



The pathological accumulation on intra-hepatocellular TAG (IHTG) is denoted as non-alcoholic fatty liver disease (NAFLD) and encompasses a spectrum of conditions from steatosis to cirrhosis. The liver is the first organ to receive insulin and it is suggested that with NAFLD, intrahepatic insulin signalling becomes aberrant. Insulin resistance (IR) is often associated with NAFLD; however, it remains unclear if obesity-associated IR causes the development of NAFLD or vice versa. Insulin signalling directly alters intra- and extrahepatic metabolism, specifically insulin can regulate hepatic fatty acid and glucose metabolism by influencing substrate availability. Here we discuss the effects that obesity, IR and/or NAFLD may have on delivery of substrates to, metabolism within and disposal from the liver and how this influences glucose and lipid metabolism.
Increasing fat mass is associated with a greater risk of metabolic disease including IR, type 2 diabetes, NAFLD (which is also known as metabolic-associated fatty liver disease(Reference Eslam, Sanyal and George1)) and CVD(Reference Despres, Carpentier and Tchernof2). The increased risk may in part be driven by a noted downregulation of fatty acid trafficking in obese adipose tissue(Reference McQuaid, Hodson and Neville3), which may lead fatty acids being stored in non-adipose tissue organs (i.e. ectopic fat deposition in liver, skeletal muscle and heart). IR is often associated with hepatic steatosis, which is the first stage of NAFLD; however, whether IR is the primary driver of steatosis or vice versa remains unclear(Reference Farese, Zechner and Newgard4).
The liver plays a key role in maintaining metabolic homeostasis, including the uptake and synthesis of glucose and fatty acids and their intracellular partitioning into storage, oxidation and secretion pathways. Insulin plays a key role in linking extra- and intrahepatic metabolism. Extrahepatically, insulin regulates the delivery of substrates to the liver and this is dependent on nutritional state. Within the liver, the action of insulin shifts cellular metabolism away from energy release and secretion towards energy storage (such as the conversion from glucose to glycogen and fatty acids to TAG)(Reference Hodson and Karpe5,Reference Lewis, Carpentier and Pereira6) . Thus, pathological accumulation of IHTG may lead to dysregulated insulin signalling which can result in elevated plasma lipid and glucose concentrations. We have recently reviewed fatty acid partitioning within the liver(Reference Hodson and Gunn7), the effect of hyperinsulinaemia on IHTG content(Reference Hodson and Karpe5), the influence of dietary fatty acids on IHTG content and metabolism(Reference Hodson, Rosqvist and Parry8) and the role of dietary carbohydrates and fats in NAFLD(Reference Yki-Jarvinen, Luukkonen and Hodson9). Therefore, the aim of this review is to discuss, from the available evidence, the effect of obesity on hepatic metabolism with a specific focus on the factors that influence glucose and lipid metabolism in human subjects.
Plasma insulin, glucose and lipids: the effect of obesity
Plasma insulin
Insulin is an important mediator in partitioning and storing substrates (e.g. lipids and glucose) within the liver and to and from tissues. In the fasting state, insulin concentrations are at their lowest and this promotes hepatic glucose, VLDL-TAG and ketone production (via substrate delivery including fatty acids and glycerol from adipose and alanine and lactate from muscle tissue) to maintain energy homeostasis. In the transition to the postprandial state, circulating plasma insulin concentrations rapidly increase in response to consumption of a mixed meal (i.e. a meal containing fat, protein and carbohydrate) leading to stimulation of nutrient storage into major peripheral tissues. For example, within the liver, glucose will be directed to glycogen synthesis pathways and in the presence of excess, glucose will be partitioned into the de novo lipogenesis (DNL) pathway for synthesis of fatty acids. Further, an increase in plasma insulin concentrations decreases plasma NEFA concentrations due to a reduction in adipose tissue TAG lipolysis and within the liver, insulin decreases glucose production. Taken together, this regulates systemic lipid and glucose concentrations between nutritional states. Thus, impairments in insulin availability, meal timing and signalling often result in a dysregulation of glucose and lipid homeostasis which have been associated with obesity, NAFLD and IR.
Hyperinsulinaemia has been suggested (although no consensus exists) to be a fasting plasma insulin ≥20 μg/ml(Reference Burchfiel, Curb and Arakaki10,Reference Gamboa-Gómez, Guerrero-Romero and Aradillas-García11) and results from an imbalance between insulin secretion and clearance by peripheral tissues and is often associated with increased adiposity(Reference Lorenzo, Hanley and Wagenknecht12). The liver is a key organ in insulin metabolism and is responsible for 40–60 % of total insulin extraction in the fasting state(Reference Harding, Bloom and Field13). Data from in vivo human and canine models suggest that a reduction in total and hepatic insulin clearance acts as the first compensatory mechanism in the early stages of weight gain(Reference Lorenzo, Hanley and Wagenknecht12,Reference Erdmann, Kallabis and Oppel14–Reference Mittelman, Van Citters and Kim16) .
Elevated systemic insulin concentrations are often associated with obesity. For example, Kim et al.(Reference Kim, Reaven and Chen17) observed an 18–28 % higher plasma insulin concentration in individuals with obesity (BMI≥30 kg/m2) compared to non-obese (BMI≤30 kg/m2) individuals. By undertaking complex physiological studies in human subjects, Smith et al.(Reference Smith, Polidori and Yoshino18) compared lean, obese and obese individuals with NAFLD and demonstrated notable step-wise increases in insulin secretion, recycling and removal by extrahepatic tissues while hepatic insulin removal decreased across the groups. In contrast, Utzschneider et al.(Reference Utzschneider, Kahn and Polidori19) reported no difference in extrahepatic and hepatic insulin extraction between obese individuals with NAFLD and age and BMI-matched controls, suggesting the dissociation of hepatic insulin extraction with steatosis. The discrepancy between these findings may be due to the duration of obesity and/or NAFLD.
