17 years. The converse is also apparent; reducing net charge decreases survival time with Asp101Asn as the archetype example. Metal-free Asp101Asn stability is indistinguishable from wild-type but disease phenotype is similar to the very strongly destabilised Ala4Val (Byström et al., Reference Byström, Andersen, Gröbner and Oliveberg2010.)
Glu100 mutations
The case of Glu100Lys is often cited as an example where net charge reduction leads to ALS. Glu100 is surface exposed at the C-terminal end of β-strand 6. Its negatively charged side-chain exists in multiple rotamers and forms no interactions with its surroundings. Glu100Lys, which reduces net charge to −4, is likely to have limited structural effects, does not reduce erythrocyte SOD1 activity, can lead to a complicated phenotype with very long progression and was shown not to co-segregate with disease in two ALS families (Felbecker et al., Reference Felbecker, Camu, Valdmanis, Sperfeld, Waibel, Steinbach, Rouleau, Ludolph and Andersen2010; Yasser et al., Reference Yasser, Fecto, Siddique, Sheikh and Athar2010; Keskin et al., Reference Keskin, Birve, Berdynski, Hjertkvist, Rofougaran, Nilsson, Glass, Marklund and Andersen2017). Conversely, the Glu100Gly mutation, which reduces SOD1 net charge only to −5, leads to a typical ALS phenotype with high penetrance (Cudkowicz et al., Reference Cudkowicz, McKenna-Yasek, Sapp, Chin, Geller, Hayden, Schoenfeld, Hosler, Horvitz and Brown1997; Aggarwal and Nicholson, Reference Aggarwal and Nicholson2005; Wang et al., Reference Wang, Johnson, Agar and Agar2008). Glu100Gly decreases monomer stability (Rumfeldt et al., Reference Rumfeldt, Stathopulos, Chakrabarrty, Lepock and Meiering2006) and reduces overall thermal stability by 10 °C in the metal-free state in comparison with Glu100Lys which only creates a 4.4 °C stability deficit (Abdolvahabi et al., Reference Abdolvahabi, Shi, Rasouli, Croom, Aliyan, Marti and Shaw2017). A similar relationship is also observed when initial folding is observed; Glu100Lys acquires resistance to proteolysis only slightly slower than wild-type, whereas Glu100Gly folding is massively reduced and it behaves more like Ala4Val SOD1 (Bruns and Kopito, Reference Bruns and Kopito2007).
SOD1 aggregation in ALS
Aggregation of protein is a common feature of neurodegenerative disease and normal ageing. This is typified by the predominantly α-synuclein-containing Lewy bodies seen in the post-mortem substantia nigra of individuals who suffered from Parkinson's disease. Bunina (Reference Bunina1962) first described intracellular inclusions, later termed Bunina bodies, surrounded by an area of clear cytoplasm within the motor neurons of fALS cases. Single and multiple intracellular hyaline, i.e. glassy or transparent, regions often with Lewy body-like cores were subsequently described by Hirano et al. (Reference Hirano, Kurland and Sayre1967), also in fALS neuronal tissues. While we do not know the content of these structures or the type of fALS under investigation in these original cases, we do see SOD1 accumulate in these structures within the cytoplasm of microglia, astrocytes and motor neurons of mutant SOD1 transgenic animal models and human ALS patients (Shibata et al., Reference Shibata, Hirano, Kobayashi, Asayama, Umahara and Ikemoto1993, Reference Shibata, Hirano, Kobayashi, Siddique, Deng, Hung, Kato and Asayama1996a; Kato et al., Reference Kato, Shimoda, Watanabe, Nakashima, Takahashi and Ohama1996, Reference Kato, Hayashi, Nakashima, Nanba, Kato, Hirano, Nakano, Asayama and Ohama1997; Bruijn et al., Reference Bruijn, Becher, Lee, Anderson, Jenkins, Copeland, Sisodia, Rothstein, Borchelt, Price and Cleveland1997, Reference Bruijn, Houseweart, Kato, Anderson, Anderson, Ohama, Reaume, Scott and Cleveland1998). Together with mutations in the sod1 gene, this is the central hallmark of SOD1-related ALS. Astrocytes have been observed with their cytoplasm almost completely replaced by 15–25 nm granule-coated fibrils. In other glia, fibrils form non-membrane-bound inclusions but are distinctly separated from the cytoplasmic constituents (Kato et al., Reference Kato, Shimoda, Watanabe, Nakashima, Takahashi and Ohama1996, Reference Kato, Hayashi, Nakashima, Nanba, Kato, Hirano, Nakano, Asayama and Ohama1997). The appearance of insoluble SOD1 and gliosis in neuronal support cells appears to facilitate neuronal death (Beers et al., Reference Beers, Henkel, Xiao, Zhao, Wang, Yen, Siklos, McKercher and Appel2006; Yamanaka et al., Reference Yamanaka, Chun, Boillee, Fujimori-Tonou, Yamashita, Gutmann, Takahashi, Misawa and Cleveland2008). SOD1 aggregation is also observed in Parkinson's disease and to a lesser degree in otherwise normal but aged neuronal tissue (Trist et al., Reference Trist, Davies, Cottam, Genoud, Ortega, Roudeau, Carmona, De Silva, Wasinger, Lewis, Sachdev, Smith, Troakes, Vance, Shaw, Al-Sarraj, Ball, Halliday, Hare and Double2017).
Content of aggregates
Despite the relatively high turnover of many SOD1 mutants, it is a major component of aggregates in mutant SOD1 transgenic mice (Jonsson et al., Reference Jonsson, Ernhill, Andersen, Bergemalm, Brännström, Gredal, Nilsson and Marklund2004). The SOD1 found in the amorphous and fibrillar aggregates in the neural tissues of human and transgenic models has been shown to be partially or completely inactive, intra-subunit disulphide reduced, metal-free and misfolded but full-length (Jonsson et al., Reference Jonsson, Graffmo, Andersen, Brännström, Lindberg, Oliveberg and Marklund2006a; Shaw et al., Reference Shaw, Lelie, Durazo, Nersissian, Xu, Chan, Gralla, Tiwari, Hayward, Borchelt, Valentine and Whitelegge2008; Karch et al., Reference Karch, Prudencio, Winkler, Hart and Borchelt2009; Bergemalm et al., Reference Bergemalm, Forsberg, Srivastava, Graffmo, Andersen, Brännström, Wingsle and Marklund2010; Lelie et al., Reference Lelie, Liba, Bourassa, Chattopadhyay, Chan, Gralla, Miller, Borchelt, Valentine and Whitelegge2011). Wild-type SOD1 is also present along with mutant protein and high molecular mass species can be linked by abnormal disulphide bonds (Jonsson et al., Reference Jonsson, Ernhill, Andersen, Bergemalm, Brännström, Gredal, Nilsson and Marklund2004; Furukawa et al., Reference Furukawa, Fu, Deng, Siddique and O'Halloran2006; Karch et al., Reference Karch, Prudencio, Winkler, Hart and Borchelt2009). Advanced glycation end products carboxymethyl-lysine, pyrraline and pentosidine co-localise with neuronal and astrocytic inclusions in human ALS and transgenic mice and are thought to be direct SOD1 modifications (Kato et al., Reference Kato, Horiuchi, Nakashima, Hirano, Shibata, Nakano, Saito, Kato, Asayama and Ohama1999, Reference Kato, Horiuchi, Liu, Cleveland, Shibata, Nakashima, Nagai, Hirano, Takikawa, Kato, Nakano and Ohama2000; Shibata et al., Reference Shibata, Hirano, Kato, Nagai, Horiuchi, Komori, Umahara, Asayama and Kobayashi1999). In mice expressing human Gly85Arg, SOD1 aggregates showed ubiquitin-positive staining only at their periphery (Bruijn et al., Reference Bruijn, Becher, Lee, Anderson, Jenkins, Copeland, Sisodia, Rothstein, Borchelt, Price and Cleveland1997). However, in a cell model, ubiquitin was found to localise throughout SOD1 aggregates (Farrawell et al., Reference Farrawell, Lambert-Smith, Mitchell, McKenna, McAlary, Ciryam, Vine, Saunders and Yerbury2018). A meta-analysis performed by Ciryam et al. (Reference Ciryam, Lambert-Smith, Bean, Freer, Cid, Tartaglia, Saunders, Wilson, Oliver, Morimoto, Dobson, Vendruscolo, Favrin and Yerbury2017) has shown that five of the 15 proteins known to interact natively with SOD1 are drawn into inclusions but the majority of the 24 proteins that are found in aggregates do not have a known metabolic association with SOD1. SOD1 co-aggregators can be divided into four broad groups which offer a snap-shot of cellular activities in response to mutant SOD1: degradation machinery, neural-specific metabolism, neural cytoskeleton components and chaperones including metallochaperones. This includes ubiquitin, αB-crystalin, β-actin, metallothionine, glutamine synthetase, tubulins, tau, S-100, Hsp-27 and 70, proteasome, vimentin, neurofilament light chain, α-internexin, synaptophysin, enolase-2, protein-disulphide isomerase, glyceraldehyde-3-phosphate dehydrogenase and the copper chaperone for SOD1 (hCCS) (Kato et al., Reference Kato, Hayashi, Nakashima, Nanba, Kato, Hirano, Nakano, Asayama and Ohama1997, Reference Kato, Horiuchi, Liu, Cleveland, Shibata, Nakashima, Nagai, Hirano, Takikawa, Kato, Nakano and Ohama2000, Reference Kato, Sumi-Akamaru, Fujimura, Sakoda, Kato, Hirano, Takikawa and Ohama2001b; Watanabe et al., Reference Watanabe, Dykes-Hoberg, Culotta, Price, Wong and Rothstein2001; Bergemalm et al., Reference Bergemalm, Forsberg, Srivastava, Graffmo, Andersen, Brännström, Wingsle and Marklund2010; Zetterström et al., Reference Zetterström, Graffmo, Andersen, Brännström and Marklund2011).
Models of aggregation
Beginning with the Gly93Ala SOD1 transgenic mice described by Gurney et al. (Reference Gurney, Pu, Chiu, Dal Canto, Polchow, Alexander, Caliendo, Hentati, Kwon and Deng1994), many strains have been produced that express various human wild-type (Wong et al., Reference Wong, Pardo, Borchelt, Lee, Copeland, Jenkins, Sisodia, Cleveland and Price1995; Graffmo et al., Reference Graffmo, Forsberg, Bergh, Birve, Zetterström, Andersen, Marklund and Brännström2013) and SOD1 ALS mutants at different levels in different genetic backgrounds. These include, but are not limited to, Gly37Arg (Wong et al., Reference Wong, Pardo, Borchelt, Lee, Copeland, Jenkins, Sisodia, Cleveland and Price1995), Gly85Arg (Bruijn et al., Reference Bruijn, Becher, Lee, Anderson, Jenkins, Copeland, Sisodia, Rothstein, Borchelt, Price and Cleveland1997), His46Arg/His48Gln (Wang et al., Reference Wang, Xu, Gonzales, Coonfield, Fromholt, Copeland, Jenkins and Borchelt2002) and Asp90Ala (Jonsson et al., Reference Jonsson, Graffmo, Brännström, Nilsson, Andersen and Marklund2006b). When comparing wild-type and Gly93Ala SOD1 transgenic mice with equal transgene expression, Graffmo et al. (Reference Graffmo, Forsberg, Bergh, Birve, Zetterström, Andersen, Marklund and Brännström2013) found mean survival time is 367 ± 56 and 155 ± 9 days, respectively, compared to an average of 2 years for non-transgenic mice. Thus, the toxic load of human SOD1 expression appears cumulative in mice.
