



Introduction
The north of Italy is rather rich in manganese deposits. Among the 391 species described so far in Italy, 16 species contain Mn3+ and all of them are from a Ligurian or Piedmontese (s.l.) manganese type-locality deposit (Table 1). Piccoliite, NaCaMn3+2(AsO4)2O(OH), was first recognised on a specimen found in 2007 by Gian Carlo Piccoli in the dumps of the Montaldo di Mondovì mine (44°19’08.9”N, 7°51.09’5”E), Borgata Oberti, Montaldo di Mondovì, Corsaglia Valley, Piedmont, Italy (Fig. 1). A preliminary characterisation of the crystal structure of the mineral was provided by Kolitsch (Reference Kolitsch2008). A second occurrence, which helped to improve the understanding of the mineral's crystal chemistry, was recognised in 2015 on specimens from the dumps of the Valletta mine (44°23’42’’N, 7°5’42’’E, 2536 m above sea level), Canosio, Maira Valley, Piedmont, Italy.

Fig. 1. Manganese ore cropping out along the Corsaglia river (Montaldo di Mondovì mine). In the photo is mineral collector Pierluigi Ambrino. Photo Marco E. Ciriotti, August 2014.
Table 1. Italian type minerals containing essential Mn3+.

* Liguria type localities; # Piedmont (and Aosta Valley) type localities.
References: 1 Kampf et al. (Reference Kampf, Carbone, Belmonte, Nash, Chiappino and Castellaro2017); 2 Cámara et al. (Reference Cámara, Baratelli, Ciriotti, Nestola, Piccoli, Bosi, Bittarello, Hålenius and Balestra2022); 3 Haidinger (Reference Haidinger1828); 4 Basso et al. (Reference Basso, Lucchetti, Zefiro and Palenzona2000); 5 Callegari et al. (Reference Callegari, Boiocchi, Ciriotti and Balestra2012); 6 Cámara et al. (Reference Cámara, Kampf, Nestola, Ciriotti, Spartà and Balestra2021b); 7 Orlandi et al. (Reference Orlandi, Biagioni, Pasero and Mellini2013); 8 Biagioni (Reference Biagioni, Bonazzi, Pasero, Zaccarini, Balestra, Bracco and Ciriotti2019); 9 Cenki-Tok et al. (Reference Cenki-Tok, Ragu, Armbruster, Chopin and Medenbach2006); 10 Lepore et al. (Reference Lepore, Bindi, Benedetto, Mugnaioli, Viti, Zanetti, Ciriotti and Bonazzi2017); 11 Basso et al. (Reference Basso, Lucchetti, Zefiro and Palenzona1993); 12 This work; 13 Kenngott (Reference Kenngott1853); 14 Bonazzi et al. (Reference Bonazzi, Menchetti and Palenzona1990); 15 Meisser et al. (Reference Meisser, Perseil, Brugger and Chiappero1999).
The name of the new mineral honours the mineral collectors Gian Carlo Piccoli (b. 1953) and his father Gian Paolo Piccoli (1928–1996) for their contribution to the knowledge of the regional mineralogy of Piedmont and Aosta Valley. Since the 1970s, father and son carried out mineralogical research in the Western Alps (Maritime and Cottian Alps, in the province of Cuneo, Piedmont), leading to the discovery of gramaccioliite-(Y) (Orlandi et al., Reference Orlandi, Pasero, Rotiroti, Olmi, Demartin and Moëlo2004) and grandaite (Cámara et al., Reference Cámara, Ciriotti, Bittarello, Nestola, Massimi, Radica, Costa, Benna and Piccoli2014), and to the creation of the mineralogical collection of the province of Cuneo, kept in the ‘Federico Eusebio’ civic museum in Alba, Cuneo Province, Piedmont. After his father's death, Gian Carlo Piccoli devoted himself to the writing of the books “Minerali delle Alpi Marittime e Cozie – Provincia di Cuneo” (Piccoli, Reference Piccoli2002) and “Minerali del Piemonte e della Valle d'Aosta” (Piccoli et al., Reference Piccoli, Maletto, Bosio and Lombardo2007b). In this latter work, more than 570 different mineral species found in Piedmont and Aosta Valley are described. Finally, Gian Carlo Piccoli is co-author of the book “Minerali in Val d'Ala” (Maletto and Piccoli, Reference Maletto and Piccoli2014).
The mineral and its name (symbol Pcc) were approved by the Commission on New Minerals, Nomenclature and Classification of the International Mineralogical Association. (IMA2017-016, Cámara et al., Reference Cámara, Biagioni, Ciriotti, Bosi, Kolitsch, Paar, Blass and Bittarello2017). Cotype material from Montaldo di Mondovì is deposited in the Mineralogical Collection of the Museo di Storia Naturale, Università di Pisa, Via Roma 79, Calci, Pisa, Italy, under catalogue number 19906 and in the mineralogical collection of the Museo civico archeologico e di scienze naturali “Federico Eusebio” di Alba, Cuneo, Italy, with catalogue numbers M00673 (Montaldo di Mondovì specimen) and M00674 (Valletta specimen).
Occurrence
The Fe–Mn ores of the Montaldo di Mondovì mine were exploited for iron (Sismonda, Reference Sismonda1841a, Reference Sismonda1841b; Conti, Reference Conti1873) from 1838 until its closure in the mid-1950s. The deposit is hosted within the ‘Klippen of Deviglia’ Unit (Vanossi, Reference Vanossi1980) belonging to the Mesozoic sequence of the innermost Briançonnais Zone (Dalla Giovanna and Vanossi, Reference Giovanna G. and Vanossi1991; Vanossi et al., Reference Vanossi, Cortesogno, Galbiati, Messiga, Piccardo and Vannucci1986). Metaquartzites and quartz-bearing marbles host the Fe–Mn ores that occur as layers, boudins, and, subordinately, in mineralised veins. Late-stage hematite-bearing veinlets crosscut both quartz arenites and marbles (Cabella et al., Reference Cabella, Gaggero and Lucchetti1992, Reference Cabella, Lucchetti and Marescotti1995, Reference Cabella, Lucchetti and Marescotti1999). The mineralisation consists of hollandite-supergroup minerals ± hematite ± braunite, with variable amounts of quartz, calcite, Mn- and Fe-rich muscovite and aegirine (Cabella et al., Reference Cabella, Lucchetti and Marescotti1999; Kolitsch et al., Reference Kolitsch, Ciriotti, Cadoni, Armellino, Piccoli, Ambrino, Blass, Odicino and Ciuffardi2011; Giai, Reference Giai2020). Further details on the geological setting are reported in Cámara et al. (Reference Cámara, Ciriotti, Kolitsch, Bosi, Bittarello, Brizio, Vignola and Blaß2021a) and references therein, which describe armellinoite-(Ce), Ca4Ce4+(AsO4)4⋅H2O, another arsenate for which the Montaldo di Mondovì mine is also the type locality. Other associated minerals include aegirine, anatase, armellinoite-(Ce), arsenogoyazite, berzeliite/manganberzeliite, braunite, calcite, chernovite-(Y), dolomite, cinnabar, cryptomelane, As-rich fluorapatite, gasparite-(Ce), gasparite-(La), goethite, hausmannite(?), hematite, hollandite, monazite-(Ce), montmorillonite, muscovite, orthoclase, pyrolusite(?), quartz, ramsdellite, ranciéite, romanèchite, rutile, spessartine, svabite, talmessite, tilasite, titanite wakefieldite-(Ce), wakefieldite-(Y), zircon, and three potential new minerals, i.e. the unnamed La-, Nd- and (Ca,Y,REE)-dominant analogues of chernovite-(Y) (Piccoli, Reference Piccoli2002, Reference Piccoli2007; Piccoli et al., Reference Piccoli, Kolitsch, Blaß and Ciriotti2007a, Reference Piccoli, Maletto, Bosio and Lombardo2007b; Ciriotti, Reference Ciriotti2007; Kolitsch et al., Reference Kolitsch, Ciriotti, Cadoni, Armellino, Piccoli, Ambrino, Blass, Odicino and Ciuffardi2011; Giai, Reference Giai2020). Piccoliite at this locality always occurs in quartz–calcite veins (Fig. 2a), associated with calcite, berzeliite/manganberzeliite and, very rarely, with chernovite and the aforementioned unnamed rare earth element (REE) analogues of chernovite-(Y).

