


Vitamin A dietary intake and deficiency
A historical perspective of vitamin A tells us that the symptoms of vitamin A deficiency and the ability to treat night blindness with animal liver have been known and documented for thousands of years (Fig. 1). Animal liver is enriched in retinyl esters (particularly retinyl palmitate, the main storage form of vitamin A). It was about 1930, in experiments in rats by T. Moore that determined that the yellow phytochemical β-carotene (pro-vitamin A) was the precursor for the active vitamin A, which was stored in the liver. Separately P. Karrer first isolated and then determined the distinct chemical structures of β-carotene and retinol( Reference Lanska 1 – Reference Wolf 3 ).

Fig. 1. (Colour online) A historical perspective of vitamin A, known and documented for thousands of years, from ancient Egypt to the early 20th century discovery of an essential ‘fat soluble factor A’( Reference Lanska 1 – Reference Wolf 3 ).
Vitamin A/retinoic acid (RA) have been shown to play essential roles in the preservation of immune function, continued promotion of good vision and the development, growth and maintenance of multiple body tissues. Animals do not have the capability to generate vitamin A via de novo synthesis and daily dietary vitamin A intake can vary. Important dietary sources of vitamin A are from richly-coloured fruit and vegetables and also fish and animal liver( Reference Grune, Lietz and Palou 4 ). More generally, richly-coloured fruit and vegetables are abundant sources of phytochemicals (such as carotenoids), which are not essential, but do have potential health benefits. Carotenoids form a large class of colourful plant pigments, which include carotenes such as β-carotene (pro-vitamin A) and lycopene and also xanthophylls such as zeaxanthin and lutein (which unlike β-carotene are not metabolised to vitamin A). Dietary and nutritional surveys performed in the USA and Europe indicate that most people's dietary intake of β-carotene is 1–2 mg/d, which is sufficient if the intake of preformed vitamin A (from animal sources) is also adequate( Reference Grune, Lietz and Palou 4 ). Potential health benefits from frequent consumption of fruit and vegetables are often ascribed to high concentrations of these phytochemicals and their relatively high antioxidant activities. However, the focus of this review is specifically how vitamin A and RA signalling can influence metabolic physiology.
Vitamin A homeostasis
Vitamin A is essential for animals, but they cannot synthesise vitamin A and also daily dietary intake can vary widely, so animals have evolved an intricate pathway to maintain precise vitamin A homeostasis( Reference Shirakami, Lee and Clugston 5 ). Firstly retinol is acquired from dietary sources in the form of preformed retinyl esters (animal sources) or the pro-vitamin β-carotene (fruit and vegetable sources), which can be enzymatically cleaved in the small intestine and converted into retinol. Secondly, to store vitamin A, retinol is coupled to fatty acids like palmitate in the liver to form retinyl esters and these are mobilised as required. Thirdly, the plasma concentration of retinol is strictly regulated and maintained at about 2–3 µm in human subjects (and about 1 µm in mice). Finally, retinol must be metabolised to RA within cells before it can enter the nucleus to regulate gene transcription via ligand induced activation of specific transcription factors.
Ingesting too much preformed vitamin A from foods (such as fish or animal liver), supplements, or prescription medications based on vitamin A can lead to hypervitaminosis A and toxicity of the liver. It may also lead to decreased bone mineral density( Reference Jackson and Sheehan 6 ) and these compounds can induce teratogenic effects in the developing embryo, thus women of child bearing age are advised to avoid such foods and drugs around conception and the first trimester of pregnancy( Reference Grune, Lietz and Palou 4 ). These problems are not an issue with intake of pro-vitamin A/β-carotene, from fruit and vegetable sources, because of the lower conversion efficiency of β-carotene to retinol equivalents (compared with preformed vitamin A)( Reference Grune, Lietz and Palou 4 ).