Mutant SOD1 transgenic mouse models were seen as a gold standard test for therapy development. The most popular is the Gly93Ala transgenic mouse created by Gurney et al. (Reference Gurney, Pu, Chiu, Dal Canto, Polchow, Alexander, Caliendo, Hentati, Kwon and Deng1994), a year after the establishment of the link between SOD1 mutants and ALS (Rosen et al., Reference Rosen, Siddique, Patterson, Figlewicz, Sapp, Hentati, Donaldson, Goto, O'Regan and Deng1993). Gurney's mouse expresses human mutant SOD1 at a high level, up to 24-fold that found in humans. These mice have relatively short disease course which is convenient for many types of experiment. Despite many therapeutic approaches having been reported to positively affect disease-related characteristics in these mice, there has been a complete failure to translate these findings into new medicines (Benatar, Reference Benatar2007). This has raised concern about the validity of SOD1 transgenic mouse experiments as a whole. It remains an open question whether the limitations arise from the transgenic models or the nature/protocols of the experiments and their limitations.
To further our understanding of the aggregation process, numerous cell and in vitro models have been developed. Mutant SOD1 aggregations are seen in cell culture models of ALS where they replicate some of the characteristics of post-mortem tissues (Durham et al., Reference Durham, Roy, Dong and Figlewicz1997; Koide et al., Reference Koide, Igarashi, Kikugawa, Nakano, Inuzuka, Yamada, Takahashi and Tsuji1998). SOD1 was first observed to aggregate in vitro in response to oxidative modification (Rakhit et al., Reference Rakhit, Cunningham, Furtos-Matei, Dahan, Qi, Crow, Cashman, Kondejewski and Chakrabartty2002). Light scattering at 350 nm by SOD1 in the presence of soluble copper (II) showed that the metal-depleted enzyme readily forms high molecular mass species. The authors posed the question ‘do these in vitro aggregates represent aggregates seen in ALS?’. This experiment illustrates the difficulty of recapitulating the disease state in vitro; any protein will aggregate if treated harshly enough, how is one to attach significance to that given that the nature of aggregates in living neuronal tissue is far from clear. Addressing this question, Lang et al. (Reference Lang, Zetterström, Brännström, Marklund, Danielsson and Oliveberg2015) showed a resemblance between in vitro aggregates and those that appear in transgenic mouse models of ALS. Using antibody probes to characterise the surface of aggregates, they found that solvent-exposed structures are maintained in vitro and in mice. Similarly, with thioflavin T binding, they showed that the dynamics of growth in both systems can be equated.
Amyloid or not?
SOD1 aggregates have been shown to bind amyloid-sensitive dyes, such as congo red or thioflavin T and S, but yield less fluorescence than is observed for amyloid-β or α-synuclein (DiDonato et al., Reference DiDonato, Craig, Huff, Thayer, Cardoso, Kassmann, Lo, Bruns, Powers, Kelly, Getzoff and Tainer2003; Chattopadhyay et al., Reference Chattopadhyay, Durazo, Sohn, Strong, Gralla, Whitelegge and Valentine2008; Furukawa et al., Reference Furukawa, Kaneko, Yamanaka, O'Halloran and Nukina2008; Oztug Durer et al., Reference Oztug Durer, Cohlberg, Dinh, Padua, Ehrenclou, Downes, Tan, Nakano, Bowman, Hoskins, Kwon, Mason, Rodriguez, Doucette, Shaw and Valentine2009). Thioflavin S stains inclusions formed by the artificial SOD1 double mutant His46Arg/His48Gln and ALS mutants Gly37Arg, Gly85Arg and Gly93Ala in transgenic mouse spinal cord tissue (Wang et al., Reference Wang, Xu, Gonzales, Coonfield, Fromholt, Copeland, Jenkins and Borchelt2002). The form of the fibres observed by Kato et al. (Reference Kato, Shimoda, Watanabe, Nakashima, Takahashi and Ohama1996, Reference Kato, Hayashi, Nakashima, Nanba, Kato, Hirano, Nakano, Asayama and Ohama1997) seems to support the notion of amyloid fibrillation. However, Kerman et al. (Reference Kerman, Liu, Croul, Bilbao, Rogaeva, Zinman, Robertson and Chakrabartty2010) have shown that SOD1 aggregates in spinal cords are non-amyloid. Instead they contain monomeric and unfolded SOD1 in a highly amorphous arrangement. To our knowledge, there are no reports of SOD1 amyloid structures found in human post-mortem ALS tissues. On the contrary, they have been shown not to bind congo red and thioflavin S (Kato et al., Reference Kato, Shimoda, Watanabe, Nakashima, Takahashi and Ohama1996). This does not mean the results of aggregation studies using thioflavin T fluorescence as a read-out for aggregation, for example, should be ignored. We should however expect the structure of SOD1 aggregates to differ from that of amyloid-β or α-synuclein. By extension, the structure or affinity of the dye-aggregate complexation will be altered and therefore the fluorescent properties of the dye itself.
Aggregate structure
For SOD1 aggregates, formation dictates form. This aphorism is true in two respects: Firstly, aggregation kinetics dictate morphology (Abdolvahabi et al., Reference Abdolvahabi, Shi, Chuprin, Rasouli and Shaw2016) (described in the following section), and secondly, the regions of the SOD1 molecule that mediate aggregation are sequestered within the aggregate structure. In the latter case, small hydrophobic side-chains occupy the core of the proto-fibril-like structure and exclude water in a manner reminiscent of the normal SOD1 folding nucleus (Nordlund and Oliveberg, Reference Nordlund and Oliveberg2006).
Insights on the structure of SOD1 fibrils can be gained from the observation of the immature state. In solution, wild-type metal-free SOD1 retains only one surface of the β-barrel (β-strands 1, 2, 3 and 6). The β-strands of the opposing face including loops constituting both metal sites, the active site channel, the disulphide sub-loop and a sizable portion of the dimer interface, all which are highly conserved across eukaryotic CuZnSODs (Fig. 4b), are significantly destabilised (Banci et al., Reference Banci, Bertini, Boca, Calderone, Cantini, Girotto and Vieru2009). Simulation of aggregation in silico also shows β-strands 1 and 2 self-associate to form β-sheet structures (Bille et al., Reference Bille, Jónsson, Akke and Irbäck2013). Binding of conformation-specific antibodies to segmented epitopes along the SOD1 primary sequence confirms the presence of β-strands 1, 2, 3 in the core of aggregates along with strand 8 (Bergh et al., Reference Bergh, Zetterström, Andersen, Brännström, Graffmo, Jonsson, Lang, Danielsson, Oliveberg and Marklund2015). β-strands 1, 2 and 6 along with strand 7 are found sequestered in aggregates as determined by protease digestion followed by mass spectrometry assignment; however, this varies along with mutation (Furukawa et al., Reference Furukawa, Kaneko, Yamanaka and Nukina2010).
Clues as to the structure of SOD1 aggregates can be gleaned from the ALS mutations themselves. C-terminal truncations at positions Leu117 (with four amino acids inserted before the stop codon), 127 (with five amino acids inserted before the stop codon), 125, 141 and 146 (Fig. 4a) are known to cause ALS in people and transgenic mice with intraneuronal SOD1-positive inclusions (Jackson et al., Reference Jackson, Al-Chalabi, Enayat, Chioza, Leigh and Morrison1997; Zu et al., Reference Zu, Deng, Lo, Mitsumoto, Ahmed, Hung, Cai, Tainer and Siddique1997; Jonsson et al., Reference Jonsson, Ernhill, Andersen, Bergemalm, Brännström, Gredal, Nilsson and Marklund2004; Wu et al., Reference Wu, Shen, Shi, Sun and Cai2012; Nakamura et al., Reference Nakamura, Bieniek, Lin, Graff-Radford, Murray, Castanedes-Casey, Desaro, Baker, Rutherford, Robertson, Rademakers, Dickson and Boylan2015). Thus, the electrostatic loop and β-strand 8 including Cys146 are not required for aggregate formation.
Bergh et al. (Reference Bergh, Zetterström, Andersen, Brännström, Graffmo, Jonsson, Lang, Danielsson, Oliveberg and Marklund2015) explored the variability of mutant SOD1 aggregate isoforms in transgenic mice and found that Asp90Ala aggregates had a distinct structure, with solvent exposure of β-strands 5, 6, 7, that was not seen in Gly93Ala, Gly85Arg and wild-type SOD1 isoforms. β-strand 4 and the disulphide sub-loop were however consistently found on the exterior of SOD1 aggregates and in a conformationally plastic state regardless of mutation. Notably, the Asp90Ala strain of SOD1 aggregation also appears in neuronal tissues of ALS patients with mutations in non-SOD1 genes whereas the aggregate strain common to other SOD1 mutations was not present (Forsberg et al., Reference Forsberg, Graffmo, Pakkenberg, Weber, Nielsen, Marklund, Brännström and Andersen2019). The two strains of SOD1 aggregate may represent the amorphous and fibrillar aggregates seen in cell-free assays (Abdolvahabi et al., Reference Abdolvahabi, Shi, Chuprin, Rasouli and Shaw2016). Switching between deposition into each form can be controlled with the presence of calcium or the method used to induce aggregation (Stathopulos et al., Reference Stathopulos, Rumfeldt, Scholz, Irani, Frey, Hallewell, Lepock and Meiering2003; Leal et al., Reference Leal, Cardoso, Valentine and Gomes2013). Solid-state NMR spectroscopy on fibrils formed by the SOD1 β-barrel core, stripped of electrostatic and zinc loops, indicates that β-strands 2, 3 and 5 retain their conformation from the free state. The rest of the molecule adopts a heterogeneous structure with multiple different conformations (Banci et al., Reference Banci, Blaževitš, Cantini, Danielsson, Lang, Luchinat, Mao, Oliveberg and Ravera2014). CD spectroscopy on fibrillar metal apo-SOD1 shows that its β-sheet content is increased with respect to soluble metalated and metal-free SOD1 (Banci et al., Reference Banci, Bertini, Durazo, Girotto, Gralla, Martinelli, Valentine, Vieru and Whitelegge2007). Thus, some of the unstructured strands and loops present in the free state reconfigure during the fibrillation process. In cells, SOD1 aggregates appear porous. This facilitates the diffuse movement of soluble proteins and small molecules throughout the inclusion (Matsumoto et al., Reference Matsumoto, Stojanovic, Holmberg, Kim and Morimoto2005; Abdolvahabi et al., Reference Abdolvahabi, Shi, Rhodes, Cook, Martí and Shaw2015). SOD1 aggregates are therefore extensively β-structured, with the first three β-strands internalised but greatly heterogeneous in both constituents and conformation when compared to classically amyloid states (Fig. 9a).
