


Introduction
Manganese oxides have long been spread ubiquitously on Earth, from the Mn deposits in Precambrian strata (Roy, Reference Roy, Nicholson, Hein, Bühn and Dasgupta1997), to the Mn nodules spread over the sea floor (Bauman, Reference Bauman1976; Jiang et al., Reference Jiang, Lin, Yao, Zhai and Guo2007) and the Mn-enriched rock or soil surface coatings (Bauman, Reference Bauman1976; Post, Reference Post1999; McKeown and Post, Reference McKeown and Post2001; Dorn, Reference Dorn, Nash and McLaren2007; Nash et al., Reference Nash, McLaren, Nash and McLaren2007; Xu et al., Reference Xu, Ding, Li, Lu, Li and Wang2018; Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a). Manganese oxides are diverse in both phase and structure and comprise mixed Mn(IV/III/II) valence states with layered, tunnelled or spinel-type structures (Post, Reference Post1999; Johnson et al., Reference Johnson, Webb, Ma and Fischer2016). In Nature, Mn oxides are widespread in coatings on the upper surface of rocks and soils (Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a), where the dominant phase in the coatings is birnessite – a layered mineral with Mn (or O) vacancy and adsorbed cations (Post, Reference Post1999; McKeown and Post, Reference McKeown and Post2001; Villalobos et al., Reference Villalobos, Toner, Bargar and Sposito2003; Webb et al., Reference Webb, Tebo and Bargat2005; Villalobos et al., Reference Villalobos, Lanson, Manceau, Toner and Sposito2006; Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a; Xu et al., Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019; Elmi, C. et al., Reference Elmi, Post, Heaney and Ilton2021). As a semiconducting mineral, birnessite shows outstanding photocatalytic reactivity (Sherman, Reference Sherman2005; Ling et al., Reference Ling, Post, Heaney, Kubicki and Santelli2017; Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a) and, as revealed by theoretical calculations, the electronic structure of birnessite is sensitive to Mn (or O) vacancy abundance as well as the metal sorption sites (Kwon et al., Reference Kwon, Refson and Sposito2008, Reference Kwon, Refson and Sposito2009; Li et al., Reference Li, Liu, Xu, Liu, Li, Ding, Chen, Yin, Lin, Wang and Lu2019a). These effects further impact the photocatalytic performance of birnessite in various photoreactions, mainly during birnessite self-reduction (Li et al., Reference Li, Liu, Xu, Liu, Li, Ding, Chen, Yin, Lin, Wang and Lu2019a) or reduction by organics (Sunda et al., Reference Sunda, Huntsman and Harvey1983; Waite et al., Reference Waite, Wrigley and Szymczak1988; Sunda and Huntsman, Reference Sunda and Huntsman1990) as well as water photocatalytic oxidation (Sherman, Reference Sherman2005; Marafatto et al., Reference Marafatto, Strader, Gonzalez-Holguera, Schwartzberg, Gilbert and Pena2015). Notably, the oxygen-evolving centre (OEC) in biological photosystem II (PSII) as Mn4CaO5 is structurally similar to birnessite (Hocking et al., Reference Hocking, Brimblecombe, Chang, Singh, Cheah, Glover, Casey and Spiccia2011; Robinson et al., Reference Robinson, Go, Mui, Gardner, Zhang, Mastrogiovanni, Garfunkel, Li, Greenblatt and Dismukes2013; Yang et al., Reference Yang, An, Zhou and Li2015), which draws the interesting parallel of the key functioning components in water oxidation and oxygen evolution between the organic and inorganic world. As a counterpart to Mn reduction, the oxidation of Mn(II) to form natural Mn oxides has been proposed through catalytic oxidation by fungi and bacteria (Northup et al., Reference Northup, Snider, Spilde, Porter, Van De Kamp, Boston, Nyberg and Bargar2010), or light-driven photo-oxidation by co-present semiconducting minerals (Madden and Hochella, Reference Madden and Hochella2005; Xu et al., Reference Xu, Ding, Li, Lu, Li and Wang2018; Xu et al., Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019). More recently, the photo-stimulated anaerobic formation of Mn oxides has been observed through the photo-oxidation of Mn(II) carbonate minerals (Liu et al., Reference Liu, Hao, Elzinga, Piotrowiak, Nanda, Yee and Falkowski2020; Lyons et al., Reference Lyons, Diamond and Konhauser2020) or biomineralisation mediated by anoxygenic photosynthetic microorganisms (Daye et al., Reference Daye, Klepac-Ceraj, Pajusalu, Rowland, Farrell-Sherman, Beukes, Tamura, Fournier and Bosak2019; Lyons et al., Reference Lyons, Diamond and Konhauser2020). These studies have implications on light-dependent Mn(II) oxidation, and Mn oxides emergence, prior to the Great Oxidation Event on Earth, especially in oceanic euphotic zones. During the oxidative formation of birnessite, sorption sites of heavy metals and the fine structure of birnessite will constantly interact with each other, which ultimately determines the biogeochemical cycle of Mn together with heavy metals.