Fig. 2. Piccoliite from the two Italian occurrences. (a) A well-developed black crystal (1 mm) on quartz from the dump of the Montaldo di Mondovì mine. Collection Gianluca Armellino, photo Pierluigi Ambrino. (b) Black crystals of piccoliite associated with grandaite from the Valletta mine dump. Field of view 5 mm. Collection and photo Roberto Bracco.
The Valletta mine was mined for iron in the 15th Century and probably hastily abandoned both due the uneconomic mixture of hematite with manganese ores and because it is located at a very high altitude (2536 m above sea level). The Fe–Mn deposit is located in the Briançonnais Zone of the Cottian Alps. Specifically, the Mn-bearing minerals are hosted in Permian quartzites overlying quartzitic conglomerates (Verrucano formation) and quartz–feldspar fine-grained schists derived from Permian rhyolitic volcanism of the so-called Axial Permian–Carboniferous Zone (Franchi and Stella, Reference Franchi and Stella1930). A major subvertical tectonic contact, Faille de Ceillac (Gidon et al., Reference Gidon, Kerchove, Michard, Tricart and Goffé1994), separates the rocks found at the Valletta mine from the Middle Triassic carbonate sequence of the Becco Grande to the north and from the Rocca La Meja ridge to the south. The rocks found at the Valletta mine are part of the Bande de Marinet, the southernmost tectonic zone of the Axial Permian–Carboniferous Zone; specifically, it is part of Unit M3, the structurally highest unit defined in the Bande de Marinet by Lefèvre (Reference Lefèvre1982). During the Alpine tectonometamorphic cycle, the rocks now exposed in the Bande de Marinet were affected by high-P, low-T metamorphism of blueschist facies, the effects of which are more evident in the metabasites interlayered in the metavolcanic quartz–feldspar schists that often display the characteristic lawsonite–glaucophane assemblage (Franchi and Stella, Reference Franchi and Stella1930; Gidon et al., Reference Gidon, Kerchove, Michard, Tricart and Goffé1994). Here, piccoliite is also always embedded in quartz matrix, associated with hematite, grandaite (Fig. 2b), tilasite/adelite, and, rarely, with thorianite. Other minerals that have been observed in the Valletta mine dump include aegirine, albite, aldomarinoite, azurite, baryte, bariopharmacosiderite, berzeliite, bosiite, braccoite, braunite, calcite, canosioite, castellaroite, coralloite, cryptomelane, diopside, fianelite, gamagarite, ganophyllite, gypsum, hollandite, ilmenite, lombardoite, magnesio-arfvedsonite, magnesio-riebeckite, magnetite, malachite, manganberzeliite, mimetite, muscovite (Mn-bearing), neotocite, opal, orthoclase, oxy-dravite (Fe3+-Mn3+-V3+-rich), palenzonaite, phlogopite, pyrobelonite, ranciéite, rhodochrosite, rhodonite, richterite, rüdlingerite, rutile, saneroite, talc, tetrahedrite-series minerals, tinnunculite, tiragalloite, titanite, tokyoite, wallkilldellite, arsenate members of the alluaudite group, as well as other possible uncharacterised amphiboles (Cámara et al. Reference Cámara, Baratelli, Ciriotti, Nestola, Piccoli, Bosi, Bittarello, Hålenius and Balestra2022 and references therein). It is worth noting that many Mn-bearing phases at the Valletta mine contain iron exclusively as Fe3+ and manganese as Mn3+.
Appearance and physical properties
Piccoliite occurs rarely at the Montaldo di Mondovì locality as black well-developed prismatic crystals within calcite–quartz veins (Fig. 2a), whereas it forms aggregates of anhedral crystals, embedded in the quartz matrix at the Valletta locality (Fig. 2b). Splinters may be very dark red. The streak is brown and the lustre is resinous to vitreous. The mineral is non-fluorescent and opaque, but transparent in thin section. Hardness was measured with Vickers indentation: four measurements were performed, with two different loads, obtaining VHN15 (two measurements) = 654 and 656 kg/mm2 and VHN25 (two measurements) = 584 and 657 kg/mm2, which corresponds to a Mohs hardness of 5–5½. Piccoliite is brittle, with no cleavage or parting observed. Fracture is irregular. Density was not measured because of the lack of heavy liquids of sufficient density. Calculated density is 4.084 g⋅cm–3 (based on the empirical formula and unit-cell volume refined from single-crystal X-ray diffraction data of the Montaldo di Mondovì specimen) and 4.011 g⋅cm–3 (Valletta sample).
Owing to the small amount of material available for the mineralogical study, the optical properties of piccoliite were determined in plane-polarised reflected light on a crystal fragment embedded in epoxy. Piccoliite is grey in colour. Under crossed polars, it is distinctly anisotropic, with rotation tints in shades of grey. Internal reflections in brownish tints are commonly observed. Reflectance was measured in air using a LEITZ microphotometer. Readings were taken for specimen and standard (Zeiss SiC Standard 079) maintained under the same focus conditions for the standard wavelengths recommended by the Commission on Ore Mineralogy of the IMA. The edge of the square-like measuring area was ⁓0.1 mm. Reflectance values (R min, R max in %) for each λ (nm) are: 7.7, 9.8 at 470 nm; 7.7, 9.5 at 546 nm; 7.4, 9.3 at 589nm and 7.4, 9.2 at 650 nm.
The mean refractive index, calculated using the Gladstone–Dale relationship (Mandarino, Reference Mandarino1979, Reference Mandarino1981), and the calculated density of the Montaldo di Mondovì material, is 1.878.
Raman spectroscopy
Unpolarised micro-Raman spectra were obtained on an unpolished sample from the Montaldo di Mondovì locality in nearly back-scattered geometry with a Jobin-Yvon Horiba XploRA Plus apparatus, equipped with a motorised x–y stage and an Olympus BX41 microscope with a 10× objective. The 532 nm line of a solid-state laser was used. The minimum lateral and depth resolution was set to a few μm. The system was calibrated using the 520.6 cm–1 Raman band of silicon before each experimental session. Spectra were collected through multiple acquisition with single counting times of 180 s. Back-scattered radiation was analysed with a 1200 mm–1 grating monochromator. Peak deconvolution was performed through the software Fityk (Wojdyr, Reference Wojdyr2010).
In the range between 100 and 1200 cm–1 (Fig. 3a), the observed Raman spectrum in piccoliite from Montaldo di Mondovì is dominated by a band at ~ 850 cm–1. Other bands are observed at (in cm–1) 160, 191 (lattice vibrations), 342, 441, 488 (bending of As–O bonds), 745 (hydroxyl deformation band) and 803 (As–O stretching vibrations, along with the band at 894 cm–1); band assignment is in agreement with Frost and Kloprogge (Reference Frost and Kloprogge2003). The Raman spectrum of piccoliite from Valletta (Fig. 3a) shows the same bands at slightly different values due to the small difference in chemical composition (in cm–1): 200, 318, 445, 518, 730, 800, 852 and 890. Differences in the intensity are probably due to the different crystallographic orientation of the grains studied.