Fig. 9. The structure of SOD1 aggregates. (a) SOD1 β-strands in the N-terminal region of the protein are found to be structured in the metal-free state by NMR (Banci et al., Reference Banci, Bertini, Boca, Calderone, Cantini, Girotto and Vieru2009). These are also commonly found to form the core of SOD1 aggregates as determined by modelling, antibody reactivity, resistance to proteolytic cleavage and solid-state NMR (green) while the absence of the C-terminal region in truncated forms of SOD1 negates its role in aggregation (red). (b) SOD1 β-strand 3 forms fibril structures with the alignment of each peptide parallel to the direction of strand elongation in contrast to traditional amyloid where the constituent protein or peptide assembles perpendicular to the direction of propagation.
Aggregate heterogeneity presents a problem for direct structural study, but much has been learnt by breaking the SOD1 molecule down into peptides which self-assemble into fibrils amenable to crystallographic structure determination. Using this approach, segments from C-terminal β-strand 8 (residues 147–153) and loop VI (residues 101–107) were shown to form steric zippers through hydrophobic side-chain interactions (Ivanova et al., Reference Ivanova, Sievers, Guenther, Johnson, Winkler, Galaleldeen, Sawaya, Hart and Eisenberg2014). Alternatively, a peptide representing β-strand 3 (residues 28–38) formed a twisted β-sheet structure through backbone hydrogen bonding with the individual β-strands aligned in the direction of polymerisation (Sangwan et al., Reference Sangwan, Zhao, Adams, Jayson, Sawaya, Guenther, Pan, Ngo, Moore, Soriaga, Do, Goldschmidt, Nelson, Bowers, Koehler, Shaw, Novitch and Eisenberg2017). This contrasts markedly with information on amyloid commonly found in cross-β structures (Fig. 9b) and further explains the subdued response from tinctorial dyes charting SOD1 aggregation.
Aggregate formation
Cellular mechanisms exist to aid protein folding or to destroy proteins which have veered irretrievably off their folding pathway. Part of this functionality separates misfolded species into different cellular compartments (Kaganovich et al., Reference Kaganovich, Kopito and Frydman2008). Intracellular stress granule formation occurs following heat-shock. These dynamic compartments harbour RNAs together with a collection of proteins with RNA-binding capacity coupled with the structural disorder. ALS mutant SOD1 has a tendency to accumulate into distinct domains around preformed stress granules with chaperones and ubiquitin. SOD1-containing stress granules are less dynamic than normal and can be transported along microtubules to JUNQ-like aggresomes (Johnston et al., Reference Johnston, Dalton, Gurney and Kopito2000; Mateju et al., Reference Mateju, Franzmann, Patel, Kopach, Boczek, Maharana, Lee, Carra, Hyman and Alberti2017). Localisation of mutant SOD1 to the JUNQ has been demonstrated in human ALS post-mortem spinal motor neurons and is toxic in cell models (Weisberg et al., Reference Weisberg, Lyakhovetsky, Werdiger, Gitler, Soen and Kaganovich2012).
SOD1 aggregates form and grow in vitro following a sigmoidal growth curve as any population does in a new environment with limited resources (Fig. 10). Aggregation must first be initiated, and this is marked by a lag period before a change in a parameter such as thioflavin T fluorescence is observed. This period before aggregation nucleation is highly variable and indicative of a stochastic process. Nucleation is followed by exponential growth which plateaus when the available substrate is exhausted (Banci et al., Reference Banci, Bertini, Durazo, Girotto, Gralla, Martinelli, Valentine, Vieru and Whitelegge2007; Chattopadhyay et al., Reference Chattopadhyay, Durazo, Sohn, Strong, Gralla, Whitelegge and Valentine2008; Abdolvahabi et al., Reference Abdolvahabi, Shi, Chuprin, Rasouli and Shaw2016). However, in vivo SOD1 translation can supplement available substrate and indefinitely postpone slowing of growth within cells. In addition, a competition exists between recruitment of substrate protomers to fibrillar or amorphous aggregates (Fig. 10) (Abdolvahabi et al., Reference Abdolvahabi, Shi, Chuprin, Rasouli and Shaw2016).
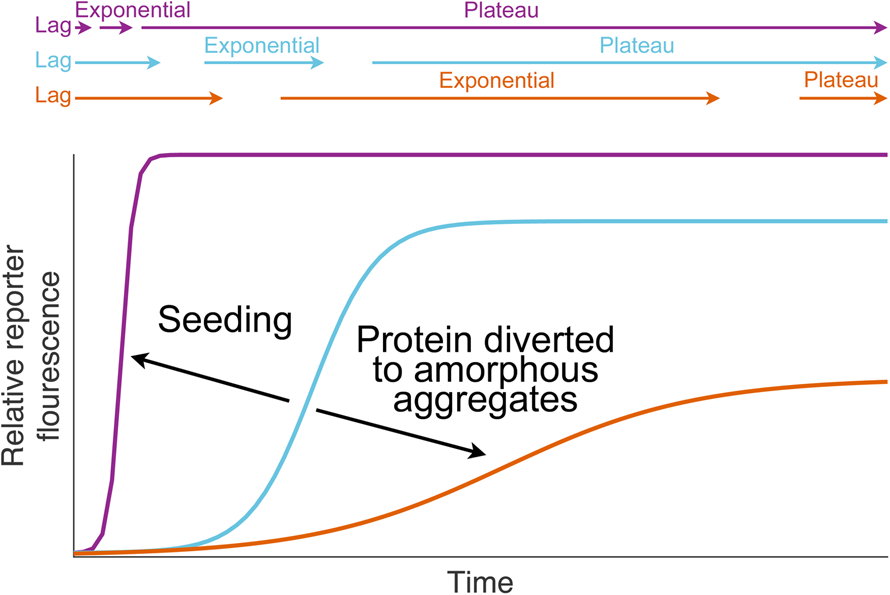
Fig. 10. SOD1 aggregate growth. SOD1 fibril growth is commonly assayed in vitro using a fluorescent dye such as thioflavin T. Fibril growth is preceded by a lag phase where little aggregation takes place. Aggregation is then nucleated in a stochastic process leading to exponential growth. Growth plateaus when all available SOD1 has been incorporated into aggregates. Each process can be modified by changing the SOD1 mutant under investigation, its concentration, adding aggregate seeds or sequestering protein in non-fibrillar aggregates.
Aggregation is a concentration-dependent process (Hofrichter et al., Reference Hofrichter, Ross and Eaton1974) and this is clearly seen in aggregate formation induced by variable expression levels of human wild-type and mutant SOD1 in transgenic mice (Wong et al., Reference Wong, Pardo, Borchelt, Lee, Copeland, Jenkins, Sisodia, Cleveland and Price1995; Graffmo et al., Reference Graffmo, Forsberg, Bergh, Birve, Zetterström, Andersen, Marklund and Brännström2013). In humans, SOD1 is posited to be supersaturated based on calculated aggregation propensity and intracellular concentration (Ciryam et al., Reference Ciryam, Lambert-Smith, Bean, Freer, Cid, Tartaglia, Saunders, Wilson, Oliver, Morimoto, Dobson, Vendruscolo, Favrin and Yerbury2017). The molecular environment also plays a role, with other solutes essentially increasing the effective concentration of SOD1 by molecular crowding thereby promoting fibrillation (Ma et al., Reference Ma, Fan, Zhou, Zhou, Meng, Hu, Chen and Liang2012). The solubility of SOD1 mutants and the ability of the cell to reduce the prevalence of toxic species through degradation are therefore a key. This may be reflected in the varying aggregation propensity of different SOD1 mutants and the lack of correlation between different experimental systems (McAlary et al., Reference McAlary, Aquilina and Yerbury2016; Abdolvahabi et al., Reference Abdolvahabi, Shi, Rasouli, Croom, Aliyan, Marti and Shaw2017).
Monomeric SOD1 had been implicated in the pathogenesis of ALS (Hough et al., Reference Hough, Grossmann, Antonyuk, Strange, Doucette, Rodriguez, Whitson, Hart, Hayward, Valentine and Hasnain2004; Rakhit et al., Reference Rakhit, Crow, Lepock, Kondejewski, Cashman and Chakrabartty2004; Ray et al., Reference Ray, Nowak, Strokovich, Brown, Walz and Lansbury2004) partly in response to the first oxidation-mediated SOD1 aggregation experiments, but the toxicity of a soluble, low molecular mass species was proven almost a decade later by Brotherton et al. (Reference Brotherton, Li and Glass2013). A SOD1 species existing as a monomeric, metal-free, disulphide-reduced, globally unfolded random coil and is enriched by SOD1 ALS-related mutations (Luchinat et al., Reference Luchinat, Barbieri, Rubino, Kozyreva, Cantini and Banci2014; Lang et al., Reference Lang, Zetterström, Brännström, Marklund, Danielsson and Oliveberg2015). However, this completely unstructured monomer must still fold into β-structure-rich fibrils. Anzai et al. (Reference Anzai, Tokuda, Mukaiyama, Akiyama, Endo, Yamanaka, Misawa and Furukawa2017) and Chattopadhyay et al. (Reference Chattopadhyay, Nwadibia, Strong, Gralla, Valentine and Whitelegge2015) proposed a destabilised and aberrantly disulphide-bonded dimer as the protomer species, while Proctor et al. (Reference Proctor, Fee, Tao, Redler, Fay, Zhang, Lv, Mercer, Deshmukh, Lyubchenko and Dokholyan2016) found, with the aid of atomic force microscopy, SEC and modelling, that SOD1 forms a trimer with non-native dimer interfaces and very substantial structural rearrangement. Banci et al. (Reference Banci, Bertini, Durazo, Girotto, Gralla, Martinelli, Valentine, Vieru and Whitelegge2007) had previously observed this phenomenon in wild-type SOD1 and we have seen it occur to monomeric and dimeric Ala4Val SOD1 (Wright et al., Reference Wright, Antonyuk, Kershaw, Strange and Samar Hasnain2013). By mutagenic stabilisation of the interfaces, Proctor et al. (Reference Proctor, Fee, Tao, Redler, Fay, Zhang, Lv, Mercer, Deshmukh, Lyubchenko and Dokholyan2016) found they could trap this trimer on route to high molecular mass aggregates. They also found a correlation between the stability of the trimer and cell death. However, no single species has been definitively demonstrated as pathogenic or to nucleate aggregation, indeed each may exert toxicity, and together they form a continuum on the path to larger aggregates.
Prion-like behaviour of SOD1
Assays of SOD1 aggregation often use mechanical agitation to speed fibrillation by fragmenting growing fibrils thus providing exponentially more termini for the incorporation of protomers. Initiation of fibrillation can also occur by seeding; transfer of wild-type or mutant fibrils from a mature reaction to a lag-phase reaction of the same or a different SOD1 variant (Fig. 10) (Chia et al., Reference Chia, Tattum, Jones, Collinge, Fisher and Jackson2010). Importantly, transfer of spinal cord homogenate from mutant SOD1 transgenic mice to an in vitro aggregation reaction has the same effect, demonstrating that these assays mimic in vivo aggregate structure and formation of kinetics.