Although there is a clear association between obesity and IR, it remains unclear how weight gain influences insulin kinetics, specifically the sequence of impairments. A number of longitudinal studies using human and canine models have explored this and demonstrated that temporal differences exist in insulin kinetics. For example, Mittelman et al.(Reference Mittelman, Van Citters and Kim16) investigated the sources of fasting hyperinsulinaemia in dogs fed a moderate fat-enriched diet for 12 weeks. They found temporal differences in changes for insulin secretion and clearance with secretion increasing at 6 weeks and clearance being reduced within 3 weeks of starting the diet(Reference Mittelman, Van Citters and Kim16). Similarly, when lean men (BMI 20⋅4–23⋅2 kg/m2, mean age 26⋅2 years) gained 2 kg/m2 during 4⋅5 months consuming a hyperenergetic diet, insulin clearance, not secretion, was the primary defect leading to basal and stimulated hyperinsulinaemia(Reference Erdmann, Kallabis and Oppel14). Overall, this suggests that an increase in fat mass primarily influences insulin clearance. However, these studies have been done in lean individuals and what remains to be elucidated is whether insulin clearance is still the primary defect in those with long-term obesity and/or NAFLD.
Plasma glucose
The liver plays a fundamental role in the regulation and maintenance of systemic glucose concentrations. In the fasted state, the liver is responsible for 75–90 % of endogenous glucose production through hepatic gluconeogenesis (GNG) and glycogenolysis to keep plasma glucose concentrations stable (3⋅5–5⋅5 mmol/l)(Reference Güemes, Rahman and Hussain20). After meal consumption, the liver receives dietary carbohydrates through direct absorption into the hepatic portal vein and retains 40–70 % of ingested glucose(Reference Moore, Coate and Winnick21). The influence of adiposity alone on hepatic and extrahepatic glucose handling remains unclear. Evidence from most(Reference Golay, Chen and Reaven22,Reference Conte, Fabbrini and Kars23) , but not all(Reference Lu, Wang and Liang24) studies has shown that obese individuals have similar fasting plasma glucose levels compared to lean individuals, despite being more IR. In contrast, high postprandial glycaemia is often observed in individuals with obesity or type 2 diabetes(Reference Carroll, Kaiser and Franks25,Reference Sandqvist, Strindberg and Lonnroth26) and may precede the deterioration of fasting glycaemic control(Reference Fysekidis, Cosson and Banu27,Reference Monnier, Colette and Dunseath28) . There are two main pathways that can lead to elevated postprandial hyperglycaemia: (1) decreased glucose uptake and disposal (via storage or oxidation of glucose) in liver, muscle and adipose tissues and (2) decreased suppression of hepatic glucose production.
It has been suggested that impaired glucose uptake into insulin-sensitive (IS) tissues, particularly skeletal muscle, is the primary defect that differentiates individuals with normal and high plasma glucose concentrations(Reference Conte, Fabbrini and Kars23,Reference Klauser, Prager and Schernthaner29,Reference Mitrou, Boutati and Lambadiari30) . Following consumption of a mixed meal (50 % carbohydrates, 40 % fat, 10 % protein total energy), Mitrou et al.(Reference Mitrou, Boutati and Lambadiari30) found skeletal muscle glucose uptake was almost 2-fold lower in obese women compared to non-obese women (5 v. 9 % fractional glucose extraction, respectively). The underlying mechanisms for these differences have not been completely resolved however, changes in expression of key GLUT(Reference Dohm, Elton and Friedman31) and reduction in skeletal muscle blood flow commonly associated with obesity(Reference Limberg, Morgan and Schrage32) have been proposed as possible defects.
The dysregulation of hepatic glucose output following nutrient availability is another factor that can potentially lead to exaggerated postprandial hyperglycaemia, especially in IR or NAFLD patients. Several studies have demonstrated persistent release of endogenous glucose despite the availability of dietary monosaccharides, suggesting diminished suppression of hepatic glucose production(Reference Guerra and Gastaldelli33). It is unclear whether decreased inhibition by insulin or increased availability of gluconeogenic substrates contributes to a greater extent towards elevated postprandial hyperglycaemia.
Plasma NEFA
Plasma NEFA are a major source of fatty acids to the liver and are predominantly derived from adipose tissue lipolysis, with a contribution from chylomicron-derived spillover in the postprandial state. As subcutaneous adipose tissue represents the majority (approximately 85 %) of all body fat(Reference Thomas, Saeed and Hajnal34) in humans, the degree of adiposity influences the storage and mobilisation of fatty acids from these respective sources to the liver. Nutritional state is also a major determinant of the metabolic tasks of energy storage and supply in adipose tissue. In fasting, adipose tissue TAG is mobilised (via a sequential series of lipases) so that there is a net release of NEFA into systemic circulation. In the transition to the postprandial state, there is a shift to net uptake/storage of (predominantly dietary) fatty acids into adipose tissue as insulin suppresses adipose tissue TAG lipolysis and upregulates lipoprotein lipase(Reference Fielding and Frayn35). Thus, plasma NEFA concentrations are highest in fasting and decrease with consumption of a mixed meal. In the postprandial state, chylomicrons are hydrolysed by lipoprotein lipase and although the majority of liberated fatty acids from chylomicron-TAG are taken up into adipose tissue for storage, some escape uptake and appear in the systemic NEFA pool and these are often referred to as spillover NEFA(Reference Frayn36). Therefore, in the postprandial period, plasma NEFA concentrations reflect fatty acids liberated from adipose tissue TAG lipolysis and chylomicron-derived spillover.