Fig. 3. Raman spectrum of piccoliite from Montaldo di Mondovì (upper, in blue) and from Valletta (lower, in orange) in the region 100–1200 cm–1 (a) and 2800–3800 cm–1 (b).
In both spectra there are two bands at ~1650 and 1770 cm–1 that may correspond to overtones or combination modes of As–O-stretching vibrations, as no structural evidence in support of the presence of H2O groups was found (see below).
Finally, in the (O–H)-stretching region (2800–3800 cm–1), a broad band at 3140 cm–1 has been observed in the spectrum of piccoliite from Montaldo di Mondovì (Fig. 3b). The same spectral feature occurs in the spectrum collected on the sample from the Valletta mine but it shows a lower intensity. By using the relation of Libowitzky (Reference Libowitzky1999), a donor–acceptor O⋅⋅⋅O distance of ~2.685 Å was calculated, in good agreement with the observed O7⋅⋅⋅O1 distance of 2.660(3) Å (see below). Similar bands are also found in the structurally related mineral carminite (Frost and Kloprogge, Reference Frost and Kloprogge2003).
Chemical composition
Preliminary energy dispersive spectroscopy analysis of piccoliite showed Na, Mg, Ca, Mn, Fe and As being the only elements present with atomic number > 8. Quantitative chemical analyses were carried out on the crystal used for the crystal-structure refinement using a CAMECA SX50 electron microprobe (wavelength dispersive spectroscopy mode, 15 kV, 15 nA and nominal beam diameter of 1 μm). The elements Al, S, K, Ti, Cr, Ni, Cu and Ba were sought but found to be below the detection limit. Counting times were 20 s on the peak and 10 s on the backgrounds for Na, and 30 s on the peak and 15 s on the background for all the other elements. Direct H2O determination was not performed because of the very small amount of material available. Chemical data are given in Table 2.
Table 2. Chemical data (in wt.%) for piccoliite