Zil'ber et al. (Reference Zil'ber, Bajdakova, Gardaš’jan, Konovalov, Bunina and Barabadze1963) showed that spinal cord and medulla oblongata homogenates from post-mortem ALS individuals could induce ALS-like symptoms in macaque monkeys. CNS homogenates or CSF fluid from these monkeys were also competent to induce ALS-like symptoms in other monkeys. Conversely, transfer of homogenates from human ALS patients to non-transgenic mice or Guinea pigs does not yield any ALS-like symptoms but transfer from human mutant SOD1 patients to mutant SOD1 transgenic mice reduces survival (Zil'ber et al., Reference Zil'ber, Bajdakova, Gardaš’jan, Konovalov, Bunina and Barabadze1963; Bidhendi et al., Reference Bidhendi, Bergh, Zetterström, Forsberg, Pakkenberg, Andersen, Marklund and Brännström2018). Similarly, seeding with mouse mutant spinal cord homogenate reduces the age at which ALS-like symptoms are first observed in a transgenic mouse recipient and their age at the time of death (Ayers et al., Reference Ayers, Fromholt, Koch, DeBosier, McMahon, Xu and Borchelt2014b; Bidhendi et al., Reference Bidhendi, Bergh, Zetterström, Andersen, Marklund and Brännström2016).
Intracellular SOD1 is actively excreted into cerebrospinal fluid where it constitutes 75% of the total SOD activity (Jacobsson et al., Reference Jacobsson, Jonsson, Andersen, Forsgren and Marklund2001). In addition, misfolded SOD1 can be secreted by motor neurons and astrocytes and can traverse the extracellular space within exosomes (Urushitani et al., Reference Urushitani, Sik, Sakurai, Nukina, Takahashi and Julien2006; Gomes et al., Reference Gomes, Keller, Altevogt and Costa2007). Aggregated SOD1 is also effectively imported by micropinocytosis facilitating transfer from dead cells to those nearby. This pathway facilitates the transfer of aggregated SOD1 between cells without direct physical contact and seeds cytoplasmic accumulation of mutant SOD1 in recipient cells which may not yet have initiated aggregation (Münch et al., Reference Münch, O'Brien and Bertolotti2011; Grad et al., Reference Grad, Yerbury, Turner, Guest, Pokrishevsky, O'Neill, Yanai, Silverman, Zeineddine, Corcoran, Kumita, Luheshi, Yousefi, Coleman, Hill, Plotkin, Mackenzie and Cashman2014). Misfolded wild-type SOD1 can be a component of the aggregates transferred from one cell to another, but once misfolding has been seeded, wild-type SOD1 can continue this function without the necessity of mutant SOD1 (Grad et al., Reference Grad, Yerbury, Turner, Guest, Pokrishevsky, O'Neill, Yanai, Silverman, Zeineddine, Corcoran, Kumita, Luheshi, Yousefi, Coleman, Hill, Plotkin, Mackenzie and Cashman2014).
Transgenic mouse experiments have shown that the site of seed inoculation largely dictates the site of initial SOD1 aggregation and the onset of disease symptoms. This is followed by the spread of aggregates along the neuroaxis and into the brainstem possibly across synaptic junctions (Ayers et al., Reference Ayers, Fromholt, O'Neal, Diamond and Borchelt2016; Bidhendi et al., Reference Bidhendi, Bergh, Zetterström, Andersen, Marklund and Brännström2016). This parallels the human ALS phenotype where symptoms of the disease arise in a single region of the body and spread as the disease progresses (Ravits and La Spada, Reference Ravits and La Spada2009). Molecular and clinical characteristics of SOD1-ALS are therefore strongly reminiscent of the prion diseases where protein misfolding is templated and pathogenic. By implication, a single misfolding or aggregation event is sufficient to trigger the disease and this may account for variability in age of onset and reduced penetrance of some SOD1 mutants. The propagated misfolding paradigm also opens the somewhat remote possibility of person-to-person transmission as has been observed for amyloid-β (Jaunmuktane et al., Reference Jaunmuktane, Mead, Ellis, Wadsworth, Nicoll, Kenny, Launchbury, Linehan, Richard-Loendt, Walker, Rudge, Collinge and Brandner2015).
Impact on disease
Intuitively one would expect the molecular characteristics of mutant SOD1 to manifest at some level in the disease phenotype in light of the relatively simple inheritance and disease penetrance of many SOD1 mutants. Armed with a cause and effect hypothesis such as increased aggregation propensity leads to shorter disease duration (Prudencio et al., Reference Prudencio, Hart, Borchelt and Andersen2009), and the ability to define this process in vitro or in cells, how good are we at predicting the disease? Lindberg et al. (Reference Lindberg, Tibell and Oliveberg2002) first related the stability of mutant, immature SOD1 to patient survival time after initial diagnosis. Wang et al. (Reference Wang, Johnson, Agar and Agar2008) developed this using the Chiti–Dobson equation and found that 69% of the variability in disease duration could be accounted for by the synergistic effect of mutant SOD1 instability and aggregation. This was subsequently disputed in light of DSC and dynamic light-scattering characterisation of several SOD1 mutants (Vassall et al., Reference Vassall, Stubbs, Primmer, Tong, Sullivan, Sobering, Srinivasan, Briere, Dunn, Colón and Meiering2011). Given the stochasticity in relatively simple microplate fluorescence assays for SOD1 aggregation (Abdolvahabi et al., Reference Abdolvahabi, Shi, Chuprin, Rasouli and Shaw2016), the small sample sizes of each SOD1 ALS mutation together with human genetic, physical and environmental heterogeneity, and the inherent differences in SOD1 mutations themselves sometimes resulting in incomplete penetrance, it will always be difficult to predict clinical milestones. In an effort to combat the latter point, a small angle X-scattering study focussed only on ALS substitutions at the Gly93 site was able to discern differences in their aggregation paralleling disease course following diagnosis (Pratt et al., Reference Pratt, Shin, Merz, Rambo, Lancaster, Dyer, Borbat, Poole, Adams, Freed, Crane, Tainer and Getzoff2014). A second option is to increase the number of data points and restrict experimental conditions by looking at the relationship between in vitro and cell or transgenic models. Lang et al. (Reference Lang, Zetterström, Brännström, Marklund, Danielsson and Oliveberg2015) showed a good inverse linear relationship between mutant transgenic mouse survival times and the ratio of unfolded aggregation precursor to total SOD1 protein. A similar effect was seen in cultured neuronal cells (McAlary et al., Reference McAlary, Aquilina and Yerbury2016).
Are aggregates toxic?
By benchmarking our model systems against the disease, it should be possible to find entities that inhibit disease characteristics in the model system and then apply that knowledge to the disease in humans. This philosophy only functions if we study the toxic species. Aggregates clearly co-segregate with SOD1 mutations and by extension SOD1-related ALS. Furthermore, their presence augurs rapid cell death (Matsumoto et al., Reference Matsumoto, Stojanovic, Holmberg, Kim and Morimoto2005). However, true insoluble aggregates only appear at late stages in transgenic mouse models, after or disconnected from the onset of ALS-like symptoms, indicating they may be a response to toxicity or innocuous bi-product rather than a cause. Highlighting this point, Gill et al. (Reference Gill, Phelan, Hatzipetros, Kidd, Tassinari, Levine, Wang, Moreno, Thompson, Maier, Grimm, Gill and Vieira2019) found that the amount of aggregated SOD1 in transgenic mouse spinal cord was inversely proportional to disease progression as measured by NeuroScore. Thus, mice fair better if they sequester more SOD1 in aggregates. The authors also found that regions most affected by disease displayed higher soluble misfolded to total soluble SOD1 ratios (Gill et al., Reference Gill, Phelan, Hatzipetros, Kidd, Tassinari, Levine, Wang, Moreno, Thompson, Maier, Grimm, Gill and Vieira2019). These low molecular mass, soluble misfolded SOD1 species, free of all PTMs are present throughout life (Jonsson et al., Reference Jonsson, Ernhill, Andersen, Bergemalm, Brännström, Gredal, Nilsson and Marklund2004; Rakhit et al., Reference Rakhit, Robertson, Vande Velde, Horne, Ruth, Griffin, Cleveland, Cashman and Chakrabartty2007; Zetterström et al., Reference Zetterström, Stewart, Bergemalm, Jonsson, Graffmo, Andersen, Brännström, Oliveberg and Marklund2007; Lee et al., Reference Lee, Kawaguchi, Li, Kapur, Choi, Kim, Park, Zhu and Yao2015; Gill et al., Reference Gill, Phelan, Hatzipetros, Kidd, Tassinari, Levine, Wang, Moreno, Thompson, Maier, Grimm, Gill and Vieira2019) and may therefore be a better candidate for the toxic species than inert aggregates.
Mitochondrial toxicity
The pathway from SOD1 mutations to destabilisation, misfolding and aggregation is well delineated. This does not however, tell us why neuronal cells die in SOD1 ALS. Accumulation of ubiquitinated SOD1 into aggregates which also sequester chaperone proteins is a potential source of toxicity as proteostatic activities become impaired. Another possible source of toxicity is the degeneration of mitochondria.
Transgenic mice expressing Gly37Arg human SOD1 develop intraneuronal vacuoles that displace cytoskeletal structures. The vacuoles appear to be mitochondria with intermembrane spaces swollen to the point of membrane structure breakdown (Wong et al., Reference Wong, Pardo, Borchelt, Lee, Copeland, Jenkins, Sisodia, Cleveland and Price1995). Vacuolisation appears in the presymptomatic stage and increases in prevalence as disease symptoms manifest and progress but their occurrence is largely restricted to axons and dendrites (Kong and Xu, Reference Kong and Xu1998). SOD1 transported into the mitochondrial IMS in the disulphide-reduced, metal-free state and is retained through hCCS-catalysed maturation. Mutant and wild-type SOD1 aggregate within mitochondria (Pasinelli et al., Reference Pasinelli, Belford, Lennon, Bacskai, Hyman, Trotti and Brown2004; Vijayvergiya et al., Reference Vijayvergiya, Beal, Buck and Manfredi2005; Deng et al., Reference Deng, Shi, Furukawa, Zhai, Fu, Liu, Gorrie, Khan, Hung, Bigio, Lukas, Dal Canto, O'Halloran and Siddique2006; Cozzolino et al., Reference Cozzolino, Pesaresi, Amori, Crosio, Ferri, Nencini and Carrì2009) leading to membrane disruption (Oladzad Abbasabadi et al., Reference Oladzad Abbasabadi, Javanian, Nikkhah, Meratan, Ghiasi and Nemat-Gorgani2013; Salehi et al., Reference Salehi, Nikkhah, Ghasemi and Arab2015; Watanabe et al., Reference Watanabe, Ilieva, Tamada, Nomura, Komine, Endo, Jin, Mancias, Kiyama and Yamanaka2016). Progressive vacuolisation may be a consequence of this aberrant accumulation. The combined effect of these changes is defective respiration, electron transport and ATP synthesis (Bowling et al., Reference Bowling, Schulz, Brown and Beal2006; Magrané et al., Reference Magrané, Sahawneh, Przedborski, Estévez and Manfredi2012).