Retinoid transport, metabolism and signalling
In the molecular era, it is through the study of genetic knockouts of the key proteins that regulate vitamin A storage and mobilisation that have elucidated how homeostasis is maintained( Reference Shirakami, Lee and Clugston 5 ). Vitamin A is a lipophilic, fat soluble molecule and therefore requires specific binding proteins in order to be transported in the circulation and within the cell. Serum retinol binding protein (RBP; gene name RBP4) is essential for mobilisation of hepatic retinol into plasma, and for the delivery of retinol to the retina of the eye (where it is required for normal vision), especially when the availability of dietary retinoid is marginal. Approximately 80–90 % of all retinoids in the body are stored as retinyl esters in the liver. Here, cellular retinol binding protein-1 and lecithin:retinol acyltransferase serve to facilitate efficient retinyl ester synthesis, metabolism and mobilisation within cells( Reference Shirakami, Lee and Clugston 5 ). Retinol derived from the hydrolysis of retinyl ester can bind to RBP and the retinol–RBP complex enters the bloodstream for transport to target tissues to meet the tissues retinoid requirements. Cellular retinol binding protein-1 is also proposed to play a role in regulating the oxidation of retinol in the two step conversion to RA( Reference Shirakami, Lee and Clugston 5 ).
RA is the active metabolite of vitamin A, and is a critical signalling molecule involved in many biological processes via ligand induced activation of specific transcription factors, nuclear hormone receptors called RA-receptors. RA-receptors form heterodimers with retinoid-X receptors and bind to RA-response elements present in the promoters of target genes via the DNA-binding domain present within each receptor( Reference McGrane 7 , Reference Bastien and Rochette-Egly 8 ). It is via this binding to DNA that enables RA to regulate gene transcription programmes. RA-signalling is well known to play essential roles in developmental biology. In contrast, within the retina of the eye, it is the metabolite 11-cis-retinal that drives photoreceptor function in the visual cycle( Reference Kiser, Golczak and Maeda 9 ).
Due to the maintenance of tight vitamin A homeostasis in adult animals, RA-mediated nuclear signalling was largely assumed that to be relatively less important, especially in the regulation of metabolic physiology. Indeed much more attention has been paid to the role of other nuclear receptors in the regulation of metabolic processes including glucose and lipid metabolism, e.g. the RA-receptor heterodimer partner retinoid-X receptors, PPAR, liver-X-receptors and farnesoid-X-receptors( Reference Evans and Mangelsdorf 10 ). However, an increasing number of ground-breaking studies have now added to the largely ignored historical data that linked vitamin A levels with body mass regulation and type-2 diabetes( Reference Basualdo, Wein and Basu 11 – Reference Abahusain, Wright and Dickerson 13 ).
Vitamin A levels and type-2 diabetes
Type-2 diabetes is a condition whereby blood glucose levels are elevated due to impaired insulin action (or insulin resistance) in muscle and liver and is commonly associated with increased fat mass and obesity. There are several reports associating increased circulating vitamin A levels or serum RBP4 (essential for transport of retinol in blood) with type-2 diabetes. Early reports of increased levels of serum and/or urine RBP4 in subjects with type-2 diabetes did not identify a cause or a down-stream effect( Reference Basualdo, Wein and Basu 11 – Reference Abahusain, Wright and Dickerson 13 ). In 2005, a role for elevated serum levels of RBP4 was first described from the laboratory of B.B. Kahn. They discovered that expression of RBP4 was elevated in adipose tissue of mice with impaired insulin action in muscle and liver in vivo ( Reference Yang, Graham and Mody 14 ). Elevated levels of this adipokine (a term used to describe hormones or paracrine factors produced by adipose tissue) were found to associate with insulin resistance in multiple genetic and diet-induced models of obesity and type-2 diabetes. Several experimental models to alter circulating RBP4 levels, either genetically or pharmacologically, suggested strongly that elevated RBP4 levels might contribute to insulin resistance in muscle and liver and thus impaired glucose homeostasis and type-2 diabetes( Reference Yang, Graham and Mody 14 ).