Fasting plasma NEFA concentrations are often reported as being elevated in obesity, NAFLD and those with type 2 diabetes(Reference Karpe, Dickmann and Frayn37,Reference Holt, Wild and Wood38) which are suggested to be due to an increase in adipose tissue mass. An alternative suggestion is that adipose tissue has become resistant to the action of insulin, which normally suppresses fatty acid mobilisation from adipose tissue, causing lipolysis to further increase(Reference Karpe, Dickmann and Frayn37). However, Karpe et al.(Reference Karpe, Dickmann and Frayn37) elegantly demonstrated that elevated NEFA concentrations are not explained by an increased adipose tissue mass as the NEFA release per kilogram of adipose tissue mass is reduced with obesity. Moreover, it has been shown that in obese adipose tissue, there is a clear downregulation in fatty acid trafficking genes(Reference McQuaid, Hodson and Neville3,Reference Klimcakova, Roussel and Marquez-Quinones39) ; adipose tissue blood flow is also notably lower and less responsive in obese compared to lean subcutaneous adipose tissue(Reference McQuaid, Hodson and Neville3). We have previously reported an impairment in the pathway for direct dietary fat storage into adipose tissue in obese, when compared to lean males, where the latter demonstrated a progressive increase in the deposition of meal fat into adipose tissue with consumption of sequential meals, while obese men failed to increase fat storage with sequential feeding(Reference McQuaid, Hodson and Neville3). Moreover, we have previously reported that contrary to general perception, spillover of chylomicron-derived fatty acids is higher, and more dynamic, in lean compared to obese individuals(Reference Piche, Parry and Karpe40), thus supporting the observed downregulation in the expression of fatty acid trafficking genes in obese adipose tissue. A potential consequence of reduced adipose tissue fatty acid trafficking with obesity/IR is that as adipose tissue cannot accommodate sufficient fat uptake, this then contributes to augmentation of IR as fatty acids are channelled towards ectopic tissues, such as the liver.
TAG-rich lipoproteins
The major contributors to plasma TAG concentrations are the TAG-rich lipoproteins, VLDL and chylomicrons, the latter only being present in the postprandial state. The liver is unique as it can export TAG in bulk through incorporation into VLDL and fasting plasma VLDL-TAG concentrations reflect a balance between substrate availability for TAG production within the liver, secretion from the liver and clearance by peripheral tissues and the liver(Reference Millar and Packard41). Fasting plasma TAG concentrations have been reported to be higher in individuals classified as obese and/or IR or have NAFLD when compared to lean and/or IS or non-NAFLD individuals in some(Reference Hodson, Bickerton and McQuaid42–Reference Smith, Shankaran and Yoshino45) but not all(Reference Smith, Shankaran and Yoshino45–Reference Lee, Lambert and Hovhannisyan47) studies.
Insulin is a key regulator in all steps of VLDL metabolism; acting directly to alter the availability of ApoB and activation of LDL-receptor which are key for production and clearance of TAG-rich lipoprotein remnant particles. Therefore, differences in inulin concentrations between phenotypes may explain the observed differences in plasma TAG concentration. VLDL kinetics have been probed in individuals with different metabolic phenotypes and a common finding is that body adiposity positively associates with VLDL-TAG concentration(Reference Mittendorfer, Yoshino and Patterson48). Higher plasma TAG concentrations observed in NAFLD patients have been explained by overproduction of VLDL-TAG(Reference Adiels, Taskinen and Packard49,Reference Fabbrini, Mohammed and Magkos50) . These studies highlight that compared to individuals without NAFLD, those with NAFLD have reduced insulin-mediated suppression of VLDL-TAG secretion. Kinetic studies have also reported lower VLDL clearance in obese IR compared to lean IS individuals(Reference Mittendorfer, Yoshino and Patterson48) which may contribute to higher plasma VLDL-TAG concentrations.
In the transition to the postprandial state, the addition of dietary fat-enriched chylomicrons into systemic circulation results in an increase of plasma TAG despite an insulin-meditated reduction in VLDL production(Reference Malmstrom, Packard and Caslake51). Even though suppressed there are approximately ten times more VLDLs than chylomicron particles in circulation with VLDLs carrying approximately 50 % of the TAG-rich lipoprotein TAG(Reference Schneeman, Kotite and Todd52). However, chylomicron TAG is a much preferred substrate for lipoprotein lipase compared with VLDL(Reference Karpe, Bickerton and Hodson53). Obese individuals typically have a greater and more exaggerated lipaemic response than lean individuals(Reference Hodson, McQuaid and Humphreys43) which may be explained by reduced suppression of VLDL-TAG production/secretion and/or reduced chylomicron clearance by obese adipose tissue. By using an intravenous fat tolerance test, Tollin et al.(Reference Tollin, Ericsson and Johnson54) demonstrated a clear inverse association between adiposity and TAG-rich lipoprotein clearance in both men and women. The lack of suppression of VLDL-TAG may be due to IR or a compensatory mechanism to lower the risk of IHTG accumulation; however, it has been suggested that VLDL-TAG secretion plateaus when IHTG exceeds 10 %(Reference Fabbrini, Mohammed and Magkos50). A possible explanation for the plateau may be due to the availability of ApoB which may be reduced with hyperinsulinaemia as insulin promotes ApoB degradation and clearance from plasma. Umpleby et al.(Reference Umpleby, Shojaee-Moradie and Fielding55) noted in NAFLD (IHTG 7⋅9–36⋅8 %) compared to non-NAFLD (IHTG 0⋅5–4⋅6 %) individuals, VLDL-ApoB concentrations were similar whilst VLDL-TAG concentrations were notably higher. In individuals unable to appropriately upregulate VLDL-TAG secretion, an increased flux of fatty acids to the liver may result in IHTG accumulation.
