


Appropriate nutritional intake and utilisation are essential for proper functioning of our organism. Indeed, deficiency or excess of certain nutrients are at the origin of many diseases, including diabetes mellitus. Nutrients can be subdivided in two main categories, micronutrients and macronutrients. Micronutrients include vitamins and minerals and are essential for the function of cells and organ systems( Reference Huskisson, Maggini and Ruf 1 , Reference Shenkin 2 ). Macronutrients include carbohydrates, amino acids and fat, and serve as energy sources and structural components of the cells( Reference Huskisson, Maggini and Ruf 1 ). The fate of the ingested macronutrients is determined by an integrated network of hormonal and neural signals which, according to the metabolic status of the organism, orchestrate the immediate use of the ingested molecules, their biochemical transformation or their long-term storage( Reference Berthoud 3 ). An in depth knowledge of the processes regulating nutrient uptake and utilisation is essential for understanding the mechanisms responsible of metabolic homeostasis and for elucidating the causes of metabolic disorders such as diabetes mellitus.
Sensory contact with food and stimulation of the oral cavity elicit salivation, gastric acid production and pancreatic exocrine and endocrine secretions( Reference Power and Schulkin 4 , Reference Giduck, Threatte and Kare 5 ). These early responses referred to as cephalic phase serve to prepare the digestive tract for digestion, absorption and utilisation of nutrients and are followed by a gastrointestinal phase when food reaches the stomach( Reference Power and Schulkin 4 – Reference Teff 6 ). During this second phase, the gastro-duodeno-jejunal mucosa and the enteric nervous system release numerous peptides, entero-hormones and neurotransmitters via paracrine, endocrine and neural mechanisms that coordinate nutrient absorption and utilisation to achieve metabolic homeostasis( Reference Konturek, Pepera and Zabielski 7 , Reference Sanger and Lee 8 ).
Insulin, a peptide hormone produced by pancreatic β-cells within the islets of Langerhans is at the core of this complex regulatory network and plays an essential role in blood glucose homeostasis and in the control of body metabolism. The amount of insulin released in the circulation is precisely tuned to prevent tissue damage caused by chronic hyperglycaemia and the life-threatening effects of prolonged hypoglycaemia( Reference Stumvoll 9 , Reference Ferrannini 10 ). The aim of this review will be to discuss the direct and indirect impacts of macronutrients on β-cells and to evaluate their contribution to the control of insulin secretion under physiological and pathological conditions.
Pancreatic β-cells as nutrient sensors
Pancreatic β-cells are highly differentiated cells that are often referred to as the ‘fuel sensors’ because of their capacity to monitor and respond to dietary nutrients( Reference Nolan and Prentki 11 , Reference Prentki, Matschinsky and Madiraju 12 ). The task of β-cells is to detect the changes in the concentration of nutrients in the bloodstream and to release appropriate amounts of insulin to ensure prompt and efficient metabolic disposal( Reference Newsholme, Cruzat and Arfuso 13 ). Insulin secretion in man and animals is pulsatile and follows the oscillatory metabolism of nutrients( Reference Gilon, Ravier and Jonas 14 , Reference Lefebvre, Paolisso and Scheen 15 ). The β-cells are able to integrate a variety of signals elicited by nutrients in the gut, brain and in the β-cells themselves permitting the fine-tuning of insulin release( Reference Zietek and Daniel 16 – Reference Torres, Noriega and Tovar 18 ) (Fig. 1).
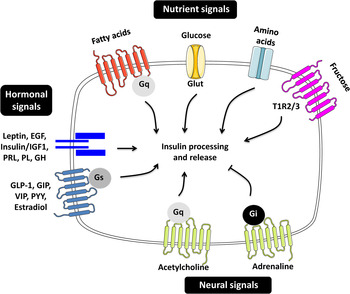
Fig. 1. (Colour online) Regulators of insulin secretion through nutrient, hormonal and neural signals. The figure summarises the different molecules contributing to the fine-tuning of insulin secretion. EGF, epidermal growth factor; IGF1, insulin-like growth factor 1; PRL, prolactin; PL, placental lactogen; GH, growth hormone; GLP-1, glucagon-like peptide 1; GIP, gastric inhibitory polypeptide; VIP, vasoactive intestinal polypeptide; PYY, peptide YY; G, guanine nucleotide-binding protein; Glut, glucose transporter; TIR, taste receptor type 1 member.