Note: s.u. = standard uncertainty; – = not detected
*The Mn4+/Mn3+ ratio was calculated on the basis of 6 cations and 10 anions pfu; **H2O was calculated on the basis of one OH group pfu.
The empirical formula of piccoliite, based on 10 (O,OH) anions per formula unit (pfu) and 6 cations pfu, is (Na0.64Ca0.35)Σ0.99(Ca0.75Na0.24)Σ0.99(Mn3+1.08Fe3+0.59Mg0.20Ca0.10)Σ1.97(As2.03V0.03Si0.01)Σ2.07O9(OH) for Montaldo di Mondovì and (Na0.53Ca0.47)Σ1.00(Ca0.76Na0.23Sr0.01)Σ1.00(Mn3+0.63Fe3+0.49Mg0.48Mn4+0.34Ca0.06)Σ2.00(As1.97P0.01Si0.01)Σ1.99O9(OH) for Valletta mine. All Fe is given as Fe3+, whereas H2O was calculated from stoichiometry to correspond to 1 (OH) pfu. See below for the discussion on oxidation state of Mn in the material studied from both localities. The ideal formula of piccoliite is NaCaMn3+2(AsO4)2O(OH), corresponding to Na2O 6.41, CaO 11.59, Mn2O3 32.63, As2O5 47.51, H2O 1.86, total 100.00 wt.%.
X-ray crystallography
Powder X-ray diffraction data were collected on Montaldo di Mondovì material using a 114.6 mm Gandolfi camera. Data (in Å, for CuKα) are listed in Table 3. Unit-cell parameters were refined from the powder X-ray data using the method of Holland and Redfern (Reference Holland and Redfern1997) based on 14 unequivocally indexed reflections. Refined unit-cell parameters are a = 8.879(2), b = 7.509(2), c = 11.707(2) Å and V = 779.7(2) Å3 with Z = 4.
Table 3. Measured and calculated powder X-ray diffraction data for piccoliite from Montaldo di Mondovì.*

* Intensity and d hkl (in Å) were calculated using the software PowderCell 2.3 (Kraus and Nolze, Reference Kraus and Nolze1996) on the basis of the structural model given in Table 5. The seven strongest reflections are given in bold. Intensities were visually estimated: vs = very strong; ms = medium–strong; m = medium; w = weak; and vw = very weak.
Single-crystal X-ray diffraction intensity data were collected on a 0.080 × 0.040 × 0.035 mm crystal from Montaldo di Mondovì using a Bruker Smart Breeze diffractometer equipped with an air-cooled CCD detector, and graphite-monochromatised MoKα radiation (Dipartimento di Scienze della Terra, Università di Pisa). The detector-to-crystal distance was 50 mm. Data were collected using ω and φ scans, in 0.5° slices, with an exposure time of 45 s per frame and corrected for Lorentz and polarisation factors and absorption using the software package Apex2 (Bruker AXS Inc., 2004). Data were obtained on a 0.243 × 0.194 × 0.119 mm crystal from Valletta mine using an Oxford Gemini R Ultra diffractometer equipped with a CCD area detector at CrisDi (Università di Torino), with graphite-monochromatised MoKα radiation. The detector-to-crystal distance was 54.8 mm. Data were collected using ω scan modes, in 1° slices, with an exposure time of 17 s per frame and corrected for Lorentz and polarisation factors and absorption using the software package CrysAlisPro (Agilent Technologies, 2015).
Observed unit-cell parameters (Table 4) are consistent with orthorhombic symmetry. The statistical tests on the distribution of |E| values (|E 2–1| = 0.872) and the systematic absences suggested the space group symmetry Pbcm. The crystal from Valletta showed significant systematic absences violations of the c-glide but further examination of structural models having a lower symmetry led to models that were equivalent to a Pbcm structure. The violations of systematic absences may be due to double diffraction phenomena. The crystal structure was solved through direct methods using SHELXS-97 (Sheldrick, Reference Sheldrick2008). After locating the heavier atoms, the structure solution was completed through successive difference-Fourier syntheses and refined using SHELXL-2014 (Sheldrick, Reference Sheldrick2015). Scattering curves for neutral atoms were taken from the International Tables for Crystallography (Wilson, Reference Wilson1992). The following curves were used: Ca vs. Na at the A1 and A2 sites; Mn vs. Mg at the Mn site; As at the As1 and As2 sites; and O at the O1–O7 sites. The As1, As2 and O sites were found to be fully occupied by As and O, respectively. The A1 and A2 sites have a mixed (Na,Ca) and (Ca,Na) occupancy, respectively. Owing to the similar scattering factors of Mn and Fe, the occupancy of the Mn site was refined based on the chemical-analytical data: mixed (Mn,Fe) occupancy with minor Mg and Ca. The H atom bonded to the oxygen atom hosted at the O7 site was found in the difference-Fourier map. After several cycles of anisotropic refinement (only H was refined isotropically), the R 1 factor converged to 0.0250 for 1554 reflections with F o > 4σ(F o) for the crystal from the Montaldo di Mondovì mine, and 0.0260 for 3242 reflections with F o > 4σ(F o) for the crystal from the Valletta mine. Details of data collection and refinement are given in Table 4. Fractional atom coordinates, site occupancies and equivalent isotropic or isotropic displacement parameters are reported in Table 5. The crystallographic information file has been deposited with the Principal Editor of Mineralogical Magazine and is available as Supplementary material (see below). Table 6 reports selected bond distances. Table 7 shows the comparison between observed site scattering and that calculated from the proposed site population. Finally, Tables 8 and 9 provide a bond-valence analysis carried out using the bond-valence parameters of Gagné and Hawthorne (Reference Gagné and Hawthorne2015).
Table 4. Crystal and experimental data for piccoliite.*