Membrane association
Misfolded SOD1 is found at high levels around vacuoles in transgenic mouse neurons in what appears to remain of the IMS (Wong et al., Reference Wong, Pardo, Borchelt, Lee, Copeland, Jenkins, Sisodia, Cleveland and Price1995; Jaarsma et al., Reference Jaarsma, Rognoni, van Duijn, Verspaget, Haasdijk and Holstege2001; Graffmo et al., Reference Graffmo, Forsberg, Bergh, Birve, Zetterström, Andersen, Marklund and Brännström2013). SOD1 associates with the intracellular face of the plasma membrane but misfolded SOD1 is also found on the outer mitochondrial membrane in ALS spinal cord cells (Petkau and Copps, Reference Petkau and Copps1979; Vande Velde et al., Reference Vande Velde, Miller, Cashman and Cleveland2008). Accumulation is progressive and, importantly, begins just prior to the onset of ALS (Pickles et al., Reference Pickles, Destroismaisons, Peyrard, Cadot, Rouleau, Brown, Julien, Arbour and Vande Velde2013). SOD1, particularly the metal-free state, incorporates within biological membranes and disorders lipid packing (Petkau and Chelack, Reference Petkau and Chelack1976; Petkau and Copps, Reference Petkau and Copps1979; Lepock et al., Reference Lepock, Arnold, Petkau and Kelly1981). Chng and Strange (Reference Chng and Strange2014) used molecular dynamics simulations to show how residues in the conformationally dynamic electrostatic and zinc-binding loops bind octanol and mediate interactions with bilayer surfaces. A C-terminal SOD1 truncation at Leu126 facilitates the adoption of a membrane-penetrant helical structure with several transmembrane helices (Lim et al., Reference Lim, Lee and Song2015), and similarly, Ala4Val SOD1 self-association created a tetramer capable of functioning as an integral membrane ion channel (Allen et al., Reference Allen, Lacroix, Ramachandran, Capone, Whitlock, Ghadge, Arnsdorf, Roos and Lal2012a).
Association of metal-free wild-type with mono- or polyunsaturated fatty acids including arachidonic, oleic and linoleic acid cause SOD1 aggregation (Kim et al., Reference Kim, Nakatomi, Akagi, Hashikawa and Takahashi2005). When incubated with phospholipids, Rasouli et al. (Reference Rasouli, Abdolvahabi, Croom, Plewman, Shi and Shaw2018) found SOD1 aggregation was influenced by the charge of the head group, lipid concentration and the SOD1 variant. For example, anionic phospholipids greatly stimulated metal-free wild-type SOD1 fibril formation over a broad range of concentrations whereas zwitterionic did not. Cationic phospholipids did catalyse SOD1 aggregation but over a much smaller range of concentration than anionic lipids. Thioflavin T binding indicated that His46Arg did not form fibrils and unusually Ala4Val also had limited fibrillation but wild-type, Gly37arg and Asp90Ala did.
Like hCCS, macrophage inhibitory factor (MIF) has a direct and high affinity interaction with both immature wild-type and mutant SOD1 (Israelson et al., Reference Israelson, Ditsworth, Sun, Song, Liang, Hruska-Plochan, McAlonis-Downes, Abu-Hamad, Zoltsman, Shani, Maldonado, Bui, Navarro, Zhou, Marsala, Kaspar, Da Cruz and Cleveland2015; Shvil et al., Reference Shvil, Banerjee, Zoltsman, Shani, Kahn, Abu-Hamad, Papo, Engel, Bernhagen and Israelson2018). MIF inhibits SOD1 deposition onto the outer mitochondrial membrane and chaperones SOD1 away from misfolding within cells and in a cell-free environment (Israelson et al., Reference Israelson, Ditsworth, Sun, Song, Liang, Hruska-Plochan, McAlonis-Downes, Abu-Hamad, Zoltsman, Shani, Maldonado, Bui, Navarro, Zhou, Marsala, Kaspar, Da Cruz and Cleveland2015). MIF over-expression in neuronal cells increases viability while MIF knockout hastens disease onset and death in SOD1 mutant transgenic mice. MIF has low abundance within motor neurons when compared with other cell types, thus this offers a rationale for motor neuron-selective toxicity despite ubiquitous SOD1 expression.
Secondary SOD1 post-translational modifications
Four PTMs contribute to the stability and function of SOD1. They are, in the sequential order in which they likely occur in the cytosol: zinc and copper acquisition, intra-subunit disulphide bond formation and dimerisation. However, SOD1 is also known to undergo a comprehensive range of secondary PTMs that are not necessary for the formation of the functional dimeric molecule (Fig. 11). These non-inheritable modifications can change the chemical and biophysical properties of SOD1 as do ALS-related mutations and so may have implications for the pathogenesis or treatment of ALS. Here we review only those PTMs that have an association with ALS.
Fig. 11. Secondary post-translational modifications and drug molecule interactions with human SOD1. Post-translational modifications observed ex vivo or in vitro are listed along with several structures of drug molecule interactions which have been validated crystallographically at Trp32 and Cys111 sites. Cysteinylation of Cys111 is the only native SOD1 post-translational modification which has been crystallographically characterised and was found to be destabilising (Auclair et al., Reference Auclair, Brodkin, D'Aquino, Petsko, Ringe and Agar2013a, Reference Auclair, Johnson, Liu, Salisbury, Rotunno, Petsko, Ringe, Brown, Bosco and Agar2013b).
Acetylation
Like many human proteins, the SOD1 N-terminus is cleaved to form acetylalanine (Barra et al., Reference Barra, Martini, Bannister, Schininà, Rotilio, Bannister and Bossa1980; Jabusch et al., Reference Jabusch, Farb, Kerschensteiner and Deutsch1980). Covalent addition of an acetyl group can also take place at accessible serine or lysine residues and changes the overall surface charge of a protein. Lysine acetylation is a global regulator of protein activity and signalling. SOD1 Lys70, which is solvent exposed on the surface of the zinc-binding loop, has been observed to be acetylated in cell culture and reduces the amount of soluble SOD1 in a manner similar to ALS mutations. Addition of deacetylase inhibitors increased the prevalence of this modification and decreased SOD1 activity by inhibiting hCCS-dependent activation. Sirtuin-1 is known to act as a deacetylase for SOD1 Lys70 and restores activation by hCCS (Lin et al., Reference Lin, Zeng, Lu, Xie, Sun, Luo, Ding, Yuan, Geng and Huang2015). Acetylation of Lys122, on the N-terminus of the electrostatic loop, does not change SOD1 activity but is found only in specific cell types and regions within the CNS indicating functional importance (Kaliszewski et al., Reference Kaliszewski, Kennedy, Blaes, Shaffer, Knott, Song, Hauser, Bossy, Huang and Bossy-Wetzel2016). Non-specific lysine acetylation of wild-type, Asp90Ala and Ala4Val SOD1 with aspirin in vitro inhibited the nucleation rate and extension of aggregates overall. As the number of acetyl groups increased, the thermal stability of metal-free SOD1 decreased. Destabilisation of acetylated fibril aggregates was also observed and thought to occur by Coulombic repulsion (Abdolvahabi et al., Reference Abdolvahabi, Shi, Rhodes, Cook, Martí and Shaw2015).
Glycation
Protein glycation is the irreversible, stochastic, non-enzymatic addition of a sugar or dicarbonyl to protein amino groups. In practice, addition is usually limited to arginine and lysine side chains. Initial addition is followed by a series of reactions which lead to heterogeneous advanced glycation end-product (AGE) formation. AGEs negatively affect proteins by inhibiting function, crosslinking, decreasing solubility and increasing protease resistance. As a result, they are associated with neurodegenerative diseases such as Alzheimer's and Parkinson's diseases (Li et al., Reference Li, Liu, Sun, Lu and Zhang2012). Arai et al. (Reference Arai, Iizuka, Tada, Oikawa and Taniguchi1987a) found glucose-modified SOD1 to constitute roughly 20% of the erythrocytic SOD1 pool and also found differences in glucosylation between SOD1 from young and old cells. In vitro glucosylation occurs at Lys3, Lys9, Lys30, Lys36, Lys122 and Lys128. These modifications reduce SOD1 activity (Arai et al., Reference Arai, Iizuka, Tada, Oikawa and Taniguchi1987a, Reference Arai, Maguchi, Fujii, Ishibashi, Oikawa and Taniguchi1987b). Modification of metalated and metal-free wild-type SOD1 with the advanced glycation end-product precursor methylglyoxal induces partial unfolding and, in the case of apoSOD1, the formation of high molecular mass oligomers (Sirangelo et al., Reference Sirangelo, Vella, Irace, Manco and Iannuzzi2016). In addition, ALS SOD1 mutant proteins are more easily glucosylated and fructosylated in vitro with respect to wild-type (Takamiya et al., Reference Takamiya, Takahashi, Myint, Park, Miyazawa, Endo, Fujiwara, Sakiyama, Misonou, Miyamoto, Fujii and Taniguchi2003). These in vitro experiments are of importance because AGE-modified SOD1 is a major component of aggregates found in post-mortem ALS neural tissues (Kato et al., Reference Kato, Horiuchi, Nakashima, Hirano, Shibata, Nakano, Saito, Kato, Asayama and Ohama1999, Reference Kato, Horiuchi, Liu, Cleveland, Shibata, Nakashima, Nagai, Hirano, Takikawa, Kato, Nakano and Ohama2000; Shibata et al., Reference Shibata, Hirano, Kato, Nagai, Horiuchi, Komori, Umahara, Asayama and Kobayashi1999).
Oxidation
Oxidative stress has long been associated with neurodegenerative disease and ALS is a typical example. SOD1 was first observed to aggregate in response to oxidative damage by Rakhit et al. (Reference Rakhit, Cunningham, Furtos-Matei, Dahan, Qi, Crow, Cashman, Kondejewski and Chakrabartty2002). Oxidation of wild-type SOD1 by hydrogen peroxide causes misfolding and aggregation in vitro and in cells. Furthermore, oxidation promotes association with ubiquitin and Hsp70, as is the case in human mutant SOD1 aggregates, and is toxic to motor neuron cultures (Ezzi et al., Reference Ezzi, Urushitani and Julien2007). There are three sites at which SOD1 is known to be oxidised: Cys111, Trp32 and the copper site histidines. Efficient formation oxo-histidine-118 bovine SOD1 (His120 human numbering) by hydrogen peroxide has been observed in vitro (Uchida and Kawakishi, Reference Uchida and Kawakishi1994). Multiple human SOD1 copper site histidines are oxidised by ascorbic acid-CuCl2, again in vitro (Rakhit et al., Reference Rakhit, Cunningham, Furtos-Matei, Dahan, Qi, Crow, Cashman, Kondejewski and Chakrabartty2002). There is no evidence of copper site modification in the cellular environment or an impact on ALS but Cys111 and Trp32 both have implications for disease pathogenesis.