Cephalic insulin release: far from anecdotic
In human subjects and rodents there is a robust cephalic phase insulin release (CPIR) that occurs in response to sensory stimulation of the oral cavity caused by mastication, tasting or food ingestion( Reference Teff 19 , Reference Powley and Berthoud 20 ). Several studies in human subjects and rodents demonstrated that blockade of the insulin response during this phase results in poor glycaemic control and reduces the immediate food intake( Reference Ahren 21 ). Insulin release during the pre-absorptive period is believed to contribute to the optimisation of postprandial glucose homeostasis by preventing a rapid rise in plasma glucose levels and an exaggerated insulin peak. CPIR is also able to inhibit glucose production in the liver and lipolysis in adipose tissue and thus to regulate energy homeostasis( Reference Picard, Naimi and Richard 22 , Reference Mitrakou, Kelley and Mokan 23 ). Obese and type 2 diabetes (T2D) subjects who are insulin resistant and hyperinsulinaemic display a reduced CPIR, resulting in elevation of postprandial glucose levels by about 40 %( Reference Ahren 21 , Reference Calles-Escandon and Robbins 24 ). Remarkably, mimicking CPIR by the infusion of tiny insulin amounts prior or during the first 10 min of food ingestion, had no effect on postprandial insulin or glucose levels in lean subjects but improved glucose control in obese( Reference Teff and Townsend 25 ) and in both T1D( Reference Kraegen, Chisholm and McNamara 26 ) and T2D patients( Reference Bruttomesso, Pianta and Mari 27 ). These data strengthen the importance of early insulin release for maintaining postprandial glucose clearance and homeostasis and suggest that defective CPIR may contribute to perturbed glucose tolerance associated with metabolic disorders and diabetes.
Gastrointestinal phase of insulin secretion
Upon arrival into the gut, the nutrients activate a regulatory signalling network between the gut and the pancreatic islets that plays a central role in metabolic homeostasis( Reference Schwartz, Seeley and Tschop 28 ). This entero-insular axis involves the release of numerous hormones by entero-endocrine cells of the gastric-duodeno-jejunal mucosa exerting an insulinotropic action( Reference Sala, Torrinhas and Giannella-Neto 29 ) (Table 1). This so-called incretin effect is responsible for the differences observed in the amount of insulin released following oral and infused food intake( Reference Perley and Kipnis 30 ). Although the list of gastric and gut-derived hormones and peptides is still expanding, the strongest candidates for the incretin effect are the glucose-dependent insulinotropic polypeptide and glucagon-like peptide 1( Reference Ma, Rayner and Jones 31 , Reference Drucker 32 ). Recently, evidence for an entero-endocrine signal attenuating insulin secretion under fasting conditions has also been provided. Indeed, the Drosophila peptide limostatin released from nutrient-sensing cells in the gut and its mammalian orthologue neuromedin U were proposed to decrease insulin production by directly targeting the β-cells( Reference Alfa, Park and Skelly 33 ).
Table 1. Gut-derived peptides and peptide hormones in the gastrointestinal tract

The passage of nutrients through the gastrointestinal tract stimulates also the secretion of numerous neuropeptides (neuropeptide Y, neuromedin, opioid-like peptides (enkephalin and endorphin), galanin, vasoactive intestinal polypeptide, calcitonin gene-related peptide, substance P, taurine, etc.) and neurotransmitters (acetylcholine, norepinephrine, serotonin, γ-aminobutyric acid, ATP, nitric oxide, etc.) by the enteric nervous system( Reference Genton and Kudsk 34 ) (Table 1). These molecules can affect pancreatic secretion through the vagal nerve and contribute to the regulation of insulin release both in the early and postprandial phases( Reference Konturek, Pepera and Zabielski 7 ). The activation of enteric neurons is a major component of the so-called gut–brain axis. The complex and fascinating connections between the gastrointestinal tract and the central and peripheral nervous systems has been extensively reviewed elsewhere( Reference Al Omran and Aziz 35 , Reference Mayer 36 ) and will not be discussed further in this paper.