* Note: for the specimen from Montaldo di Mondovì, Kolitsch (Reference Kolitsch2008) gives the following unit-cell parameters (space group Pbcm) from his preliminary crystal-structure determination [R(F) = 0.0195]: a = 8.885(2), b = 7.535(2), c = 11.707(2) Å and V = 783.8(3) Å3. The weighting scheme is defined as follows: w = q / [ σ2(F o2) + (a×P)2 + b×P + d + e×sin(θ)/λ] where P = [0.33333 × Maximum of (0 or F o2) + (1–0.333333) × F c2]; used values are a = 0.0210 and b = 0.5539 for the Montaldo di Mondovì sample and a = 0.0121 and b = 0.6188 for the Valletta sample.
Table 5. Atomic sites, multiplicity (m) and Wyckoff letter (W), site occupancy (s.o.), atom fractional coordinates, and equivalent isotropic or isotropic (*) displacement parameters (in Å2) for piccoliite.

Table 6. Selected bond lengths (in Å) for piccoliite.

* D.I. = Distortion index of bond lengths (Baur, Reference Baur1974)
Table 7. Refined vs. calculated site-scattering values (electrons per formula unit) and site population for piccoliite.

* Calculated using ionic radii from Shannon (Reference Shannon1976); a radius of O2– was taken as 1.37 Å considering average coordination number of bonding oxygen atoms.
Table 8. Weighted bond-valences (vu) for piccoliite.*

* Notes: Left and right superscripts indicate the number of equivalent bonds involving anions and cations, respectively. Bond valence at mixed-occupancy sites were weighted and their BVS (Σcations) compared with the weighted atomic valences (WAV) from the empirical formula. BVS of anions (Σanions) have been corrected considering hydrogen bonds.
Table 9. Weighted bond-valences (vu) for pilawite-(Y) (data from Pieczka et al. Reference Pieczka, Hawthorne, Cooper, Szełeg, Szuszkiewicz, Turniak, Nejbert and Ilnicki2015).*

* Notes: Bond valence at mixed-occupancy sites were weighted and their BVS (Σcations) compared with the weighted atomic valences (WAV) from the empirical formula. BVS of anions (Σanions) have been corrected considering hydrogen bonds.
X-ray absorption spectroscopy
X-ray absorption spectroscopy (XAS) measurements at the K-edge of Mn were performed at the LISA beamline (BM-08) (d'Acapito et al., Reference d'Acapito, Lepore, Puri, Laloni, La Mannna, Dettona, De Luisa and Martin2019) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Samples were measured using a pair of Si (111) flat crystals; Si-coated focusing mirrors (E cutoff ≈ 16 KeV) were used for harmonic rejection. Measurements were performed on a single crystal in the fluorescence mode using a four-channel Silicon Drift Detector (SDD) ARDESIA (Hafizh et al., Reference Hafizh, Bellotti, Carminati, Utica, Gugiatti, Balerna, Tullio, Lepore, Borghi, Ficorella, Picciotto, Zorzi, Capsoni, Coelli, Bombelli and Fiorini2019); Mn model compounds (rhodochrosite, bixbyite-(Mn) and pyrolusite) were measured in transmission mode. The spectra were acquired with a fixed k step of 0.05 Å–1 up to a maximum value of 12 Å–1. Samples were measured at room temperature. Standard procedures (Lee et al., Reference Lee, Citrin, Eisenberger and Kincaid1981) were followed to extract the structural extended X-ray absorption fine structure (EXAFS) signal (k•χ(k)): pre-edge background removal, spline modelling of bare atomic background, edge step normalisation using a region far above the edge, and energy calibration using the software ATHENA (Ravel and Newville, Reference Ravel and Newville2005). Model atomic clusters centred on the absorber atom were obtained by ATOMS (Ravel, Reference Ravel2001), using atomic coordinates taken from the crystal-structure determination reported in this study; theoretical amplitude and phase functions were generated using the FEFF8 code (Ankudinov et al., Reference Ankudinov, Ravel, Rehr and Conradson1998). EXAFS spectra were fitted through the ARTEMIS software (Ravel and Newville, Reference Ravel and Newville2005) in the Fourier-Transform (FT) space.
The XAS spectrum in the X-ray absorption near edge structure (XANES) region of piccoliite is in Fig. 4, together with those of rhodochrosite, bixbyite-(Mn), pyrolusite and hollandite. An accurate quantification of Mn valence state is rather difficult, especially without comparison with well-known model compounds belonging to the same structural family, as different octahedral arrangements and distortions, especially related to Jahn-Teller effects, may affect the pre-edge peak intensity and the overall XANES shape, i.e. the features commonly used to evaluate Mn valence (see Farges, Reference Farges2005 and Manceau et al., Reference Manceau, Marcus and Grangeon2012 for a detailed discussion). It is, therefore, safer to limit the discussion to a purely qualitative assessment. The position of the main absorption edge of piccoliite (main inflection point at 6548.4 eV), although lying close to that of bixbyite-(Mn) (6547.4 eV), is shifted towards higher energy values, clearly indicating the presence of a moderate quantity of Mn4+. As a comparison, the edge position of hollandite (6551.4 eV, data from Manceau et al., Reference Manceau, Marcus and Grangeon2012), where the average Mn valence is close to 4+, lies much closer to that of pyrolusite (6552.4 eV).