Human SOD1 Cys111 residues are solvent exposed but oppose each other 9 Å apart across the sub-unit interface when the protein is in the dimeric form. Their side-chain sulphydryl groups are reactive (Calabrese et al., Reference Calabrese, Federici, Bannister, Bannister, Rotilio and Finazzi-Agrò1975). Cysteine is used sparingly in nature due to its reactivity and a cysteine at position 111 is uncommon in the primary sequence of SOD1 orthologues. Its persistence in different phylogenetic groups however could point to a functional role, possibly environmental redox sensing (Go et al., Reference Go, Chandler and Jones2015). Cys111 is known to be a key modulator of SOD1 expression level, folding, stability, aggregation and toxicity (Jabusch et al., Reference Jabusch, Farb, Kerschensteiner and Deutsch1980; Lepock et al., Reference Lepock, Frey and Hallewell1990; Hallewell et al., Reference Hallewell, Imlay, Lee, Fong, Gallegos, Getzoff, Tainer, Cabelli, Tekamp-Olson, Mullenbach and Cousens1991; Suzuki et al., Reference Suzuki, Yasui, Ishikawai, Nomura, Watanabe, Mikami, Yamano and Ono2011; Chen et al., Reference Chen, Shang, Qiu, Fujiwara, Cui, Li, Gao and Kong2012; Zhang et al., Reference Zhang, Zhang, Zi, Xue and Wan2017). A potential mechanism for this may be found in the asymmetry and hydrogen bonding network of SOD1 loop VI within which Cys111 is found (Ihara et al., Reference Ihara, Fujiwara, Yamaguchi, Torigoe, Wakatsuki, Taniguchi and Suzuki2012). The Cys111 side-chain sulphydryl is amenable to a host of post-translational modification reactions, the simplest of which is reversible oxidation to sulphenic acid (Xu et al., Reference Xu, Liang, Li, He, Yuan, Huang, Liu, Tang, Pang, Du, Yang, Chen, Wang, Zhang and Liang2018). Successive addition of oxygen by hydrogen peroxide or incubation in the air creates sulphinylated and sulphonylated SOD1. Sulphonylation of wild-type SOD1 Cys111 predisposes it to adopt a Gly93Ala mutant-like conformation and inhibits fast axonal transport (Bosco et al., Reference Bosco, Morfini, Karabacak, Song, Gros-Louis, Pasinelli, Goolsby, Fontaine, Lemay, McKenna-Yasek, Frosch, Agar, Julien, Brady and Brown2010). The sulphonylated form increases in prevalence as the disease progresses and it also localises to hyaline inclusions found within spinal cord neurons of mutant SOD1 transgenic mice (Fujiwara et al., Reference Fujiwara, Nakano, Kato, Yoshihara, Ookawara, Eguchi, Taniguchi and Suzuki2007; Chen et al., Reference Chen, Shang, Qiu, Fujiwara, Cui, Li, Gao and Kong2012; Xu et al., Reference Xu, Liang, Li, He, Yuan, Huang, Liu, Tang, Pang, Du, Yang, Chen, Wang, Zhang and Liang2018). Finally, Bosco et al. (Reference Bosco, Morfini, Karabacak, Song, Gros-Louis, Pasinelli, Goolsby, Fontaine, Lemay, McKenna-Yasek, Frosch, Agar, Julien, Brady and Brown2010) observed immunoreactivity of a conformation-specific antibody which recognises sulphonylated wild-type SOD1 in four of nine sporadic ALS cases tested but not in control individuals or ALS cases with mutations in other genes. This highlights the potentially disastrous effect of irreversible oxidation on SOD1 behaviour.
Mass spectrometry analysis of erythrocytic SOD1 from Gly93Ala and wild-type transgenic mice together with native human wild-type SOD1 showed Trp32 to be post-translationally oxidised (Taylor et al., Reference Taylor, Gibbs, Kabashi, Minotti, Durham and Agar2007). Like Cys111, the presence of tryptophan in the mid-point of β-strand 3, where it is unusually solvent exposed for a large non-polar amino acid, is uncommon and again suggests some functional importance. In vitro Trp32 is oxidised by hydrogen peroxide in the presence of bicarbonate to N-formylkynurenine and kynurenine or a di-tryptophan species. This promotes misfolding and non-disulphide, covalently linked dimerisation ultimately leading to aggregation (Zhang et al., Reference Zhang, Andrekopoulos, Joseph, Chandran, Karoui, Crow and Kalyanaraman2003, Reference Zhang, Joseph, Crow and Kalyanaraman2004; Medinas et al., Reference Medinas, Gozzo, Santos, Iglesias and Augusto2010; Coelho et al., Reference Coelho, Iqbal, Linares, Silva, Lima, Cuccovia and Augusto2014). Trp32 also facilitates the conversion of wild-type SOD1 to a misfolded form by mutant SOD1; a process which is hindered by the substitution of serine at position 32 (Grad et al., Reference Grad, Guest, Yanai, Pokrishevsky, O'Neill, Gibbs, Semenchenko, Yousefi, Wishart, Plotkin and Cashman2011). Similarly, substitution of phenylalanine inhibits SOD1 aggregation and prolongs survival of SOD1 mutant cultured neurons (Zhang et al., Reference Zhang, Andrekopoulos, Joseph, Chandran, Karoui, Crow and Kalyanaraman2003; Taylor et al., Reference Taylor, Gibbs, Kabashi, Minotti, Durham and Agar2007).
Thiolation
S-thiolation is the reversible formation of mixed disulphide bonds between free protein sulphydryls and low molecular mass cellular thiols such as glutathione or cysteine. These modifications are known to protect cysteine from irreversible oxidation to sulphinate and sulphonate. Modification of SOD1 with cysteine or glutathione results in addition to at most one Cys111 per dimer likely due to lack of space in the inter-dimer groove (Wilcox et al., Reference Wilcox, Zhou, Jordon, Huang, Yu, Redler, Chen, Caplow and Dokholyan2009; Auclair et al., Reference Auclair, Brodkin, D'Aquino, Petsko, Ringe and Agar2013a). The preponderance of glutathionylation versus cysteinylation in vivo has been disputed. Furthermore, sample preparation under oxidative conditions has been shown to increase the extent of thiolation (Auclair et al., Reference Auclair, Salisbury, Johnson, Petsko, Ringe, Bosco, Agar, Santagata, Durham and Agar2014). S-glutathionylation can be spontaneous or glutathione-S-transferase catalysed and often results from oxidative stress. Covalent linkage of glutathione to SOD1 was first observed in human erythrocyte SOD1 with the Asp90Ala mutation (Marklund et al., Reference Marklund, Andersen, Forsgren, Nilsson, Ohlsson, Wikander and Oberg1997). The site of glutathione adduct formation was subsequently found to be Cys111 (Wilcox et al., Reference Wilcox, Zhou, Jordon, Huang, Yu, Redler, Chen, Caplow and Dokholyan2009). Glutathionylation of mutant and wild-type SOD1 reduces dimer affinity (Redler et al., Reference Redler, Wilcox, Proctor, Fee, Caplow and Dokholyan2011; McAlary et al., Reference McAlary, Yerbury and Aquilina2013). Analysing wild-type SOD1 purified from nerve tissue Auclair et al., Reference Auclair, Johnson, Liu, Salisbury, Rotunno, Petsko, Ringe, Brown, Bosco and Agar2013b found roughly 40% to be cysteinylated at Cys111 in comparison with little cysteinylation in red blood cell SOD1 (Auclair et al., Reference Auclair, Salisbury, Johnson, Petsko, Ringe, Bosco, Agar, Santagata, Durham and Agar2014). This modification protected Cys111 from sulphinylation and sulphonylation in response to hydrogen peroxide but decreased the protein's overall thermal stability by 5 °C (Auclair et al., Reference Auclair, Brodkin, D'Aquino, Petsko, Ringe and Agar2013a, Reference Auclair, Johnson, Liu, Salisbury, Rotunno, Petsko, Ringe, Brown, Bosco and Agar2013b).
Acylation
S-acylation is the addition of a lipid group to a protein cysteine. Palmitoylation is the most common S-acylation and involves the catalysed formation of a covalent but reversible thioester bond between a free sulphydryl and the fatty acid palmitate. As a result, the hydrophobicity of the target protein is increased and this can facilitate partitioning of otherwise soluble proteins with lipid membranes. Disulphide-reduced SOD1 has been shown to be palmitoylated in cells, transgenic spinal cord lysate and human spinal cord. Modification takes place primarily at Cys6 but to a lesser extent at Cys111 and even Cys57 and Cys146. SOD1 mutants were also shown to have a higher degree of palmitoylation which may be a consequence of their propensity to exist in the disulphide-reduced state. In addition, wild-type SOD1 also showed a higher degree of palmitoylation in sporadic ALS cases. The degree of palmitoylation correlated with the amount of SOD1 associated with cell membranes (Antinone et al., Reference Antinone, Ghadge, Lam, Wang, Roos and Green2013, Reference Antinone, Ghadge, Ostrow, Roos and Green2017). Further analysis found hCCS to be palmitoylated in the opposite manner to SOD1 with decreasing levels in mutant SOD1-related ALS and sporadic ALS (Antinone et al., Reference Antinone, Ghadge, Ostrow, Roos and Green2017). Reversible palmitoylation could therefore play a role in reducing the three-dimensional search space of hCCS for zinc-bound, disulphide-reduced SOD1 by facilitating membrane association prior to copper and disulphide transfer.
Phosphorylation
SOD1 was first observed to be phosphorylated in myeloid cells after exposure to granulocyte-colony stimulating factor (Csar et al., Reference Csar, Wilson, Strike, Sparrow, McMahon, Ward and Hamilton2001). It has subsequently been shown to be phosphorylated at Thr2, Ser59 and Ser98 (Wilcox et al., Reference Wilcox, Zhou, Jordon, Huang, Yu, Redler, Chen, Caplow and Dokholyan2009; Tsang et al., Reference Tsang, Liu, Thomas, Zhang and Zheng2014). Ser59 and Ser98 are phosphorylated through a direct interaction with Dun1 in response to oxidative stress and this directs SOD1 to the nucleus where it acts as a transcription factor for genes involved in the oxidative stress response (Tsang et al., Reference Tsang, Liu, Thomas, Zhang and Zheng2014). While production of phosphorylated SOD1 in amounts necessary for biophysical characterisation is challenging, Fay et al. (Reference Fay, Zhu, Proctor, Tao, Cui, Ke and Dokholyan2016) constructed and crystallised a SOD1 Thr2Asp phosphomimetic mutant. The wild-type SOD1 structure is maintained by the Thr2Asp substitution but strengthens the dimer interaction and slows dissociation rates even when the Ala4Val mutation was present along with glutathionylation at Cys111. In addition, the authors predicted that Thr2Asp destabilises a non-native trimeric structure. As a result, the Thr2Asp substitution reduces cell death resulting from Ala4Val SOD1 expression (Fay et al., Reference Fay, Zhu, Proctor, Tao, Cui, Ke and Dokholyan2016).
Deamidation
Deamidation is the removal of the side-chain amide from a glutamine or asparagine residue. Under physiological conditions, the reaction is not enzymatic but slow and may act as a molecular clock (Robinson and Robinson, Reference Robinson and Robinson2001). The sequential mechanism leads to a mixture of aspartic and isoaspartic acids. Deamidation therefore recapitulates a single amino acid missense mutation. SOD1 deamidation has been observed in erythrocytic SOD1 and occurs naturally to a SOD1 truncation comprising β-strands 1–4 and the disulphide sub-loop (amino acids 1–61 incorporating Cys6/57Ser mutations) in vitro during protein purification at Asn26 (Shi et al., Reference Shi, Rhodes, Abdolvahabi, Kohn, Cook, Martí and Shaw2013b; Bierczyńska-Krzysik et al., Reference Bierczyńska-Krzysik, Łopaciuk, Pawlak-Morka and Stadnik2014). Asn26 is positioned in SOD1 loop II within a tripeptide, Ser-Asn-Gly, known to predispose the central residue to deamidation (Wright, Reference Wright1991). Furthermore, single and compound deamidation replicating Asn/Asp amino acid substitutions created modest reductions in SOD1 thermal stability and increased rate of aggregation (Shi et al., Reference Shi, Rhodes, Abdolvahabi, Kohn, Cook, Martí and Shaw2013b).