However, human studies of obesity, insulin resistance or type-2 diabetes have been more controversial since not all of these have reported elevations in circulating RBP4 levels. Technical problems using enzyme-linked immunoassays may have led to inaccurate measurements of serum RBP4 concentrations( Reference Kotnik, Fischer-Posovszky and Wabitsch 15 ). Other confounding issues such as genetic background, age, sex, adipose distribution and kidney function should also be considered( Reference Kotnik, Fischer-Posovszky and Wabitsch 15 ). The exact mechanism by which elevated RBP4 levels contribute to insulin resistance and impaired glucose homeostasis remains unclear, but will be considered next. Serum RBP was first reported to induce hepatic expression of the key gluconeogenic enzyme phosphoenolpyruvate carboxykinase and thus increase circulating blood glucose levels via increased hepatic glucose production( Reference Yang, Graham and Mody 14 ). This effect could be directly via increased RA-signalling to the RA-response elements in the gene promotor of phosphoenolpyruvate carboxykinase (also see next section). Elevated levels of serum RBP were also reported to impair insulin signalling in skeletal muscle by an unknown mechanism( Reference Yang, Graham and Mody 14 ).
The early findings, implicating elevated circulating levels of RBP4 with insulin resistance, had used the available transgenic mouse models at the time, the whole body knockout of RBP4 and muscle-specific overexpression of RBP4( Reference Yang, Graham and Mody 14 , Reference Quadro, Blaner and Salchow 16 , Reference Quadro, Blaner and Hamberger 17 ). Very recent advances in the field have been reported with the generation of mice with adipocyte-specific RBP4 overexpression( Reference Lee, Yuen and Jiang 18 ) and mice with hepatocyte-specific deletion of RBP4( Reference Thompson, Sargsyan and Lee 19 ). These complementary studies concluded that adipocyte RBP4 is not a significant source of circulating RBP4 since circulating RBP4 was undetectable in mice lacking hepatocyte RBP4( Reference Thompson, Sargsyan and Lee 19 ). However, in the setting of insulin resistance, adipocyte RBP4 may have a more important paracrine function that is confined within the adipose tissue compartment. Indeed, adipocyte-specific overexpression of RBP4 increased proinflammatory markers and genes encoding lipases associated with lipolysis in adipose tissue( Reference Lee, Yuen and Jiang 18 ). These alterations in adipose tissue were also associated with increased circulating NEFA and an increase in hepatic lipid accumulation without alterations in hepatic retinoid levels or genes.
In some tissues such as muscle and adipose, retinol-RBP4 is recognised by a plasma membrane receptor termed ‘stimulated by retinoic acid 6’ that transports retinol from the binding protein into cells( Reference Noy 20 ). Stimulated by retinoic acid 6 also appears to signal via a phosphorylation cascade involving Janus kinases and their associated transcription factors called signal transducers and activators of transcription( Reference Noy 20 ). These observations suggest more than one possible mechanism through which RBP4 may control insulin action but until a more complete network of signalling downstream of RBP4 and/or stimulated by retinoic acid 6 is determined, the role of RBP4 in metabolic disease will remain unclear.
Dietary, pharmacological or genetic manipulation of vitamin A/retinoic acid homeostasis
In contrast, dietary vitamin A deficiency or genetic intervention to decrease endogenous production of RA and thus reduce RA-signalling in mice leads to decreased fasting blood glucose levels and reduced hepatic gluconeogenesis( Reference Shin, Odom and Scribner 21 , Reference Kiefer, Orasanu and Nallamshetty 22 ). Specifically, mice lacking the RA-synthesizing enzyme retinaldehyde dehydrogenase-1 have reduced expression of key gluconeogenic enzymes glucose-6-phosphatase and phosphoenolpyruvate carboxykinase, which is a bona fide RA-inducible target gene since the phosphoenolpyruvate carboxykinase gene promoter has been shown to contain a specific RA-receptor-binding site i.e. a RA-response element( Reference Shin, Odom and Scribner 21 ).