Taken together, insulin plays a key role in regulating plasma glucose, NEFA and TAG-rich lipoprotein levels across nutritional states which is mediated through extrahepatic and intrahepatic metabolic cascades that rapidly transition cellular task from energy supply to energy storage. With increasing fat mass, there is an elevated risk of hyperinsulinaemia and IR; this may co-exist with (i) an increased level of plasma NEFA in the fasting state, and an attenuated suppression in the postprandial state, (ii) dysregulated post-prandial glycaemic excursion, and/or (iii) increased plasma TAG concentrations in the fasting and post-prandial states (Fig. 1a–c).
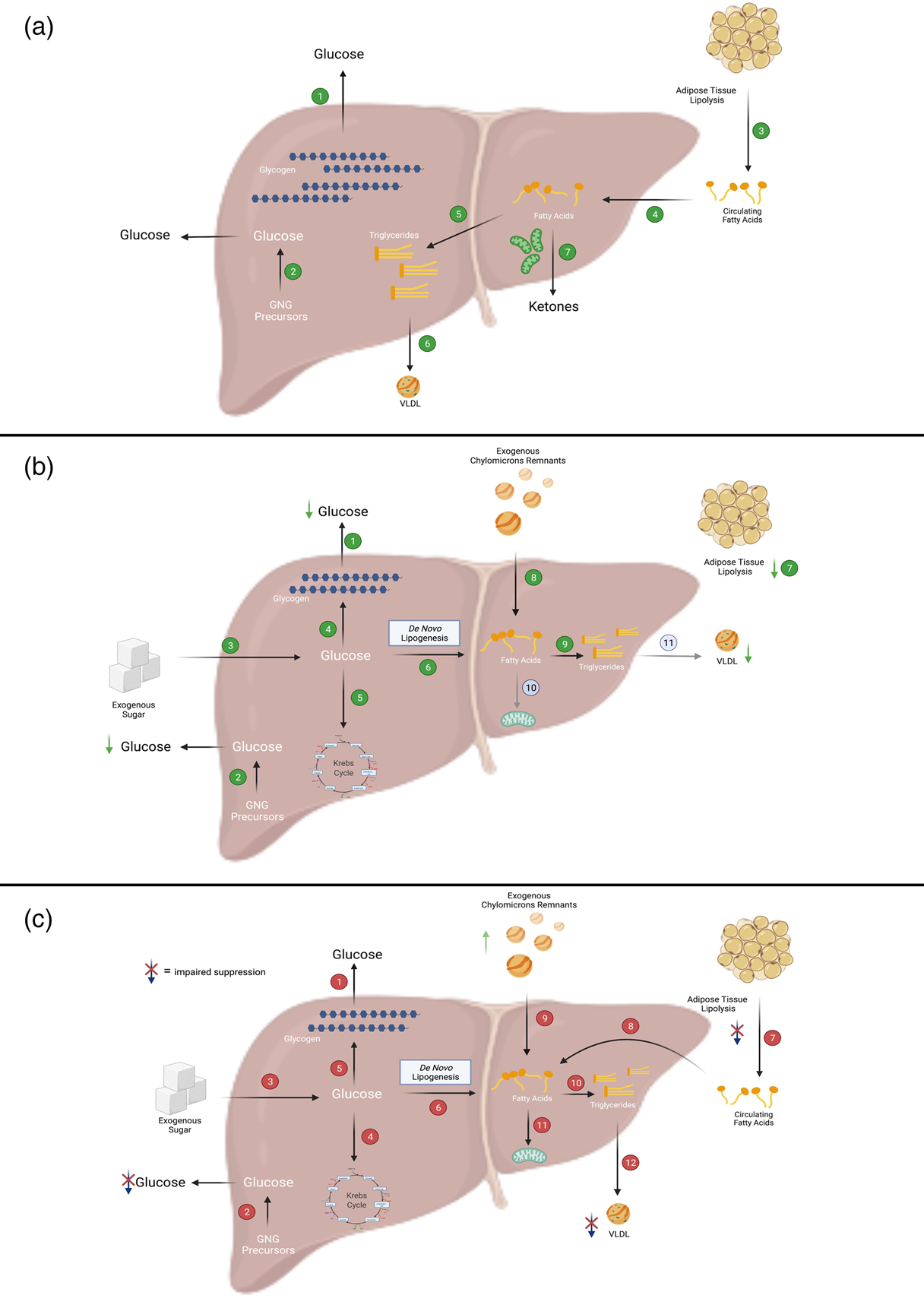
Fig. 1. Overview of fatty acid and glucose metabolic pathways in the liver. (a) In the fasted state, when insulin levels are low, the liver secretes glucose, TAG and ketone bodies into the circulation for delivery to peripheral tissues. In the liver, (1) glucose is produced primarily via the breakdown of glycogen or (2) through the synthesis of new glucose from precursors such as lactate, alanine and glycerol (referred to as gluconeogenesis (GNG)). The main source of fatty acids to the liver is (3) adipose-derived NEFA which can be taken up by the liver via (4) fatty acid transporters. Within the liver, fatty acids can then be partitioned into esterification pathways to form TAG which can be either (5) stored in cytosolic lipid droplets or (6) secreted in VLDL. Alternatively, fatty acids can enter (7) oxidation pathways (i.e. the TCA cycle or ketogenesis). (b) In insulin-sensitive individuals, after consumption of a mixed meal, (1 and 2) hepatic glucose output (i.e. GNG and glycogenolysis) is suppressed and (3) dietary sugars taken up by the liver can be (4) stored as glycogen, (5) broken down via glycolysis or (6) used to form fatty acids via de novo lipogenesis (DNL). As insulin suppresses (7) adipose tissue TAG mediated lipolysis, there is decreased flux of NEFA to the liver. Dietary fatty acids enter the liver as either spillover NEFA or as (8) chylomicron remnants. Within the liver, fatty acids preferentially enter (9) esterification rather than (10) oxidation pathways. During the postprandial period, (11) insulin-mediated VLDL secretion is suppressed. (c) In individuals with insulin resistance (which may be due to obesity and/or NAFLD), after consumption of a mixed meal, (1 and 2) hepatic glucose output (which may be due to increased GNG and/or glycogenolysis) is not suppressed, whilst (3) dietary sugars enter the liver. Within the liver, sugars may then enter (4) glycolytic or (5) storage pathways, and (6) the DNL pathway which is upregulated in these individuals. As adipose tissue fatty acid trafficking is downregulated in insulin resistance/obesity, insulin-mediated suppression of (7) adipose tissue lipolysis is not suppressed to the same extent as in insulin-sensitive individuals and thus there is plausibly a greater a flux of (8) NEFA to the liver. Additionally, there is a lower flux of chylomicron-derived spillover fatty acids to the liver but likely greater delivery of (9) TAG-rich chylomicron remnants to the liver. Within the liver, fatty acids are preferentially partitioned towards (10) esterification rather than (11) oxidation pathways. VLDL secretion may not be (12) suppressed to the same extent as in insulin-sensitive individuals. Figure created with BioRender.com.
Delivery of fatty acids to and synthesis within the liver