Impact of nutrients on β-cell signalling
The metabolic signalling pathways elicited in β-cells by nutrients and culminating in insulin secretion have been intensively investigated for decades. Because of space constrains, in this section we will only highlight the main molecular mechanisms governing the effects of macronutrients on β-cells and we refer the reader to other excellent reviews that have extensively addressed this matter( Reference Nolan and Prentki 11 , Reference Prentki, Matschinsky and Madiraju 12 , Reference Rutter, Pullen and Hodson 37 ). Nutrient-induced insulin secretion from β-cells occurs through a unique signal transduction system, which differs considerably from that of neuromodulators or peptide hormones. Indeed, nutrients must be metabolised in the β-cell to cause insulin secretion( Reference Prentki, Matschinsky and Madiraju 12 ). In contrast, other secretagogues, such as incretins, cytokines, neurotransmitters, etc. modulate insulin secretion by binding to specific cell-surface receptors and by activating signalling cascades that involve the production of classical second messengers such as Ca2+ and cAMP( Reference Donath, Storling and Berchtold 38 , Reference Rutter and Hodson 39 ). Nutrients, such as glucose and fatty acids, have a dual effect on β-cell function. Acute exposure of β-cells to elevated glucose or fatty acid concentrations stimulates insulin secretion, while prolonged exposure to these same nutrients causes impaired insulin secretion, characterised by excessive hormone release at low glucose concentrations and no further increase upon glucose rise( Reference Eizirik, Korbutt and Hellerstrom 40 , Reference Davalli, Ricordi and Socci 41 ). Glucose enters the β-cells and is metabolised, resulting in an increase in the ATP:ADP ratio, closure of ATP-sensitive K+ channels and membrane depolarisation. This will in turn trigger the opening of L-type Ca2+ channels, causing a rapid rise in intracellular Ca2+ concentration and the fusion of insulin-containing granules with the plasma membrane( Reference Henquin 42 ). Although glucose is unequivocally the principal factor triggering insulin release, several other macro- and micronutrients act synergistically with glucose to potentiate secretion. Until recently, the dietary monosaccharide fructose was believed to be unable to stimulate insulin secretion because β-cells do not express the fructose transporter GLUT5( Reference Curry 43 , Reference Sato, Ito and Udaka 44 ). However, recent work has provided evidence that fructose can potentiate glucose-induced insulin secretion by binding to the sweet taste receptors that are present both in mouse and human β-cells( Reference Kyriazis, Soundarapandian and Tyrberg 45 – Reference Malaisse 47 ). NEFA have also the capacity to amplify insulin secretion, through three interdependent processes, defined as the ‘trident model’ of β-cell signalling( Reference Nolan, Madiraju and Delghingaro-Augusto 48 ). Two pathways involve intracellular fatty acid metabolism, whereas the last one relies on the activation of a membrane-bound NEFA receptor. NEFA potentiation of glucose-induced insulin secretion is particularly effective and vital under conditions of insulin resistance, when β-cells are called to compensate for the increased insulin needs( Reference Nolan, Leahy and Delghingaro-Augusto 49 , Reference Prentki and Nolan 50 ). Although dietary proteins by themselves do not provoke a frank insulin excursion, co-ingested with carbohydrates they can markedly potentiate the insulin response. However, their impact on insulin secretion varies depending on the quality and quantity of proteins present in the meals( Reference Esteves de Oliveira, Pinheiro Volp and Alfenas 51 ). Diets with low protein content induce a mild insulin secretion, whereas a high-protein meal potentiates the insulinaemic response. The reduced glycaemic excursion in response to proteins and fat added on top of carbohydrates appear to be lost or attenuated in diabetic subjects( Reference Gannon, Ercan and Westphal 52 ). The amino acid composition will determine how insulin secretion is induced( Reference Newsholme, Bender and Kiely 53 ). The cationic amino acid L-arginine, induces plasma membrane depolarisation and triggers insulin granule exocytosis upon Ca2+ entry through voltage-gated channels. L-alanine is co-transported with Na+ and induces cell membrane depolarisation, voltage-dependent Ca2+ channel opening and, consequently, insulin granules exocytosis. The metabolism of alanine results in increased intracellular ATP levels and activation of a signalling cascade leading to insulin exocytosis. Aspartate and glutamate are key components of the NADH shuttles, a primordial mechanism to achieve efficient glucose oxidation( Reference Newsholme, Bender and Kiely 53 ).
In addition to being insulin secretagogues, nutrients regulate proliferation and survival of β-cells( Reference Bouwens and Rooman 54 , Reference Oh 55 ) and exposure to nutrients can affect β-cell fate and characteristics. A recent study allowed the identification in larval zebrafish of a compensatory mechanism in which β-cells promote differentiation of new endocrine precursor cells in response to overnutrition and to the resulting insufficient insulin secretory capacity( Reference Li, Maddison and Page-McCaw 56 ). Similar results were found in the mature zebrafish pancreas with the identification of active nutrient-sensitive progenitors and β-cell differentiation in response to metabolic cues( Reference Ninov, Hesselson and Gut 57 ). The same authors observed also a dramatic increase in β-cell proliferation in response to a high-energy diet. Both, β-cell proliferation and differentiation were associated with the down-regulation of the Notch signalling pathway and to the activation of mammalian target of rapamycin-dependent signalling( Reference Ninov, Hesselson and Gut 57 ). Dor and co-workers have further characterised the tight regulation of β-cell maturation and function in response to nutrient stimuli and food composition( Reference Stolovich-Rain, Enk and Vikesa 58 ). Indeed, in mice, the transition from the high-fat mother milk to a carbohydrate-rich chow diet, enables the β-cells to acquire their glucose responsiveness and their capacity to proliferate under conditions of increased insulin demand( Reference Stolovich-Rain, Enk and Vikesa 58 ). The nutritional transition occurring in this critical developmental window are therefore essential for the acquisition of an appropriate functional β-cell mass.