Moreover, mice without retinaldehyde dehydrogenase-1 are protected from high-fat diet-induced obesity-insulin resistance. The mechanism of this protection was proposed to be from the accumulation of retinaldehyde (the sole precursor to RA). It was also suggested that retinaldehyde can increase the expression of mitochondrial uncoupling protein1 to drive uncoupled respiration and adaptive thermogenesis in white adipose tissue, essentially promoting a brown adipose phenotype to increase energy expenditure and inhibit weight gain. This effect is not unlike the well-characterised pharmacological effects of RA to also induce uncoupling protein-1( Reference Alvarez, de Andres and Yubero 23 – Reference Kumar, Sunvold and Scarpace 26 ). RA has also been shown to inhibit the cellular process of adipocyte differentiation and prevent obesity and insulin resistance in rodents( Reference Murray and Russell 27 – Reference Schwarz, Reginato and Shao 30 ).
The overall beneficial metabolic effects of RA have also been reported to include changes in hepatic lipid metabolism leading to repartitioning of fatty acids away from TAG storage and towards oxidation( Reference Berry and Noy 31 ). However, liver toxicity with large doses and/or prolonged exposure, restricts the use of RA as a potential therapy for obesity and type-2 diabetes. Moreover, hypertriglyceridemia is a relatively frequent side effect of retinoid therapy (e.g. for dermatological disorders)( Reference Bershad, Rubinstein and Paterniti 32 ). Retinoid-induced hypertriglyceridemia in human subjects has also been modelled in a number of rodent studies and has been reported to occur in response to high doses of vitamin A (as retinol or retinyl palmitate), RA isomers (including RA, 9-cis RA and 13-cis RA) and synthetic retinoid-X receptors-specific agonists (rexinoids)( Reference Standeven, Beard and Johnson 33 , Reference Standeven, Thacher and Yuan 34 ). These and other seemingly contradictory findings are reviewed in more detail elsewhere, and may depend on the balance between nuclear hormone heterodimers and the complex interplay with other transcription factors controlling adaptations to nutrient intake and metabolic needs( Reference Bonet, Ribot and Palou 35 ).
Beneficial metabolic effect of the synthetic retinoid Fenretinide
We have shown that Fenretinide, a synthetic retinoid with decreased toxicological profile, can reduce obesity and prevent insulin-resistance in both muscle and liver in the high-fat diet-induced obese mouse model, via major effects on adipose and liver gene expression. Fenretinide was originally developed as a chemotherapeutic agent, has been widely studied in clinical trials of breast cancer chemoprevention( Reference Mody and Mcilroy 36 ). Fenretinide can strongly stimulate expression of classic RA-induced targets, decrease levels of serum retinol binding protein, inhibit adipocyte differentiation and decrease expression of the adipocyte secreted satiety hormone leptin( Reference Yang, Graham and Mody 14 , Reference Preitner, Mody and Graham 37 – Reference Shearer, Morrice and Henderson 39 ). These effects are in contrast to the subtle effects of specifically altering RBP4 levels in the whole body or in tissue-specific manner, which as mentioned earlier, do not lead to alterations in hepatic retinoid levels or classic RA-induced target genes that are involved in the tight control of retinoid homeostasis.
Fenretinide also has effects independent of RA-signalling due to its chemical structure (addition of a bulky hydroxyphenyl group compared with RA). These effects have been attributed to the inhibition of ceramide biosynthesis, a sphingolipid that has been implicated in the pathogenesis of insulin resistance( Reference Bikman, Guan and Shui 40 , Reference Mcilroy, Tammireddy and Maskrey 41 ). An excess supply of NEFA such as palmitate with a high-fat, high-sugar diet and/or obesity are thought to lead to increased ceramide production, which can impair mitochondrial function at the level of metabolic flux via fatty-acid β-oxidation and the tricarboxylic acid cycle( Reference Rodriguez-Cuenca, Barbarroja and Vidal-Puig 42 , Reference Chaurasia and Summers 43 ). This impairment is evident with an accumulation of acylcarnitines (fatty-acid β-oxidation intermediates) and tricarboxylic acid cycle intermediates. In mice, Fenretinide treatment completely normalised the accumulation of these intermediates in adipose tissue despite these mice continuing to feed a high-fat, high-sugar diet( Reference Mcilroy, Tammireddy and Maskrey 41 ).