Changes in fatty acid fluxes to the liver, fatty acid and TAG synthesis within and disposal from the liver determine both IHTG content and circulating lipoprotein concentrations. Within the liver, FAs are broadly partitioned between oxidation (to form ketones (e.g. 3-hydroxybutyrate) and CO2) and esterification (to form predominantly TAG) pathways(Reference Hodson and Frayn56). In the transition from the fasting to postprandial state, a number of metabolic pathways mediated by a rise in insulin concentrations will influence the fatty acid flux to the liver. Changes in the delivery of fatty acids from adipose tissue and diet to the liver and those synthesised within the liver may contribute to IHTG accumulation and hypertriglyceridemia.
Adipose tissue-derived NEFA
NEFA turnover is a key determinant of VLDL-TAG production(Reference Lewis57,Reference Nielsen and Karpe58) in both the fasting and postprandial states, with stable isotope tracer studies demonstrating that adipose tissue-derived NEFA contribute to the largest proportion of VLDL-TAG. Indeed, in lean, IS individuals, between 75 and 84 % of VLDL-TAG is derived from systemic NEFA (i.e. fatty acids predominantly from subcutaneous adipose tissue)(Reference Hodson, McQuaid and Humphreys43,Reference Fabbrini, Mohammed and Magkos50,Reference Barrows and Parks59–Reference Vedala, Wang and Neese61) . Although the relative contribution of plasma NEFA to VLDL-TAG is 42–72 % in obese/IR(Reference Hodson, McQuaid and Humphreys43,Reference Fabbrini, Mohammed and Magkos50,Reference Fabbrini, Magkos and Mohammed60–Reference Donnelly, Smith and Schwarzenberg62) , when expressed as an absolute contribution given that these individuals typically have higher plasma VLDL-TAG concentrations, it is likely there would be no notable difference between groups. Alternatively, differences in study protocols and labelling times may also influence these results. The remaining fatty acids will be derived from non-systemic NEFA (including those from visceral adipose tissue and those already present in the liver). Visceral adiposity has been suggested to be a culprit for higher NEFA flux directly to the liver via the portal vein, as waist circumference is strongly associated with IHTG content(Reference Kotronen, Westerbacka and Bergholm63,Reference Nielsen, Guo and Johnson64) . In line with this, Meek et al. (Reference Meek, Nair and Jensen65) observed greater proportions of fatty acids derived from visceral fat reaching the liver under conditions of moderate hyperinsulinaemia.
In the transition to the postprandial state, the relative contribution of plasma NEFA to VLDL-TAG is decreased (to between 28 and 57 % in lean and obese IS and IR individuals)(Reference Hodson, Bickerton and McQuaid42,Reference Hodson, McQuaid and Humphreys43,Reference Barrows and Parks59,Reference Donnelly, Smith and Schwarzenberg62) due to the anti-lipolytic action of insulin on adipose tissue TAG(Reference Frayn, Arner and Yki-Jarvinen66). The degree to which adipose tissue hydrolysis is suppressed by insulin may be influenced by phenotype. For example, in the postprandial state, there may be a greater delivery of NEFA to the liver in IR compared to IS individuals due to a lower suppression of insulin-mediated adipose tissue TAG hydrolysis(Reference McQuaid, Hodson and Neville3,Reference Coppack, Evans and Fisher67) . However, we have previously found in studies feeding a single and sequential meals (5 h apart over a 24 hour period) that there was no difference in the relative or absolute contribution of systemic NEFA to VLDL-TAG in lean IS and abdominally obese IR males(Reference Hodson, Bickerton and McQuaid42,Reference Hodson, McQuaid and Humphreys43) . In the postprandial state, the plasma NEFA pool is a combination of fatty acids liberated from adipose tissue TAG and chylomicron-derived spillover NEFA and thus, it is plausible that the lack of difference between the groups was due to differences in the contributions from these sources as there was no difference in total plasma NEFA concentrations(Reference Hodson, McQuaid and Humphreys43).