Impact of nutrients on β-cell gene expression
The pancreatic islets are highly vascularised structures and nutrients and other circulating factors impact not only on β-cell secretory functions, but also on other β-cell activities such as proliferation and survival. Indeed, glucose sensing regulates both basal β-cell proliferation rate and their capacity to regenerate following injury. Dor and co-workers provided evidence that glucose-induced proliferation requires the activation of glucokinase, the enzyme that catalyses the initial step of glucose utilisation in β-cells( Reference Porat, Weinberg-Corem and Tornovsky-Babaey 59 ). Further investigations revealed a β-cell-specific regulation of the level of cyclin D2 mRNA driven by glucose metabolism exerted through the activation of a Ca2+-dependent pathway( Reference Salpeter, Klochendler and Weinberg-Corem 60 ).
However, chronic exposure to elevated concentrations of glucose and lipids is detrimental for β-cells. The excess of nutrients can lead to deterioration of β-cell function through the modification of transcriptional, post-transcriptional and translational events. Multiple independent studies that analysed the transcriptomic profile of human or rodent diabetic islets or of islets exposed to a diabetogenic environment, identified alterations in the expression of genes associated with insulin processing and secretion (e.g. Pcsk1/2, GLP1R), lipid metabolism (e.g. stearoyl-CoA desaturase 1, stearoyl-CoA desaturase 2 and fatty acid desaturase-2) oxidative stress (e.g. Cdkn1b, Tmem27, Pax6, Cat, Prdx4 and Txnip), cell proliferation and islet cell differentiation (e.g. Cdkn1b, Tmem27 and Pax6)( Reference Dreja, Jovanovic and Rasche 61 – Reference Imai, Patel and Doliba 63 ). A better understanding of the role of these differentially expressed genes in obese- and diabetes-associated settings may help understanding the factors linking obesity to impaired islet-cell activity. Reduced expression of the glucose-transporters GLUT1 and GLUT2 (encoded by Slc1a1 and Slc2a2 genes, respectively) have been reported in human islets isolated from hyperglycaemic T2D donors( Reference Del Guerra, Lupi and Marselli 64 ). In vitro investigations in an insulin-secreting β-cell line demonstrated that, in the presence of chronically elevated extracellular glucose concentrations, GLUT2 is either directly degraded at the plasma membrane or undergoes endocytosis followed by a rapid degradation( Reference Hou, Williams and Vicogne 65 ). The glucose-dependent degradation of GLUT2 suggests that systemic nutrient overload can directly contribute to impaired β-cell glucose sensing and to the consequent loss of metabolic homeostasis. Glucose controls also the binding of several key transcription factors (e.g. MafA, NeuroD and PDX1) to the insulin gene promoter and is thus a major physiologic modulator of insulin gene expression( Reference Poitout, Hagman and Stein 66 ). Impaired PDX1 and MafA binding to the insulin promoter is also observed upon prolonged exposure to the SFA palmitate( Reference Hagman, Hays and Parazzoli 67 ). Hence, both, glucose and palmitate are able to affect the binding of transcription factors that control the activity of the insulin promoter, pointing to an involvement of these regulators of gene expression in the mechanisms of glucolipotoxicity( Reference Poitout, Amyot and Semache 68 ).
Emerging evidence suggests an impact of diet-dependent epigenetic modifications in the aetiology of metabolic disorders( Reference Schwenk, Vogel and Schurmann 69 ). Hu et al.( Reference Hu, Xu and He 70 ) observed an aberrant DNA methylation profile in β-cells cultured for 1 month in a medium containing high glucose and lipid concentrations. Although, DNA methylation is a key event associated with gene silencing, TCF7L2 mRNA was unexpectedly increased, whereas the protein Tcf7l2 was reduced in islets under conditions of glucolipotoxicity. The transcription factor Tcf7l2 has been shown to regulate both β-cell proliferation and insulin secretion( Reference Welters and Kulkarni 71 ). Elevated mRNA levels of Tcf7l2 and reduced protein levels have also been observed in islets of T2D patients and in diabetic animal models( Reference Hu, Xu and He 70 , Reference Pradas-Juni, Nicod and Fernandez-Rebollo 72 , Reference Morrison, Locke and Arif 73 ), emphasising the potential involvement of nutrient-induced gene expression changes in metabolic disturbances. Ling and colleagues analysed the genome-wide expression profile of palmitate-treated human islets and identified 1860 differentially expressed genes. Among them, thirty-seven genes were also differentially expressed in islets from T2D individuals. Interestingly, this study identified changes in the DNA methylation pattern of multiple diabetes candidate genes, such as TCF7L2 and GLIS3( Reference Hall, Volkov and Dayeh 74 ). These results suggest that lipid-induced epigenetic modifications may affect glucose-stimulated insulin secretion and/or β-cell survival by impacting on gene expression.