Ceramides can also inhibit insulin-signalling by directly increasing the phosphatase activity towards protein kinase B/Akt( Reference Stratford, Hoehn and Liu 44 ). Thus, probably through its effect on ceramides, Fenretinide can increase protein kinase B/Akt phosphorylation in adipocytes, even in the absence of insulin( Reference Mcilroy, Delibegovic and Owen 38 ). Moreover, the primary oxidised catabolite of Fenretinide, which is a very poor inducer of RA-signalling, can also inhibit the enzyme responsible for the final step of ceramide synthesis and so similarly to Fenretinide, can also increase protein kinase B/Akt phosphorylation in adipocytes( Reference Mcilroy, Tammireddy and Maskrey 41 ).
Fibroblast growth factor 21: a new physiological target of vitamin A/retinoic acid
Most recently, Fenretinide and RA have been reported to repress the levels of the liver derived hormone fibroblast growth factor (FGF) 21( Reference Morrice, Mcilroy and Tammireddy 45 ). Hepatic Fgf21 expression and circulating levels of FGF21 are increased by stimuli such as fasting, protein/amino acid restriction and fructose ingestion via a range of transcription factors, including PPARα and carbohydrate response-element binding protein( Reference Giralt, Gavalda-Navarro and Villarroya 46 , Reference Fisher and Maratos-Flier 47 ). Thus, FGF21 has emerged as an important beneficial regulator of glucose and lipid homeostasis but its levels are also abnormally increased in insulin-resistant states in rodents and human subjects. Interestingly, Fenretinide can normalise elevated levels of FGF21 in both high-fat diet-induced obese mice and in genetically obese-diabetic mice in association with the wide-range of beneficial effects on metabolic parameters reported previously and summarised earlier (Fig. 2)( Reference Morrice, Mcilroy and Tammireddy 45 ). Furthermore, diverse treatments to improve glycaemic control in human subjects can decrease elevated levels of FGF21 e.g. with metformin, rosiglitazone, insulin, insulinotropic agents, bariatric surgery, lifestyle modification, fish oil supplements or exercise( Reference Fan, Sun and Zhang 48 – Reference Gomez-Ambrosi, Gallego-Escuredo and Catalan 54 ). Since Fenretinide has been widely studied as a cancer therapeutic due to its favourable toxicological profile and is undergoing phase II clinical trials for treatment of insulin resistance in obese human subjects with hepatic steatosis, these new findings should be of considerable interest to the field of nutrition, obesity and diabetes research.
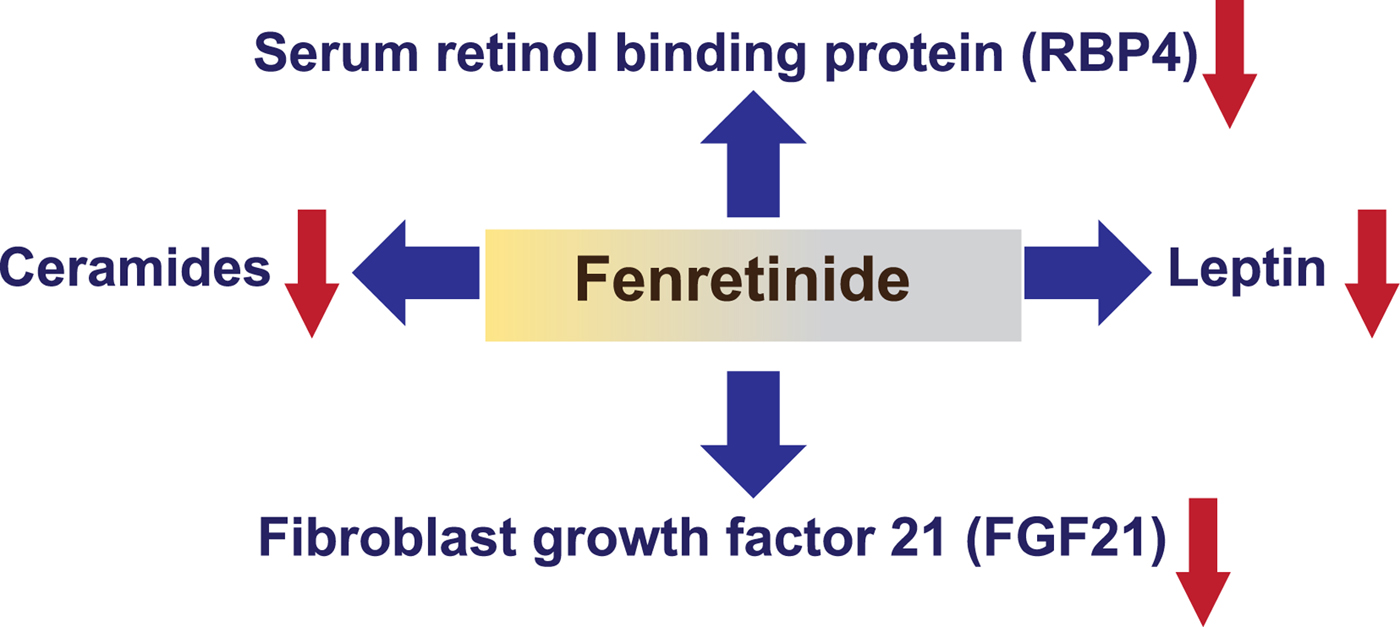