Dietary-derived fatty acids
Dietary fat can enter the liver either as chylomicron remnants or chylomicron-derived spillover NEFA(Reference Hodson and Fielding68) and the contribution to VLDL-TAG is dependent on the amount and frequency of fat intake with the majority of studies reporting total dietary fat contribution. The relative contribution of dietary fatty acids to VLDL-TAG is reported as 19–39 % in lean, IS, non-NAFLD, and 12–28 % in obese, IR, NAFLD(Reference Hodson, Bickerton and McQuaid42,Reference Hodson, McQuaid and Humphreys43,Reference Barrows and Parks59,Reference Donnelly, Smith and Schwarzenberg62) . As few studies have compared individuals with varying degrees of IR after feeding the same test meal, it is challenging to know if differences in the contribution of dietary fatty acids to VLDL-TAG exist. We observed no difference in absolute contribution of dietary fatty acids to VLDL-TAG after a mixed test meal or three sequential test meal spaced 4 h apart in IS and IR individuals(Reference Hodson, Bickerton and McQuaid42,Reference Hodson, McQuaid and Humphreys43) .
The contribution of the different dietary sources (i.e. spillover v. remnants) is not often considered, with the chylomicron remnant pathway being suggested to play a minor role in TAG synthesis compared to other fatty acid sources(Reference Lambert, Ramos-Roman and Browning46). We have previously found no difference in the relative or absolute contribution of either spillover NEFA or chylomicron remnant TAG to VLDL-TAG between IS and IR men(Reference Hodson, Bickerton and McQuaid42). However, it is plausible that as fatty acid trafficking is downregulated in obese adipose tissue, that when given a high-fat meal(s), a decreased chylomicron hydrolysis leading to decreased spillover-derived fatty acids(Reference Piche, Parry and Karpe40), would result in a greater TAG-rich chylomicron remnant flux to the liver. Once at the liver, these remnants are taken up by receptor-mediated endocytosis by the lipoprotein receptor-related protein and then intracellularly, remnants are channelled through a lysosomal pathway to liberate fatty acids. Thus, it is plausible that the lack of reported difference in the contribution of chylomicron remnant-TAG to VLDL-TAG can be explained by poor temporal resolution, as lysosomal function may be attenuated with either IR or IHTG accumulation; the effects of these on lysosomes are not well understood. Equally, the low remnant TAG contribution could in part be explained by differences in the amount and type of fat consumed in the test meal, the frequency of meals and the phenotype of the individual. Given the varying contribution of dietary fat to VLDL-TAG (and potentially IHTG content), it would be of interest to compare individuals across the spectrum of adiposity and plasma insulin concentrations fed a high-fat test meal(s) to determine the relevance of chylomicron remnant TAG on VLDL-TAG production.
Newly synthesised fatty acids (de novo lipogenesis)
In human subjects, DNL occurs predominantly in the liver and the end product is often considered to be the C16 SFA, palmitate. The expression and activity of key lipogenic genes are mainly regulated at the transcriptional level by insulin-stimulated SREBP-1c and glucose-stimulated ChREBP. Thus, the contribution of DNL-fatty acids to VLDL-TAG in lean IS individuals is typically low (<10 % of newly-synthesised palmitate) in the fasting state and upregulated in the fed state(Reference Pramfalk, Pavlides and Banerjee44,Reference Barrows and Parks59,Reference Marques-Lopes, Ansorena and Astiasaran69–Reference Timlin and Parks71) . It is suggested that increased DNL is the underlying pathogenesis of IR(Reference Zammit72), however direct evidence for this is lacking in human subjects.
The contribution of DNL-derived fatty acids to the TAG pool is typically measured using stable isotope tracers (e.g. 13C-acetate, 2H2O, or labelled sugars)(Reference Hodson, Parry and Cornfield73). In cross-sectional or observational studies that have compared obese NAFLD/IR to obese non-NAFLD/IR individuals, hepatic DNL is typically higher(Reference Lambert, Ramos-Roman and Browning46,Reference Diraison, Moulin and Beylot74) . By comparing obese and lean individuals differing in insulin sensitivity who consumed an isoenergetic high-fat diet for 5 d, fasting hepatic DNL was 4-fold higher in the obese, and this was found to be driven primarily by hyperinsulinaemia(Reference Schwarz, Linfoot and Dare75). In support of this observation, there was no difference between the contribution of hepatic DNL in lean and obese individuals (1⋅5 v. 2 %, respectively) with similar fasting insulin concentrations(Reference Schwarz, Linfoot and Dare75). By defining individuals as normo- or hyperinsulinaemic and matching for BMI (26⋅9 v. 27⋅5 kg/m2) and IHTG content (3⋅4 v. 3⋅4 %), we observed a greater contribution of DNL-derived fatty acids to VLDL-TAG in the fasting state which was further increased in the postprandial state in the hyper- compared to normo-insulinaemic group(Reference Pramfalk, Pavlides and Banerjee44). Thus, it is plausible that fasting hyperinsulinaemia may be a stronger predictor of hepatic DNL than either body fatness or liver fat per se (Reference Pramfalk, Pavlides and Banerjee44). In contrast, Smith et al.(Reference Smith, Shankaran and Yoshino45) reported that hepatic DNL was significantly higher in obese non-NAFLD compared to lean subjects despite no significant difference in fasting plasma insulin concentrations. Typically, in the transition from fasting to the fed state, there is a notable increase in the contribution of DNL-derived fatty acids to VLDL-TAG(Reference Pramfalk, Pavlides and Banerjee44,Reference Barrows and Parks59,Reference Pramfalk, Pavlides and Banerjee70,Reference Timlin and Parks71) . However, Donnelly et al.(Reference Donnelly, Smith and Schwarzenberg62) observed a lack of change in DNL between nutritional states in individuals with NAFLD (n = 9) and therefore it could be speculated that the DNL pathway in these individuals was constitutively activated.