In addition to the effect of the nutritional environment on metabolic health in the adulthood, numerous studies have attempted to elucidate the influence and the long-term consequences of a perturbed prenatal metabolic status. Maternal obesity and excessive energy intake during pregnancy were found to expose the fetus to nutrient surfeit and to substantially increase the likelihood of an individual to become obese and develop metabolic disorders( Reference Vaag, Brons and Gillberg 75 , Reference Heerwagen, Miller and Barbour 76 ). Indeed, the offspring of mothers fed a high-fat diet (HFD) throughout pregnancy and lactation exhibits a remarkably similar obesogenic phenotype, characterised by increased fat mass, hyperinsulinaemia and hyperleptinaemia( Reference Howie, Sloboda and Kamal 77 ). Strikingly, these phenotypic traits are associated with the abnormal expression of regulatory genes in the pancreas of adult offspring, including increased mRNA levels of INS1, INS2, proinflammatory cytokines (TNF-α, CD68 and IL1-R1), STAT3 and reduced expression of PI3K. These changes have been reported in the adult pancreas of offspring from mothers exposed to an obesogenic diet either pre-conception and throughout pregnancy and lactation, or merely during pregnancy and lactation periods( Reference Howie, Sloboda and Reynolds 78 ).
Protein-restricted diets during pregnancy have also a major impact on fetal pancreas and modify the expression of more than 10 % of the islet genes. The alterations concern mainly genes associated with the tricarboxylic acid cycle, ATP production, cell proliferation and anti-oxidative defence pathways and are prevented by maternal taurine supplementation throughout pregnancy( Reference Reusens, Sparre and Kalbe 79 ). Epigenetic marks in the pancreatic genome of the offspring are believed to link the maternal diet to the susceptibility of developing obesity and diabetes, a phenomenon known as cellular memory. Indeed, maternal protein restriction elicited permanent modifications in histone marks in the enhancer region of the Hnf4a locus, resulting in the down-regulation of Hnf4a in the islets of the offspring( Reference Sandovici, Smith and Nitert 80 ). Impaired expression of this transcription factor can have a major impact on β-cell activities. Indeed, mutations in the Hnf4a gene result in the development of a particular form of diabetes (maturity onset diabetes of the young type 1)( Reference Kapoor, Locke and Colclough 81 , Reference Pearson, Boj and Steele 82 ).
So far, most studies on the influence of the parental diet on metabolism and glucose homeostasis in the offspring have focused on the role of the mothers. Nonetheless, accumulating evidence suggest that the paternal diet has also an impact on the offspring metabolism. Morris and co-workers studied the transgenerational effect of paternal obesity and high-fat feeding on rat progeny. Their study revealed that chronic exposure of male rats to HFD programmes the dysfunction of β-cells in their female offspring causing the appearance of an early phenotype of glucose intolerance. The impaired glucose clearance worsens with ageing and is associated with altered expression of 642 genes in the islets of adult female offspring( Reference Ng, Lin and Laybutt 83 ). Gene ontology and KEGG pathway analysis highlighted an enrichment of dysregulated genes involved in insulin and glucose metabolism, as well as calcium-, MAPK- and Wnt-signalling, in the control of apoptosis and of the cell cycle. These transcriptomic changes were also accompanied by epigenetic alterations such as hypomethylation of the Il13ra2 gene encoding for the IL-13 receptor subunit α−2( Reference Ng, Lin and Laybutt 83 ). In a second survey, the same group extended this study by comparing the gene networks affected in retroperitoneal white adipose tissue and in pancreatic islets( Reference Ng, Lin and Maloney 84 ). Their analysis revealed that paternal diet-induced obesity modifies the same cellular processes and signalling pathways in the fat tissue and in islets. In particular, they found that many genes encoding olfactory receptors are down-regulated in the progeny. These results suggest that paternal HFD exerts transgenerational regulation of the nutrient-sensing machinery and causes impaired glucose homeostasis in F1 generation( Reference Ng, Lin and Maloney 84 ).
Another study carried out in fruit flies reported that increased sugar in the diet of males for just 1 or 2 d before mating can lead to obesity in the next generation( Reference Ost, Lempradl and Casas 85 ). High dietary sugar led to modifications in gene expression through epigenetic changes, without affecting growth and development in the progeny. Specifically, the transcriptomic profile of the offspring was characterised by an active deposition of the histones H3K9me3 and H3K27me3 and by changes in the expression of genes involved in key metabolic pathways, including glycolysis, Krebs cycle, mitochondrial metabolism and polysaccharide metabolism( Reference Ost, Lempradl and Casas 85 ). These findings strengthen the conclusion that nutrient-induced transgenerational DNA methylation and their consequent modifications in gene expression can act as an endocrine disruptor and promote the development of impaired metabolic phenotypes.