Fig. 4. Mn K-edge XANES of piccoliite from Valletta mine, together with rhodochrosite, bixbyite-(Mn), pyrolusite and hollandite (hollandite spectrum from Manceau et al., Reference Manceau, Marcus and Grangeon2012).
The Mn K-edge EXAFS spectrum and Fourier transform of piccoliite are shown in Fig. 5a and b, respectively; results of the EXAFS multiparameter fit are reported in Table 10. Attempts to fit the first shell using an average Mn–O distance calculated from the starting model were unsuccessful, leading to a poor fit and an extremely high Debye–Waller factor (σ2) [0.02(1) Å–2]; only the use of six different Mn–O paths, as suggested by the crystal-structure data, gave a suitable fit for the data and resulted in a drop of σ2 of a factor 5, thus confirming that Mn is hosted in a very distorted site. The set of six distances is in excellent agreement with the crystal-structure model (Table 6), resulting in a value of <Mn–O> = 2.03 Å. The second shell signal is dominated by the contribution of the four closest Mn–As scattering paths. Resulting Mn–As distances are in good agreement with those obtained by the crystal-structure refinement. Neither the contribution of the Mn–Mn/Mg scattering paths nor that of the Mn–Ca/Na ones was observable during the fit procedure. The lack of these contributions is probably due to destructive interferences occurring between the signals corresponding to the aforementioned paths as suggested in the Supplementary figure 1. It is noticeable, however, that Mn–As paths are not affected by this interference, and show higher amplitude at higher k values.

Fig. 5. Mn K-edge EXAFS (a) and Fourier transform, uncorrected for phase-shift (b) of piccoliite from Valletta mine.
Table 10. Structural parameters from the Mn K-edge EXAFS analysis of piccoliite from Valletta mine.*

* S02 = Many-body amplitude reduction factor, R= interatomic distance, σ2= Debye-Waller factor. Errors as reported in ARTEMIS.
Structure description
The crystal structure of piccoliite (Fig. 6a) is characterised by MnO5(OH) octahedra forming edge-shared [Mφ10] dimers; these dimers are connected through corner-sharing, forming two-up-two-down [[6]M 2([4]TO4)4φ2] chains [M = Mn, T = As and φ = O(OH)] running along c (Fig. 6b, c). These chains are bonded in the a and b directions by sharing corners with AsO4 tetrahedra, giving rise to a framework of tetrahedra and octahedra hosting Ca2+ and Na+ cations.