Targeting the SOD1 molecule for therapeutic benefit
Small molecules
Small molecules, produced synthetically or derived from natural sources, formed the mainstay of a revolution in drug discovery that occurred in the latter half of the 20th century and still constitute roughly ¾ of new chemical entities registered by the US Food & Drug Administration. Like ALS mutations or natural amino acid modifications, interactions with exogenous small molecules could have positive or negative effects on SOD1 behaviours associated with ALS. Several small-molecule strategies have been developed to address the underlying folding, metalation, stability and aggregation characteristics associated with ALS mutant SOD1 and target-specific sites on the SOD1 molecule.
The Trp32 site
Human SOD1 has a solvent-exposed tryptophan residue on the surface of β-strand 3. This residue is poorly conserved with threonine, serine, asparagine, glutamate, lysine and arginine all preferred across other eukaryotic CuZnSOD analogues. Indeed, only a few simian species and the African elephant are known to incorporate tryptophan at this position while a conservative phenylalanine substitution is found at the equivalent position in human extracellular CuZnSOD (SOD3) indicating hydrophobicity at this site has some functionality. Accordingly, Pokrishevsky et al. (Reference Pokrishevsky, McAlary, Farrawell, Zhao, Sher, Yerbury and Cashman2018) showed that substitution of Trp32 for the more frequently found serine decreases mutant and wild-type SOD1 stability and resistance to proteolytic digestion.
Despite reduced stability, the Trp32Ser substitution confers a protective effect on SOD1 aggregation. This includes slowed aggregation kinetics, an inability to propagate Trp32 SOD1 seeded aggregation, and a propensity to form amorphous rather than fibrillar aggregates (Grad et al., Reference Grad, Guest, Yanai, Pokrishevsky, O'Neill, Gibbs, Semenchenko, Yousefi, Wishart, Plotkin and Cashman2011; Pokrishevsky et al., Reference Pokrishevsky, McAlary, Farrawell, Zhao, Sher, Yerbury and Cashman2018). β-strand 3 has been found in the core of SOD1 aggregates (Bergh et al., Reference Bergh, Zetterström, Andersen, Brännström, Graffmo, Jonsson, Lang, Danielsson, Oliveberg and Marklund2015) so removing a hydrophobic interface may hinder fibril formation. All likely contribute to reduced loss of motor function on the expression of Trp32Ser human SOD1 in zebra fish in comparison with human wild-type (DuVal et al., Reference DuVal, Hinge, Snyder, Kanyo, Bratvold, Pokrishevsky, Cashman, Blinov, Kovalenko and Allison2019). Thus, inhibiting abnormal SOD1 self-interactions at Trp32 is a possible strategy to prevent aggregation and toxicity.
Several small molecules have been shown crystallographically to bind at or near the Trp32 site. This includes a series of aromatics which exploit a hydrophobic interaction with the Trp32 indole ring system (Fig. 11) (Antonyuk et al., Reference Antonyuk, Strange and Hasnain2010; Kershaw et al., Reference Kershaw, Wright, Sharma, Antonyuk, Strange, Berry, O'Neill and Hasnain2013; Ramu et al., Reference Ramu, Unni, Wright, Srinivas and Padmanabhan2018) and four catecholamines that interact with a groove in the SOD1 surface created by loop II close to Trp32 (Wright et al., Reference Wright, Antonyuk, Kershaw, Strange and Samar Hasnain2013). A fragment-linking approach created one compound which was able to bind the Trp32-loop II site with low micromolar affinity and inhibit Trp32 oxidation (Manjula et al., Reference Manjula, Wright, Strange and Padmanabhan2018), a post-translational modification known to facilitate aggregation (Taylor et al., Reference Taylor, Gibbs, Kabashi, Minotti, Durham and Agar2007).
5-Fluorouridine was also shown crystallographically to interact with the SOD1 Trp32 site (Fig. 11) (Wright et al., Reference Wright, Antonyuk, Kershaw, Strange and Samar Hasnain2013). Inclusion of 5-fluorouridine in cell culture media prior to exposure to human SOD1-ALS spinal cord homogenates could significantly reduce induced SOD1 aggregation (Pokrishevsky et al., Reference Pokrishevsky, Hong, Mackenzie and Cashman2017). 5-Fluorouridine, uridine and telbivudine have also been shown to have positive effects in cell and animal model systems including postponing disease onset and increasing survival in Gly93Ala transgenic mice (Pokrishevsky et al., Reference Pokrishevsky, McAlary, Farrawell, Zhao, Sher, Yerbury and Cashman2018; DuVal et al., Reference DuVal, Hinge, Snyder, Kanyo, Bratvold, Pokrishevsky, Cashman, Blinov, Kovalenko and Allison2019; Rando et al., Reference Rando, de la Torre, Martinez-Muriana, Zaragoza, Musaro, Hernández, Navarro, Toivonen and Osta2019).
The Cys111 site
Like Trp32, SOD1 Cys111 is poorly conserved and oxidation of its sulphydryl has a role in aggregation (Chen et al., Reference Chen, Shang, Qiu, Fujiwara, Cui, Li, Gao and Kong2012; Xu et al., Reference Xu, Liang, Li, He, Yuan, Huang, Liu, Tang, Pang, Du, Yang, Chen, Wang, Zhang and Liang2018). Opposing Cys111 residues are separated by a short distance across the SOD1 dimer interface. Covalent tethering of Cys111 side-chain sulphydryls by maleimide cross-linking was shown to substantially increase mutant SOD1 thermal stability (Auclair et al., Reference Auclair, Boggio, Petsko, Ringe and Agar2010). This experiment is reminiscent of covalent tethering of the SOD1 dimers by the introduction of a Val148Cys substitution performed by Ray et al. (Reference Ray, Nowak, Strokovich, Brown, Walz and Lansbury2004) which, taking cues from lysozyme and thymidylate synthase research, was able to reinforce Ala4Val SOD1 dimer affinity and prevent in vitro aggregation. We were able to mimic these effects by binding the small, antioxidant molecule ebselen to opposing Cys111 residues where they increased dimer affinity of Ala4Val SOD1 nearly 60-fold. In a second mode of action, ebselen was also an efficient catalyst for SOD1 disulphide bond formation which restored normal folding to Ala4Val and Gly93Ala mutants within cells (Capper et al., Reference Capper, Wright, Barbieri, Luchinat, Mercatelli, McAlary, Yerbury, O'Neill, Antonyuk, Banci and Hasnain2018).
Cisplatin addition to Cys111 prevents and even reverses aggregation of metal-free SOD1 in cell-free and cell-based models. The affinity of this interaction was found to be 37 µM and cisplatin was able to increase metal-free SOD1 thermal stability by 8 °C (Banci et al., Reference Banci, Bertini, Blaževitš, Calderone, Cantini, Mao, Trapananti, Vieru, Amori, Cozzolino and Carrì2012b). Crystallographic determination of the SOD1-cisplatin structure shows the ligand positioned at Cys111 (Fig. 11) but full ligand occupancy is not possible due to steric clashes across the dimer interface and cisplatin geometry is significantly distorted (Banci et al., Reference Banci, Bertini, Blaževitš, Calderone, Cantini, Mao, Trapananti, Vieru, Amori, Cozzolino and Carrì2012b; Shabalin et al., Reference Shabalin, Dauter, Jaskolski, Minor and Wlodawer2015).
Metal-coordinating sites
An inability to correctly metalate is a recurring characteristic of SOD1 mutants and increasing the availability of zinc, copper or both is a logical response. High doses of zinc are toxic to Gly93Ala transgenic mice but doses up to 60 mg kg−1 day−1 were tolerated. A higher dose of 90 mg kg−1 day−1 was also tolerated when supplemented with 0.3 mg kg−1 day−1 copper. In each case, mouse survival was increased by roughly 10 days on average (Ermilova et al., Reference Ermilova, Ermilov, Levy, Ho, Pereira and Beckman2005). A subsequent 2011 ALS clinical trial assessed a similar mixture of zinc and copper in humans for toxicity but details of progression beyond phase I/II are sparse.
Taking the alternative approach of increasing copper availability, diacetylbis(N(4)-methylthiosemicarbazonato) copper (II) (CuATSM) was shown to increase survival in Gly93Ala, Gly37Arg and Gly93Ala/hCCS overexpressing transgenic mice in a dose-dependent fashion (Soon et al., Reference Soon, Donnelly, Turner, Hung, Crouch, Sherratt, Tan, Lim, Lam, Bica, Lim, Hickey, Morizzi, Powell, Finkelstein, Culvenor, Masters, Duce, White, Barnham and Li2011; McAllum et al., Reference McAllum, Lim, Hickey, Paterson, Donnelly, Li, Liddell, Barnham, White and Crouch2013; Williams et al., Reference Williams, Trias, Beilby, Lopez, Labut, Samuel Bradford, Roberts, McAllum, Crouch, Rhoads, Pereira, Son, Elliott, Franco, Estévez, Barbeito and Beckman2016; Vieira et al., Reference Vieira, Hatzipetros, Thompson, Moreno, Kidd, Tassinari, Levine, Perrin and Gill2017). In a cell model, CuATSM increases SOD1 abundance and activity while reducing aggregation and cell death but only for non-metal-binding site mutants (Farrawell et al., Reference Farrawell, Yerbury, Plotkin, McAlary and Yerbury2019). CuATSM has recently completed a phase I clinical trials with a further phase I dose escalation trial underway and a phase II trial began on 30 September with completion date of 30 December 2020.
Promoting production of stable SOD1 over unfolded or aggregated species with pharmaceutical chaperones has been one of the two most explored avenues for SOD1-specific therapeutic development. What holds these strategies back as effective treatments for SOD1 ALS are combinations of toxicity, specificity, affinity and delivery; problems often associated with drug discovery but especially difficult to overcome when the target is in the central nervous system.
Biologics
Biologics, including antibodies and engineered proteins, offer a solution to the specificity problem above. The SOD1 interactome is relatively small but several interaction partners have the potential for therapeutic repurposing. This repertoire has been broadened by surface display techniques, directed evolution and novel antibody generation providing us with many proteins which are able to specifically interact with disulphide-reduced, metal-free, aggregated or soluble misfolded SOD1.
Antibodies
The production of antibodies which react specifically with SOD1 structural conformations commonly found in ALS has revolutionised our understanding of the SOD1 structure–misfunction relationship in the last decade or so. These antibodies are often termed misfolding-specific because they preferentially bind epitopes that are not accessible when SOD1 is mature, either through internalisation or reduced conformational dynamics. The first such antibody was a polyclonal generated against a peptide found in β-strand 8 including the disulphide-forming Cys146 and specifically recognised monomeric SOD1 (Rakhit et al., Reference Rakhit, Robertson, Vande Velde, Horne, Ruth, Griffin, Cleveland, Cashman and Chakrabartty2007). Together with analytical or diagnostic use, conformation or mutant-specific antibodies may also have therapeutic applications. While SOD1 aggregates are found within cells, there is strong evidence that aggregates or small misfolded species can traverse the extracellular space and seed aggregation in surrounding cells. If this toxic transport could be inhibited, the progressive nature of ALS could be arrested. Justification for this comes in part from work showing the presence of native SOD1 reactive antibodies in sporadic ALS sufferers with longer than mean survival (van Blitterswijk et al., Reference van Blitterswijk, Gulati, Smoot, Jaffa, Maher, Hyman, Ivinson, Scherzer, Schoenfeld, Cudkowicz, Brown and Bosco2011).