Overall, these studies carried out in mammals and flies highlight the role of non-genetic factors in the transgenerational susceptibility to metabolic disorders. For an in depth description of the mechanisms through which the parental diet influences the metabolic phenotype in the offspring, we refer the reader to a recent review by Rando and Simons( Reference Rando and Simmons 86 ).
Impact of nutrients on non-coding RNA expression in β-cells
The human genome contains about 21 000 protein-coding genes but the DNA sequences driving protein expression represent only about 2 % of the 3·2 billion base pairs constituting our genome. Until recently, the sequences not involved in protein expression were considered evolutionary relic, irrelevant for the control of cellular activities. The advent of new high-throughput sequencing techniques permitting a systematic analysis of all RNA present in the cells has dramatically changed this view. Indeed, the results of the ENCODE (Encyclopedia of DNA Elements) project, an initiative launched in 2003 to identify all functional elements in the human genome, revealed that most DNA sequences can be transcribed to RNA giving rise to thousands of RNA molecules that are not translated to protein sequences( 87 , Reference Genomes Project, Abecasis and Altshuler 88 ). These non-coding RNA transcripts fall in distinct categories according to their length and functional characteristics. Small non-coding RNA that are shorter than 200 nucleotides include well-described molecules such as transfer RNA, small nucleolar RNA and small nuclear RNA, but also two large classes of newly discovered molecules, the Piwi-associated RNA and the microRNA (miRNA)( Reference Naqvi, Islam and Choudhury 89 , Reference Dozmorov, Giles and Koelsch 90 ). Piwi-associated RNA are particularly abundant in the germline where they contribute to maintain genome integrity by preventing transposon movement( Reference Siomi, Sato and Pezic 91 , Reference Grivna, Beyret and Wang 92 ). MiRNA are expressed in virtually all cells and are involved in a variety of physiological processes, including cell differentiation, proliferation, apoptosis and in the development of many diseases, including cancer and diabetes( Reference Dumortier, Hinault and Van Obberghen 93 – Reference Guay, Roggli and Nesca 95 ). These non-coding small RNA that are typically 21–23 nucleotide long are major regulators of gene expression. Each miRNA can partially pair to 3′untraslated regions of more than one hundred different target mRNA leading to translational repression and/or messenger degradation( Reference Bartel 96 ). The newly discovered long non-coding RNA (lncRNA) are longer than 200 nucleotides and can be involved in numerous gene regulatory activities such as transcription, splicing, protein degradation and chromatin modifications( Reference Wilusz, Sunwoo and Spector 97 , Reference Ulitsky and Bartel 98 ).
There is increasing evidence that non-coding RNA actively contribute to the control of vital functions in the organism, including the maintenance of metabolic homeostasis. Indeed, many non-coding RNA have been shown to be mis-expressed in human metabolic disorders. Among the different classes of non-coding RNA, most studies focused on the role of miRNA. Both glucose and lipids are able to regulate miRNA expression (Fig. 2). Indeed, miR-34a and miR-146a expressions are increased in response to prolonged exposure of the β-cell line MIN6 and pancreatic islets to palmitate( Reference Lovis, Roggli and Laybutt 99 ). Altered levels of miR-34a were found to sensitise β-cells to apoptosis and to cause defective glucose-induced insulin secretion, whereas the rise in miR-146a promoted stress-induced β-cell death( Reference Lovis, Roggli and Laybutt 99 ). Another screen carried out in MIN6 cells led to the identification of sixty-one miRNA regulated by glucose( Reference Tang, Muniappan and Tang 100 ). Detailed analysis of the function of one of these miRNA revealed that the increase of miR-30d occurring in the presence of elevated glucose concentrations induces the expression of the transcription factor MafA and, consequently, of the insulin gene( Reference Tang, Muniappan and Tang 100 , Reference Zhao, Mohan and Ozcan 101 ). In another in vitro study, primary islet cells and insulin-secreting cell lines incubated with stimulatory glucose concentrations displayed reduced levels of miR-375( Reference El Ouaamari, Baroukh and Martens 102 ), a key regulator of insulin production, insulin secretion and β-cell proliferation( Reference Poy, Eliasson and Krutzfeldt 103 , Reference Poy, Hausser and Trajkovski 104 ). In contrast, prolonged exposure of human islets to high glucose concentrations caused the induction of miR-133a, a miRNA targeting the mRNA of polypirimidine tract-binding protein that is required for insulin mRNA stabilisation( Reference Fred, Bang-Berthelsen and Mandrup-Poulsen 105 ). Blockade of miR-133a was able to prevent the decrease of polypirimidine tract-binding protein and in insulin biosynthesis rates observed upon chronic exposure to high-glucose, suggesting that this miRNA contributes to β-cell dysfunction under hyperglycaemic conditions. Elevated glucose concentration was also found to up-regulate miR-29a expression in human and rat islets, resulting in impaired glucose-induced insulin release( Reference Bagge, Clausen and Larsen 106 ). The increase of miR-29 leads to direct translational repression of the plasma membrane monocarboxylate transporter, preventing the leakage of glycolytic intermediates out of the oxidative pathway and the entry of pyruvate-lactate in β-cells during exercise( Reference Pullen, da Silva Xavier and Kelsey 107 ).