Fig. 6. Crystal structure of piccoliite, as seen down c (a). In (b) and (c), the chains of octahedra, decorated on both sides by (AsO4) tetrahedra and running along c, are shown along b and a, respectively. Symbols: light pink polyhedra = As-centred tetrahedra; purple polyhedra = Mn-centred polyhedra; light blue ellipsoids = A1 site; yellow ellipsoids = A2 site; red ellipsoids = O1–O6 sites; blue ellipsoid: O7 site; white sphere: H7 site; dashed lines represent hydrogen bonds. Figure obtained with Vesta 3 (Momma and Izumi, Reference Momma and Izumi2011).
Arsenic is coordinated tetrahedrally by oxygen atoms, with <As–O> = 1.690 and 1.683 Å for the As1 and As2 sites, respectively (Montaldo di Mondovì data). The observed <T–O> value is in good agreement with the grand mean value reported for arsenates in the literature [1.687(27) Å, Gagné and Hawthorne, Reference Gagné and Hawthorne2018]. The bond-valence sum (BVS) values at As1 and As2 are 4.96 and 5.06 valence units (vu), respectively. Trivalent Mn at the Mn site, partially replaced by Fe3+ and minor Mg2+, and possibly Ca2+, is hosted within a Jahn-Teller distorted octahedra, with bond distances ranging from 1.804(1) to 2.172(1) Å, and a <Mn–O> distance of 2.023 Å. Calculated BVS at the Mn site is 3.03 vu (Table 8), slightly larger than the expected atomic valence from site population (2.82 vu; Table 7). Finally, Ca and Na are hosted in the cavities of the octahedra and tetrahedra framework. Calcium is partitioned preferentially at the seven-fold coordinated A2 site, with average bond distance of 2.479 Å, whereas Na occurs at the seven-fold coordinated A1 site, having a slightly larger average bond distance (2.501 Å). The BVS at the A1 and A2 sites are 1.33 and 1.71 vu, respectively, in agreement with mixed (Na,Ca) and (Ca,Na) site populations, respectively (Table 8). The hydrogen atom is bonded to the oxygen hosted at the O7 site, forming a O7–H⋅⋅⋅O1 hydrogen bond. Its bond strength, calculated from the relation of Ferraris and Ivaldi (Reference Ferraris and Ivaldi1988), is 0.25 vu.
The blackish colouration of the mineral suggests the presence of both Mn3+ and Mn4+ and XANES data indicate the presence of Mn4+ in piccoliite from the Valletta mine. Both mineralisations have a strongly oxidised character, with Fe always occurring as Fe3+ (e.g. hematite) and Mn as Mn3+ and Mn4+ (e.g. members of the hollandite supergroup). In agreement with this, the calculated bond valence incident at the Mn site for the Valletta specimen is larger than the expected from a formula without Mn4+ (2.89 vu vs. 2.73 vu, respectively), thus supporting a partial amount of more highly charged cations ordered at that site. The presence of Mn4+ is needed to charge balance the presence of Mg2+ at the M sites. It could be also balanced either by dehydroxylation at the O7 anion site or by an increase of Na (> 1 apfu) at the A1,2 sites. If we assume that part of the Mn is present as Mn4+ (likewise for the Montaldo di Mondovì sample), and we calculate 41% of Mn4+ in the bulk Mn, the average charge becomes +3.025, closer to the corresponding BVS calculated at the M sites (3.035 vu). Moreover, the calculated bond length would be 2.015 Å, closer to the observed value (2.023 Å) than the value calculated if all Mn is considered Mn3+ (2.041 Å). Besides, if all the Mn is considered as Mn3+ there is an O site with a relatively high BVS of ⁓2.33 vu (see Table 8). The assumed presence of 41% of Mn4+ would reduce the BVS to 2.24 vu, still high and thus showing a weakness for this topology. This remanent high BVS is due to the presence of a very short distance (M–O6 = 1.804 Å) compared to the average equatorial bond distance of the MO6 octahedron (2.093 Å). This distance is even shorter than the one observed by Gagné and Hawthorne (Reference Gagné and Hawthorne2020) for both six-fold coordinated Mn3+ and Mn4+, 1.843 and 1.841 Å, respectively. The BVS values reported in Table 8 for the crystal from Valletta when considering 35% of Mn4+ reduces the BVS at O6 to 2.145 vu, and yields a calculated averaged bond length of 2.024 Å, very close to the one observed (2.029 Å); moreover, the observed BVS at the M site (2.897 vu) is almost coincident with the average charge (+2.900). In any case, both XANES data and crystal-chemical considerations indicate that Mn in the Valletta specimen is mainly present at its trivalent state, thus Mn3+ is the dominant cation of the dominant charge (+3). The ideal-charge cation formula is thus A11+ A22+ M 3+(T 5+O4)O(OH).
The hydrogen-bonding topology is rather constrained and shows a O–H⋅⋅⋅O angle of 175(5)°. The donor–acceptor (O7⋅⋅⋅O1) distance is short (2.67 Å) but, using that donor–acceptor distance, the corresponding wavenumber value calculated using the relation of Libowitzky (Reference Libowitzky1999), 3066 cm–1, is in good agreement with the observed (OH)-stretching Raman shifts (3124 and 3252 cm–1). The presence of two bands could agree with two local arrangements with two Mn3+ or one Mg2+ and Mn4+ in agreement with the proposed presence of Mn4+ at the M sites, as the O7 site is shared by two edge-sharing MO6 octahedra. Further shifts can be attributed to the mixed valence at the A1 site, also coordinating the O7 anion site.
Relation to other species
The topology of the piccoliite structure is not new. The mineral pilawite-(Y), ideally CaYAl2(SiO4)2O(OH) (Pieczka et al., Reference Pieczka, Hawthorne, Cooper, Szełeg, Szuszkiewicz, Turniak, Nejbert and Ilnicki2015), has the same topology (see Fig. 7b), although its symmetry is monoclinic (P21/c), with a β angle of 90.61(4)°. Consequently, pilawite-(Y) has two symmetrically independent M sites and two anions split in two non-symmetrically equivalent positions (O1, O2 and O7, O8, corresponding, respectively, to O5 and O2 in the piccoliite structure).
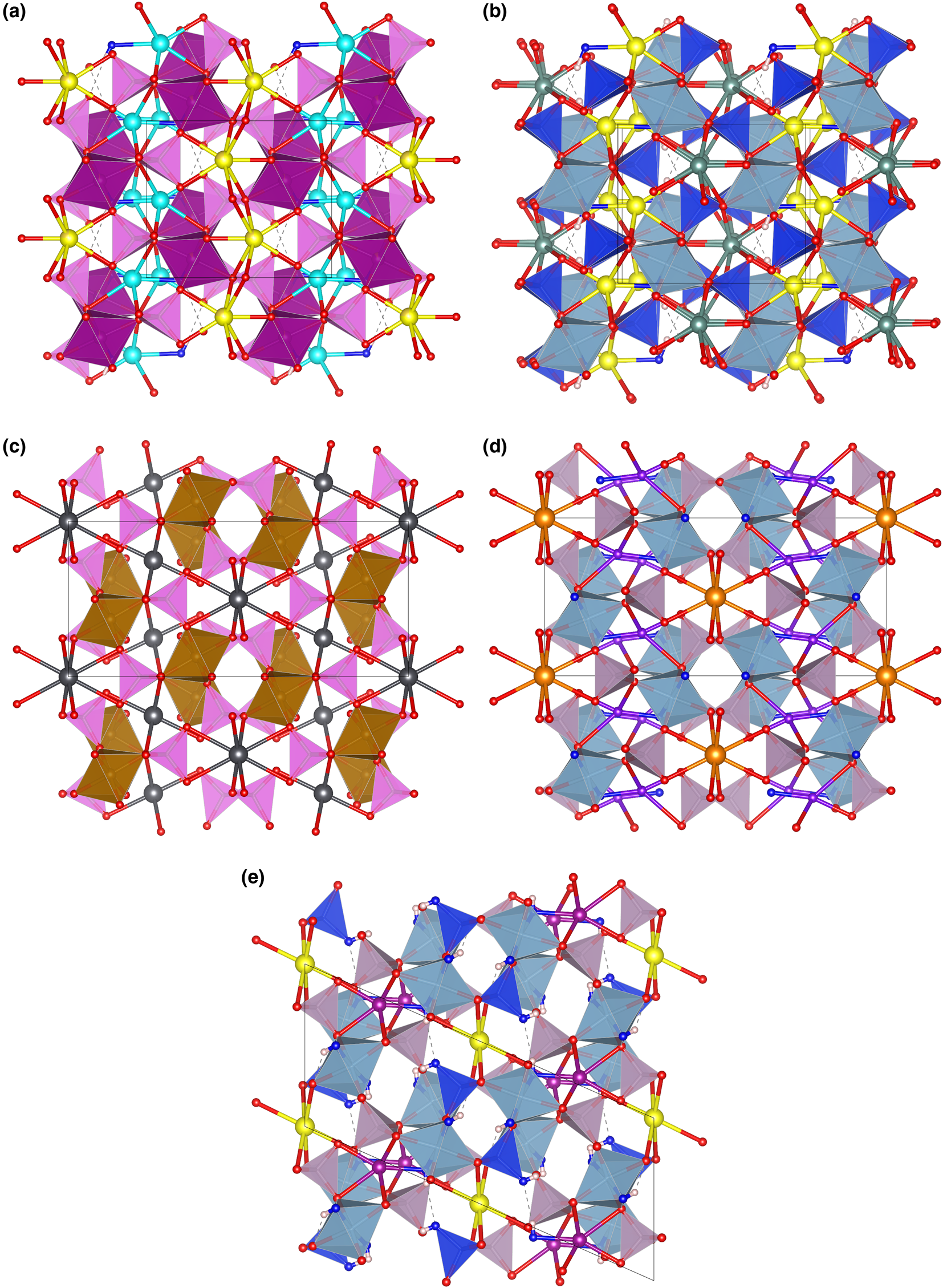
Fig. 7. The framework of octahedra and tetrahedra in piccoliite (a), pilawite-(Y) (b), carminite (c), palermoite (d) and attakolite (e), as seen along the chain direction. Seven-fold to eight-fold coordinated cations and their bonds are shown as ball-and-stick. Symbols: light pink polyhedra = As-centred tetrahedra; violet polyhedra = P-centred tetrahedra; blue polyhedra = Si-centred tetrahedra; purple polyhedra = Mn-centred polyhedra; cyan polyhedra = Al-centred octahedra; spheres: light blue = Na; yellow = Ca; red = O; purple = Mn; greyish blue = Y; dark grey = Pb; orange = Sr; violet = Li; blue = anion site hosting (OH) groups; white = H; dashed lines represent hydrogen bonds. Figure obtained with Vesta 3 (Momma and Izumi, Reference Momma and Izumi2011).
A particularity of the structure of piccoliite is the high U eq value observed in both crystals for the anion at the O1 site (Table 5), coordinating the As1O4 tetrahedron, which receives further bonding from the H bonded to O7. The observed value is three times the mean value observed for the other anion sites. The anisotropic displacement values also show a dynamic disordering of this anion site along [001] in the (010) plane. This is a particularly striking feature considering the stressed nature of the topology of the hydrogen bond and could be related to a particular instability of the structure that could relax at lower temperatures, lowering the symmetry to monoclinic by a shear in the (010) plane. Interestingly, the U eq value of the corresponding O6 site in the pilawite-(Y) structure is still 50% higher than the average anion-site value, but much lower, in agreement with a monoclinic relaxation of a possible high-T orthorhombic structure, as suggested by Pieczka et al. (Reference Pieczka, Hawthorne, Cooper, Szełeg, Szuszkiewicz, Turniak, Nejbert and Ilnicki2015). Likewise, no anion site has a BVS > 2.1 vu in pilawite-(Y) (see Table 9). Both facts, along with the possible charge partitioning of Mn at the M site, may explain the weak violations of the c-glide observed in piccoliite from the Valletta Mine.
Other minerals related to piccoliite are reported in Table 11. The structure of carminite, palermoite and attakolite are reported in Fig. 7c, d and e, respectively, for comparison. These structures are not topologically congruent, as the [[6]M 2([4]TO4)4φ2] chains cross-link in different ways in the a direction (Pieczka et al., Reference Pieczka, Hawthorne, Cooper, Szełeg, Szuszkiewicz, Turniak, Nejbert and Ilnicki2015), or in the b direction for palermoite (different cell setting; see Table 11). There is also a relative ½ shift of the chains along the chain direction between palermoite (and attakolite) and carminite. Palermoite and attakolite keep the same topology; attakolite relaxes to monoclinic due to the presence of HSiO4 groups that point to an empty site alternating with Mn along [001]. This allows for the ordering of Si in 1 out of 4 sites with tetrahedral coordination, whereas high-charge cations (P in this case) order in tetrahedra without (OH) groups. Though these structures can be considered as having different polytypic stacking of the [[6]M 2([4]TO4)4φ2] chains, the extra framework cavities are occupied by large cations (Sr, Pb, Y, Ca and Na) or smaller cations (Mn2+, Li and H) with different bonding topologies, leading to different stoichiometries (from 4 to 3 apfu) and may not be strictly considered polytypes.
Table 11. Mineral species related to piccoliite.