The D3H5 IgG antibody was generated against recombinant, metal-free Gly93Ala SOD1 and when delivered directly to the cerebral ventricles was able to postpone weight loss and increase life span by small amounts. Increasing the period over which the antibody was administered and starting the treatment earlier increased survival from +6 to +9 days compared to control Gly93Ala transgenic mice (Gros-Louis et al., Reference Gros-Louis, Soucy, Larivière and Julien2010).
Active immunisation with peptides designed to create a native immunogenic response specific for misfolded SOD1 has also been shown to increase life-span and slow disease progression in transgenic mice (Urushitani et al., Reference Urushitani, Ezzi and Julien2007; Liu et al., Reference Liu, Tjostheim, Dasilva, Taylor, Zhao, Rakhit, Brown, Chakrabartty, McLaurin and Robertson2012). However, selection of the immunising peptide has to be carefully considered to not induce an immune response which in itself can be life threatening (Sábado et al., Reference Sábado, Casanovas, Rodrigo, Arqué and Esquerda2015).
Single-chain recognition proteins
High-affinity antibodies can now be generated through traditional protocols cheaply and efficiently. However, their size, flexibility and quaternary structure present a problem for structural study, rational redesign and intracellular delivery. Single-chain recognition domains circumvent these problems and have found interesting uses in relation to SOD1.
Patents have been filed for SOD1-reactive nanobodies; small, single-chain antibodies often generated by immunisation of camelids. Micro-nanomolar binding affinity for wild-type and mutant SOD1 has been described which was able to prevent the formation of large Ala4Val SOD1 fibrils in vitro with some protein forming small amorphous aggregates. Expression of the nanobody within cells reduced SOD1 aggregation and a single direct delivery of nanobody to Gly93Ala SOD1 transgenic mouse brain delayed disease onset and prolonged survival (Robberecht et al., Reference Robberecht, Rousseau and Schymkowitz2016).
A thermophilic variant of the immunoglobin-binding protein G B1 domain (HTB1) was optimised for SOD1 binding by directing mutations to putative stable regions on the surface of the protein followed by yeast surface display (Banerjee et al., Reference Banerjee, Oren, Ben-Zeev, Taube, Engel and Papo2017). Unmodified HTB1 could not bind SOD1 while the product of this process was a binding domain with 300 and 674 nM dissociation constants for Gly93Ala and Gly37Arg, respectively. In vitro aggregation in the presence of modified HTB1 showed SOD1 forming small amorphous aggregates rather than large fibrils, reduced intracellular SOD1 misfolding and increased cell viability. The versatility of this technique is illustrated by the successful retargeting of HTB1 to bind amyloid-β peptide with low micromolar affinity with positive effects on aggregation and toxicity (Banerjee et al., Reference Banerjee, Oren, Dagan, Taube, Engel and Papo2018).
Using phage display in bacteria, Ghadge et al. (Reference Ghadge, Pavlovic, Parthasarathy, Kay and Roos2013) created two single chain variable fragments (scFv) from a pre-existing library which were able to interact with wild-type, Ala4Val and Val148Gly SOD1 but not Gly93Ala SOD1 possibly indicating Gly93 forms part of the protein–protein interface. When co-expressed with Ala4Val SOD1 in neuronal-like cells, SOD1 aggregation was reduced and cell viability was increased. Ghadge et al. (Reference Ghadge, Kay, Drigotas and Roos2018) then showed that scFV adeno-associated viral (AAV) delivery to Gly93Ala transgenic mice increased survival when administered neonatally and just prior to the onset of symptoms. On the whole, single chain recognition domains do not replicate the very high target affinity that can be found with well-selected antibodies. However, when combined with reliable, efficient and targeted gene delivery by AAV vectors, these small proteins are a very promising new strategy to engage and change the fate of many cellular proteins including SOD1.
Conclusion
After more than 50 years of concerted effort, we have gained a very detailed understanding of SOD1 biology. Despite this body of work, new SOD1 functions and behaviour are regularly discovered including, in recent years, a role in signalling, stress granule formation and transcriptional regulation. Perhaps the most enigmatic behaviour of SOD1 discovered over these years is its propensity to aggregate and the ramifications this has on ALS.
We now understand how ALS mutations cause aggregation in SOD1-ALS. Generally, mutations to conserved residues in the metal-binding, disulphide, dimer interface half of the SOD1 β-barrel prevent adequate folding and acquisition of a stable structure. These unfolded structures predispose mutant SOD1 to degradation but also lead to the accumulation of the non-conserved part of the barrel into non-amyloid fibrils or amorphous aggregates. While the exact structure of these aggregates remains unresolved, the research community is developing the tools with which to arrest their formation. Small-molecule therapies including CuATSM and ebselen confer order on the functional part of the SOD1 β-barrel while Trp32-targeting compounds bind in the region known to form the core of SOD1 aggregates. What currently limits the usefulness of these compounds as therapeutics, either singly or as a cocktail, is a combination of affinity, specificity and delivery. Honing delivery of zinc and copper, the specificity of disulphide formation and the affinity of anti-aggregation compounds will be necessary for this strategy to succeed in the clinic.
The presence of SOD1 aggregates in mutant SOD1 cases of ALS is unquestionable but there is also strong evidence that wild-type SOD1 is a component of aggregates in at least some sporadic ALS cases (Shibata et al., Reference Shibata, Hirano, Kobayashi, Sasaki, Kato, Matsumoto, Shiozawa, Komori, Ikemoto and Umahara1994, Reference Shibata, Asayama, Hirano and Kobayashi1996b). There are also many reports of misfolded SOD1 in sporadic ALS cases (Bosco et al., Reference Bosco, Morfini, Karabacak, Song, Gros-Louis, Pasinelli, Goolsby, Fontaine, Lemay, McKenna-Yasek, Frosch, Agar, Julien, Brady and Brown2010; Forsberg et al., Reference Forsberg, Jonsson, Andersen, Bergemalm, Graffmo, Hultdin, Jacobsson, Rosquist, Marklund and Brännström2010; Pokrishevsky et al., Reference Pokrishevsky, Grad, Yousefi, Wang, Mackenzie and Cashman2012; Grad et al., Reference Grad, Yerbury, Turner, Guest, Pokrishevsky, O'Neill, Yanai, Silverman, Zeineddine, Corcoran, Kumita, Luheshi, Yousefi, Coleman, Hill, Plotkin, Mackenzie and Cashman2014; Paré et al., Reference Paré, Lehmann, Beaudin, Nordström, Saikali, Julien, Gilthorpe, Marklund, Cashman, Andersen, Forsberg, Dupré, Gould, Brännström and Gros-Louis2018). Some of the secondary post-translational modifications described in ‘Secondary SOD1 post-translational modifications’ section may be responsible for destabilising wild-type SOD1 and lead to misfolding (Bosco et al., Reference Bosco, Morfini, Karabacak, Song, Gros-Louis, Pasinelli, Goolsby, Fontaine, Lemay, McKenna-Yasek, Frosch, Agar, Julien, Brady and Brown2010). This topic is however contentious; Da Cruz et al. (Reference Da Cruz, Bui, Saberi, Lee, Stauffer, McAlonis-Downes, Schulte, Pizzo, Parone, Cleveland and Ravits2017) found no SOD1 misfolding in sporadic ALS samples. If differences in detection are eliminated, this discrepancy must arise from the tissue source, inherent variability of patient samples and subjective data interpretation.
Heterogeneity is common in ALS and we should not be surprised that some sporadic cases, or tissues therein, exhibit SOD1 misfolding and aggregation while others do not. The size of aggregates, the abundance of misfolded SOD1 and the proportion of cells that display these characteristics is also key here. Discovery of misfolded SOD1 in non-SOD1 fALS cases, Parkinson's disease and aged but healthy individuals supports the notion that SOD1 aggregates are a commonality in ageing and neurodegenerative disease (Nishiyama et al., Reference Nishiyama, Murayama, Shimizu, Ohya, Kwak, Asayama and Kanazawa1995; Trist et al., Reference Trist, Davies, Cottam, Genoud, Ortega, Roudeau, Carmona, De Silva, Wasinger, Lewis, Sachdev, Smith, Troakes, Vance, Shaw, Al-Sarraj, Ball, Halliday, Hare and Double2017; Forsberg et al., Reference Forsberg, Graffmo, Pakkenberg, Weber, Nielsen, Marklund, Brännström and Andersen2019). In each case, the size and number of these inclusions was lower than is common in SOD1-ALS patients, thus it may be that investigators expect to see large mutant SOD1-like aggregates when much smaller, punctate forms are missed in non-SOD1 ALS tissue samples.
If SOD1 aggregation is common to mutant SOD1-ALS, non-SOD1 ALS, neurodegenerative disease in general and ageing, is it toxic? The toxicity of protein or peptide aggregates has been questioned recently across neurodegenerative disease research. Low molecular mass misfolded species including SOD1 trimers with non-native secondary, tertiary and quaternary structure have been posited to be toxic (Zhu et al., Reference Zhu, Beck, Griffith, Deshmukh and Dokholyan2018). These species may exist within cells but there is currently no clear rationale or mechanistic understanding as to their toxicity. Basic science examining this concept is of the upmost importance if we are to create new, viable therapeutics to specifically address these species.
The linkage of SOD1 with ALS in 1993 stimulated a proliferation in research and understanding of SOD1 biology. Throughout, biophysics has been at the forefront, and as we find answers to the questions highlighted above, biophysics will also doubtless continue to be instrumental.
Acknowledgements
We would like to thank the current and past members of the Molecular Biophysics Group particularly Drs Michael Capper, Michael Hough, Gunter Grossmann, Lorreta Murphy and Richard Strange for their contributions over many years of research on SOD1 and SOD1-ALS. We would also like to acknowledge discussions with members of the ICOSA, an international consortium on SOD-ALS that was established in September 2001 bringing together the laboratories of Hart, Hasnain, Hayward and Valentine to work on understanding SOD1-ALS. This group was further extended to include several other laboratories that functioned as a consortium until 2012. Our programme on ALS has been supported by the Motor Neurone Disease Association for several years (Oct02/092; 06/6015; Mar09/6055; Apr15/833-791) and more recently by MRC's MRF (MRF-060-0002-RG-HASNA) and ALSA foundation (20-DDC-492). Recent award to G.S.A.W. for a MNDA fellowship (Wright/Oct18/969-799) is also acknowledged. We have benefited from extensive use of synchrotron radiation facilities including the SRS (Daresbury), Diamond Light Source (Didcot), Swiss Light Source (Villigen) and synchrotron Soleil (Paris).
Conflict of interest
The authors declare no conflicts of interest.












 Open access
Open access