Fig. 2. (Colour online) Glucose and fatty acids modify function and mass of β-cells by altering the miRNA levels. Expression of several miRNA is up- or down-regulated (up and down arrows) upon exposure to glucose or fatty acids. Glucose regulates the expression of miR-29a, miR-30d, miR-133a and miR-375. Fatty acids (palmitate or a high-fat diet) regulate the expression of miR-34a, miR-132, miR-146a, miR-184, miR-203, miR-210, miR-338-3p and miR-383. MiRNA in green have a positive effect on insulin synthesis and secretion, proliferation and survival, while those in red have a negative effect. MiRNA marked with a blue square are implicated in two or more cellular processes. Gq, guanine nucleotide-binding protein q; Glut, glucose transporter.
Beside these studies carried out in vitro, several research teams investigated the impact of nutrients on β-cells in animal models. Global profiling of islets isolated from Goto-Kakizaki rats, an animal model characterised by chronic hyperglycaemia led to the identification of thirty differentially expressed miRNA in pancreatic islets( Reference Esguerra, Bolmeson and Cilio 108 ). The level of at least four of them, miR-130a, miR-132, miR-212 and miR-335 was found to be directly regulated by glucose. The islet miRNA expression profile is also strongly influenced by the diet. Indeed, mice maintained on a HFD for several weeks display major changes in islet miRNA expression( Reference Nesca, Guay and Jacovetti 109 , Reference Tattikota, Rathjen and McAnulty 110 ). Detailed analysis of the functional role of these miRNA revealed that part of the changes elicited by the HFD have a positive impact on β-cells and result in the expansion of the β-cell mass and in improved insulin secretion( Reference Nesca, Guay and Jacovetti 109 ). In fact, up-regulation of miR-132 increases the secretory capacity of β-cells, induces a higher proliferation rate and causes better survival in the presence of apoptotic stimuli( Reference Nesca, Guay and Jacovetti 109 ). The islets of mice on a HFD express also lower levels of miR-184( Reference Nesca, Guay and Jacovetti 109 , Reference Tattikota, Rathjen and McAnulty 110 ). Down-regulation of this miRNA promotes the proliferation of β-cells and protects them from palmitate-induced apoptosis( Reference Nesca, Guay and Jacovetti 109 , Reference Tattikota, Rathjen and McAnulty 110 ). Moreover, HFD causes a decrease in the expression of miR-338-3p( Reference Nesca, Guay and Jacovetti 109 ), a miRNA that is also down-regulated during compensatory β-cell mass expansion in pregnant rats( Reference Jacovetti, Abderrahmani and Parnaud 111 ). Blockade of this miRNA using antisense oligonucleotides or with a viral construct capable of sequestering this non-coding RNA results in β-cell proliferation both in vitro and in vivo ( Reference Jacovetti, Abderrahmani and Parnaud 111 , Reference Jacovetti, Jimenez and Ayuso 112 ). Taken together, these findings suggest that the changes in the level of these miRNA are part of the mechanisms that allow β-cells to adapt to the rise in the insulin demand occurring under conditions of obesity and insulin resistance. However, not all changes in islet miRNA expression elicited in response to HFD are beneficial for the activity of insulin-secreting cells. In fact, part of them have a deleterious impact on β-cell functions and may contribute to β-cell failure and to the development of diabetes. Indeed, down-regulation of miR-203, miR-210 and miR-383 resulted in an increase in apoptosis both in rat and human β-cells( Reference Nesca, Guay and Jacovetti 109 ).
Altered expression in islets from diabetic donors of miR-7a, miR-187 and a cluster of miRNA in an imprinted locus on human chromosome 14q32 have also been reported and are associated with impaired insulin secretion and β-cell dysfunction( Reference Locke, da Silva Xavier and Dawe 113 – Reference Kameswaran, Bramswig and McKenna 115 ). Moreover, the expression of several miRNA was also modified in the islets of ob/ob and db/db obese mice that are deficient in leptin or leptin receptor, respectively( Reference Poy, Hausser and Trajkovski 104 , Reference Nesca, Guay and Jacovetti 109 , Reference Belgardt, Ahmed and Spranger 116 , Reference Zhao, Keller and Rabaglia 117 ). Although the precise mechanisms regulating the expression of most of these miRNA are not yet known, at least part of the changes in the level of these non-coding RNA is likely to be caused by the elevated plasma concentrations of TAG and NEFA observed in these obese animals.