Dietary macronutrient composition may also influence hepatic DNL. For example, consumption of a high carbohydrate diet has been shown to significantly upregulate the contribution of DNL-derived fatty acids to VLDL-TAG(Reference Marques-Lopes, Ansorena and Astiasaran69,Reference Hudgins, Hellerstein and Seidman76,Reference Wilke, French and Goh77) . In individuals with and without NAFLD, palmitate production was significantly elevated in individuals with NAFLD after consumption of a diet enriched with free sugars compared to a low sugar diet, whereas there was no notable change in palmitate production in BMI- and age-matched controls(Reference Umpleby, Shojaee-Moradie and Fielding55). Although glucose consumption has been shown to stimulate hepatic DNL, consumption of glucose and fructose combined exacerbates DNL even further(Reference Hudgins, Parker and Levine78).
The role of DNL in the pathogenesis of NAFLD continues to be debated. The discourse appears to be based on evidence from observational studies that have reported the contribution of DNL-derived fatty acids to VLDL-TAG to be notably higher when compared to non-NAFLD participants (14–22 v. about 5–10 %)(Reference Smith, Shankaran and Yoshino45–Reference Lee, Lambert and Hovhannisyan47,Reference Diraison, Moulin and Beylot74) . For example, Lambert et al.(Reference Lambert, Ramos-Roman and Browning46) reported that obese individuals with high liver fat (mean 18⋅4 %) had greater fasting hepatic DNL compared to obese individuals with low liver fat (mean 3⋅1 %). Moreover, the hepatic expression of lipogenic genes (SREBP-1c and FASN) has been found to be notably higher (33 and 70 %, respectively) in liver biopsies from obese (n = 11) compared to non-obese (n = 8) patients, notably CPT1-a (a rate-limiting enzyme for mitochondrial fatty acid oxidation) was 65 % lower in the obese livers(Reference Pettinelli, Del Pozo and Araya79).
Schwarz et al.(Reference Schwarz, Linfoot and Dare75) demonstrated that greater stimulation of DNL in the fasted state by hyperinsulinaemia may occur independently of obesity. However, it is plausible that the coexistence of IR and obesity/NAFLD further exacerbate hepatic DNL. In support of this hypothesis, studies by Lambert et al.(Reference Lambert, Ramos-Roman and Browning46) and Smith et al.(Reference Smith, Shankaran and Yoshino45) have reported greater fasting hepatic DNL in obese individuals with NAFLD compared to BMI-matched non-NAFLD individuals. Although DNL appears to be higher in individuals with obesity or IR or IHTG accumulation, it remains unclear what the primary contributor to the initial upregulation in hepatic DNL is and is likely to be multifactorial.
The contribution of the different fatty acid sources to VLDL-TAG (and presumably IHTG) is influenced by an individual's phenotype and the macronutrient composition of their diet. Adipose tissue-derived NEFA contribute the majority of fatty acids to VLDL-TAG in both fasting and the postprandial state and across a range of adiposity. Overall the evidence for the contribution of dietary fatty acids to VLDL-TAG across different phenotypes is inconsistent and it is plausible that the contribution has been underestimated due to methodological differences such as meal composition and timing of measurements post-test meal consumption. Across studies assessing the contribution of DNL fatty acids to VLDL-TAG, findings are more consistent with a notably higher contribution in individuals with IR and/or NAFLD compared to those who are IS and /or non-NAFLD (Fig. 1a–c). However, although IR and NAFLD often co-exist, it remains elucidated if IR-mediated DNL causes the accumulation of IHTG or vice versa.
Insulin resistance: effects on liver metabolism
Obesity is strongly associated with impaired insulin signalling and/or plasma clearance which are key features of IR and the aetiology and molecular landscape has recently been comprehensively reviewed(Reference James, Stockli and Birnbaum80). Briefly, within the liver, insulin typically binds to the insulin receptor causing the activation of insulin receptor substrates and subsequent phosphorylation of Akt; a key regulator of downstream gluconeogenic (via FOXO1) and lipogenic (via SREBP-1c) pathways. Impairments in insulin signalling appear to be pathway-selective, with insulin-mediated suppression of GNG being blunted while insulin stimulation of DNL remains intact or amplified; these observations led to the concept of selective hepatic IR.