References: [1] this work; [2] Pieczka et al. (Reference Pieczka, Hawthorne, Cooper, Szełeg, Szuszkiewicz, Turniak, Nejbert and Ilnicki2015); [3] Kharisun et al. (Reference Kharisun, Bevan and Pring1996); [4] Kampf et al. (Reference Kampf, Adams, Mills and Nash2016); [5] Roberts et al. (Reference Roberts, Cooper, Hawthorne, Criddle and Stirling2002); [6] Blomstrand (Reference Blomstrand1868), redefined by Grice and Dunn (Reference Grice and Dunn1992); [7] Hatert et al. (Reference Hatert, Lefèvre and Fransolet2011); [8] Moore and Araki (Reference Moore and Araki1975); [9] Schumer et al. (Reference Schumer, Yang and Downs2017).
* s.g. = space-group type; # Mead and Mrose (Reference Mead and Mrose1968) described a monoclinic cell a = 11.45, b = 15.69, c = 7.30 Å, β = 91.5° which resembles a relaxed cell of palermoite that would correspond to a I2/m s.g.
Acknowledgements
Reviewers Anthony R. Kampf, Adam Pieczka and Pete Leverett are thanked for useful suggestions. F.C. acknowledges financial support by the grant Ricerca Locale 2020, Università di Milano, and from the Italian Ministry of Education (MUR) through the project “Dipartimenti di Eccellenza 2018–2022”. M.E.C. acknowledges financial support of AMI–Associazione Micromineralogica Italiana. Francesco d'Acapito and “LISA” CRG staff at ESRF (The European Synchrotron) are kindly acknowledged for the useful discussions and for the provision of in-house beamtime.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2022.129
Competing interests
The authors declare none.





















 Open access
Open access