The level of several pancreatic miRNA is also altered in rats born from mothers fed a low-protein diet during pregnancy( Reference Dumortier, Hinault and Gautier 118 ). In particular, prenatal protein restriction resulted in the overexpression of miR-375 in the fetuses. The level of this miRNA remained augmented in the islets of adult rats, likely contributing to the reduced β-cell mass and function typically observed in the progeny of mothers fed a low-protein diet. Recently, changes in the level of several miRNA were found to contribute also to post-natal β-cell maturation and to the acquisition of a fully mature β-cell phenotype( Reference Jacovetti, Matkovich and Rodriguez-Trejo 119 ). The miRNA changes and the maturation of β-cells were shown to be driven by the nutritional shift occurring at weaning and to be prevented if the pups are fed a HFD instead of a carbohydrate-rich diet.
Oligonutrients can also regulate the expression of miRNA within the endocrine pancreas. Grape seed procyanidin extracts have been demonstrated to directly act on islet cells and to modulate insulin production by down-regulating insulin gene expression as well as insulin exocytosis-related genes and by inhibiting insulin biosynthesis( Reference Castell-Auvi, Cedo and Movassat 120 ). The miRNA expression profile of rat islets exposed to grape seed procyanidin extracts for 45 d revealed a significant down-regulation of miR-1249, miR-483, miR-30c-1* and up-regulation miR-3544. Gene ontology analysis of the predicted targets of these miRNA, revealed an enrichment of genes coding for components of the insulin-signalling pathway such as protein kinase B and extracellular signal-regulated kinases, suggesting that procyanidins exert their bioactivity on pancreatic islets by modifying the expression of a group of miRNA( Reference Castell-Auvi, Cedo and Movassat 120 ).
LncRNA have emerged as a novel class of functional RNA and are strongly suspected to regulate genome activities through a broad spectrum of mechanisms( Reference Ulitsky and Bartel 98 , Reference Kornfeld and Bruning 121 , Reference Knoll, Lodish and Sun 122 ). Pioneering studies by Ferrer and colleagues led to the identification of numerous conserved β-cell-specific lncRNA that are dysregulated in islets of hyperglycaemic T2D donors and often mapping to genomic regions enriched in islet protein-coding genes( Reference Moran, Akerman and van de Bunt 123 ). A very elegant genetic screening of eighty-nine human pancreatic islet samples has recently unveiled numerous genetic variants including many lncRNA suggested to regulate gene expression and exon use. Among the coding-genes and the 493 lncRNA detected in islet cells, multiple SNP were associated with known T2D-associated genes differing according to the normoglycaemic or hyperglycaemic status of the patients( Reference Fadista, Vikman and Laakso 124 ).
The imprinted lncRNA H19 that is generated from the Igf2 locus has been involved in the transgenerational transmission of epigenetic changes in germ cells in a mouse model of gestational diabetes mellitus( Reference Ding, Wang and Shu 125 ). This study unveiled that intrauterine hyperglycaemia alters the methylation of the gene and reduces the expression of Igf2/H19 in the islets of Langerhans. Igf2 and H19 expressions were also altered in the sperm of adult progeny from hyperglycaemic mothers, indicating that epigenetic changes in germ cells contribute to transgenerational transmission of metabolic disorders( Reference Ding, Wang and Shu 125 ).
Conclusion
Pancreatic β-cells and insulin, their main secretory product, are at the core of a complex regulatory network that governs body metabolism and energy expenditure. Carbohydrates, lipids and proteins are all capable of generating signals inside β-cells eliciting immediate insulin release or engendering changes in the expression of protein-coding and non-coding RNA that allow the β-cells to adapt their activities to conditions of increased insulin demand. The excess of dietary nutrients within critical developmental windows or in adulthood can, however, have deleterious consequences on β-cell functions, causing metabolic perturbations and the manifestation of different forms of diabetes mellitus. Therefore, a better understanding of the events elicited by dietary nutrients in β-cells will be of paramount importance for the design of new therapeutic approaches to prevent and treat these metabolic disorders that are reaching epidemic proportions.
Financial Support
Research of the authors is supported by the Swiss National Science Foundation, by the Foundation Francophone pour la Recherche sur le Diabète and by the Société Francophone du Diabète.
Conflicts of Interest
None.
Authorship
R. R., A. R. T. and C. J. searched the literature and wrote the manuscript.