To date, the majority of studies investigating this concept have been undertaken using rodent models, with a focus on identifying potential branch points in the insulin signalling pathway where sensitivity may diverge. A study by Biddinger et al.(Reference Biddinger, Hernandez-Ono and Rask-Madsen81) demonstrated that deletion of the liver-specific insulin receptor in rodent models resulted in both hyperglycaemia and hyperinsulinaemia whilst IHTG and plasma TAG remained unchanged. This work provided the basis for distinguishing between total (i.e. insulin suppression of GNG and stimulation of DNL are both impaired) and selective IR (i.e. only insulin suppression of GNG is impaired)(Reference Biddinger, Hernandez-Ono and Rask-Madsen81). Human subjects with mutations in the insulin receptor exhibit a similar phenotype to total IR in the rodent models (low hepatic DNL and TAG-depleted VLDL)(Reference Semple, Sleigh and Murgatroyd82). Although these findings suggest that the dichotomy in selective IR occurs distal to the insulin receptor, evidence from human genetic(Reference Semple, Sleigh and Murgatroyd82,Reference Honma, Sawada and Ueno83) and knockout rodent studies on multiple isoforms of insulin receptor substrates(Reference Dong, Copps and Guo84,Reference Valverde, Burks and Fabregat85) and Akt(Reference Lu, Wan and Leavens86, Reference Titchenell, Quinn and Lu87) are inconclusive. Thus, insulin signalling at the whole-body and tissue-specific level is a more complex cascade of pathways than has previously been proposed by loss-of-function/deletion studies. In support of this concept, protein regulation of metabolic pathways may be additionally governed by distinct insulin sensitivity thresholds(Reference Cook, Langlet and Kido88), multiple phosphorylation sites(Reference Wu and Williams89) and differential expression patterns(Reference Kubota, Kubota and Kajiwara90,Reference Li, Lai and Christiano91) .
It is often suggested that patients with NAFLD have selective hepatic IR; however, evidence for a mechanism underpinning this in human subjects is lacking. Recent data on insulin-dependent and independent regulation of hepatic metabolism have highlighted the importance of substrates (e.g. monosaccharides) that may, in part, explain selective IR. To disentangle this hypothesis, Ter Horst et al. (Reference Ter Horst, Vatner and Zhang92) investigated the contribution of insulin-mediated (i.e. using glucose) v. substrate-mediated (i.e. using fructose) regulation of hepatic DNL and glucose production in obese individuals with (n = 16) and without (n = 15) NAFLD, using stable isotope tracer methodologies. The authors found that fasting hepatic DNL was higher in NAFLD v. non-NAFLD individuals; however, insulin-mediated hepatic DNL was greater in non-NAFLD compared to NAFLD individuals. In contrast, substrate-mediated DNL was robustly upregulated in both NAFLD and non-NAFLD individuals(Reference Ter Horst, Vatner and Zhang92). By taking liver biopsies from a subset of the NAFLD (n = 8) and non-NAFLD (n = 8) groups, the authors found decreased phosphorylation of insulin signalling proteins and increased mRNA expression of ChREBPβ, in obese NAFLD patients(Reference Ter Horst, Vatner and Zhang92). On the basis of these observations, the authors concluded that obese individuals with resistance to insulin's ability to suppress hepatic glucose production are also resistant to insulin's ability to suppress DNL, suggesting that IR may not be selective in human livers. Due to the methodological challenges in measuring GNG and/or DNL, these pathways are not often specifically probed in human subjects.
Given the paucity of evidence from human studies, and on the basis of what has been reported in rodent models, an alternative suggestion is that hepatic insulin signalling remains intact and that IR is peripheral to the liver. In support of this, by deleting insulin signalling pathways within the liver of rodents, it was shown that hepatic glucose production was modulated via adipose tissue-derived glycerol suggesting adipose tissue IR contributes to impaired hepatic glucose production(Reference Perry, Camporez and Kursawe93). However, Jin et al.(Reference Jin, Browning and Murphy94) measured the contribution of adipose-derived glycerol to glucose production, and found that the livers of NAFLD patients maintained the ability to compensate for increased availability of precursor substrate (e.g. decreased glucose release from glycogen stores in response to increased delivery of glycerol). On the basis of this work, it could be speculated that even with IHTG accumulation, the liver maintains some degree of metabolic flexibility; however, this may be dependent on other factors such as duration and severity of NAFLD.
In summary, there is little evidence that selective hepatic IR exists in human subjects. Rather, recent evidence suggests that features of IR may be driven by either: (i) peripheral IR which results in greater delivery of GNG substrates being delivered to the liver, or (ii) total hepatic IR, with DNL appearing intact due to increased substrate availability to leading to the pathway being constitutively activated.
Conclusions
Studies have consistently shown that obesity is associated with ectopic fat deposition and increasing plasma insulin concentrations. The liver is a key metabolic organ involved in the metabolism of glucose and lipid handling and insulin regulates substrate fluxes to the liver, processes within the liver and secretion out of the liver. Therefore, the pathological accumulation of intracellular TAG often leads to dysregulation in glucose and lipid metabolism which may precede or occur with IR and NAFLD. Although rodent models have suggested a phenomenon of selective tissue-specific IR, the evidence for this in human subjects specifically for the liver is limited and inconsistent. Some of the obstacles in understanding if and how selective hepatic IR manifests in human subjects include the challenges of undertaking longitudinal studies to elucidate if weight gain or hyperinsulinaemia is the primary hit and identifying what the underlying primary defect would be. Notably in the liver, the catabolic and anabolic pathways are difficult to separate as they share intermediate metabolites and receptor signalling. Therefore, developing methodologies and relevant models that allow the dissociation of the catabolic and anabolic pathways will provide much needed insight into the underlying aetiology of hepatic IR.
Acknowledgements
The authors thank their colleagues and peers for useful and informative discussions whilst putting this review together.
Financial Support
L. H. is a British Heart Foundation Senior Research Fellow in Basic Science. S. R. N. is supported by a Novo Nordisk Postdoctoral Fellowship run in partnership with the University of Oxford.
Conflict of Interest
None.
Authorship
The authors had sole responsibility for all aspects of preparation of this paper.