Fig. 2. (Colour online) Identified targets of synthetic retinoid Fenretinide via classic nuclear hormone signalling or via non-nuclear protein interaction. Fenretinide was originally found to decrease levels of serum retinol binding protein (RBP) 4 in cancer chemoprevention trials and in models of obesity and insulin resistance( Reference Yang, Graham and Mody 14 , Reference Mody and Mcilroy 36 , Reference Preitner, Mody and Graham 37 ). Fenretinide and retinoic acid (RA) can decrease expression of the adipocyte secreted satiety hormone leptin in cells and in vivo ( Reference Mcilroy, Delibegovic and Owen 38 , Reference Shearer, Morrice and Henderson 39 ) and repress levels of the liver derived hormone fibroblast growth factor (FGF) 21( Reference Morrice, Mcilroy and Tammireddy 45 ). Independently of RA-signalling, Fenretinide can also inhibit the enzyme responsible for the final step of ceramide synthesis( Reference Bikman, Guan and Shui 40 , Reference Mcilroy, Tammireddy and Maskrey 41 , Reference Morrice, Mcilroy and Tammireddy 45 ).
Conclusions
There is now considerable evidence to implicate vitamin A homeostasis in the regulation of body fat and blood glucose levels. It appears that altered levels of circulating retinol (bound to RBP) has a completely distinct role compared with RA, in regulating endocrine hormones and gene transcription programmes to alter lipid and glucose metabolism. Powerful new approaches combining the techniques of phenotyping of transgenic mice with metabolomics, lipidomics and next-generation DNA sequencing to profile epigenomic changes and nuclear receptor occupancy across the genome will aid us to understand more about these interactions in the future. Moreover, these approaches are likely necessary to understand more about the potential of phytochemical and pharmaceutical strategies to treat obesity and type 2 diabetes in preclinical models and to be able to translate positive findings to human subjects.
Acknowledgements
The author would like to thank M. Delibegovic and P. McCaffery (both University of Aberdeen) for their helpful comments and proof reading in preparation of this review.
Financial Support
Work in the author's laboratory was supported by the British Heart Foundation Intermediate Basic Research Fellowship FS/09/026, The Royal Society (of London) grant, Tenovus Scotland grants G10/04 and G14/14 to N. Mody, University of Aberdeen Centre for Genome Enabled Biology and Medicine (CGEBM) PhD studentship to Nicola Morrice and Biotechnology and Biological Sciences Research Council (BBSRC) studentship to George D. Mcilroy.
Conflict of Interest
None.
Authorship
The author was solely responsible for all aspects of preparation of the paper.