This review first looks back on the mineral evolution processes of Mn oxides throughout geological history and the vast occurrence of Mn oxides in various modern natural settings. Notably, Mn coatings widespread on the Earth's surface, together with its remarkable performance of photon-to-electron conversion under solar irradiation are highlighted. Further, the effect of Mn (or O) vacancies and metal cation sorption on the electronic structure and photocatalytic activity of Mn oxides as revealed by density function theory are summarised. Manganese oxides participate actively in photocatalytic redox cycling on the Earth's surface, involving both the photo-oxidation of Mn(II) and the photoreductive dissolution of Mn oxides. During these processes, heavy metal sorption/desorption, organic oxidation, and even water splitting and oxygen evolution can occur. This study provides novel insights into the photogeochemical cycling of natural Mn oxides as well as its environmental implications.
Natural occurrence of Mn oxides
Coevolution of Mn valence states within the geosphere and biosphere
Mineral ecology of manganese
Manganese is concentrated mainly in the Earth's mantle (~1200 ppm; Turekian and Wedepohl, Reference Turekian and Wedepohl1961; Jackson and Dasgupta, Reference Jackson and Dasgupta2008), whereas the average content of Mn in the Earth's crust is 950 ppm (Taylor, Reference Taylor1964). The oxidation state of Mn-bearing minerals has increased in an episodic manner from II to IV since 2.5 Ga to the present (Hazen et al., Reference Hazen, Downs, Eleish, Fox, Gagné, Golden, Grew, Hummer, Hystad, Krivovichev, Li, Liu, Ma, Morrison, Pan, Pires, Prabhu, Ralph, Runyon and Zhong2019). The majority of Mn oxides with high-valence Mn appeared after the major oxidation events (Hazen et al., Reference Hazen, Downs, Eleish, Fox, Gagné, Golden, Grew, Hummer, Hystad, Krivovichev, Li, Liu, Ma, Morrison, Pan, Pires, Prabhu, Ralph, Runyon and Zhong2019; Fig. 1). The highest and lowest valence states of Mn oxides observed in Nature are IV and II, respectively (Owen and Bricker, 1965). Manganese deposits were produced mainly in the Precambrian such as Moanda, Gabon (Gauthier-Lafaye et al., Reference Gauthier-Lafaye, Holliger and Blanc1996) and Hunan Province, South China (Yu et al., Reference Yu, Xu, Zhao, Huang and Zou2020), while high-quality Mn oxide ores were enriched in the Ordovician, Carboniferous and Quaternary such as at Molango, Mexico (Martino, Reference Martino1986). Manganese ore is generally formed during deep sea sedimentation, where the main ore is manganese carbonates such as rhodochrosite (Roy, Reference Roy2006).

Fig. 1. Evolution of average oxidation state of Mn-bearing minerals on the Earth (from Hazen et al., Reference Hazen, Downs, Eleish, Fox, Gagné, Golden, Grew, Hummer, Hystad, Krivovichev, Li, Liu, Ma, Morrison, Pan, Pires, Prabhu, Ralph, Runyon and Zhong2019). GOE – Great Oxidation Event.
There are three kinds of common Mn oxides, which have different arrangements of Mn–O octahedra, i.e. tunnel structures (pyrolusite, ramsdellite, nsutite, hollandite, romanèchite, todorokite and manganite); layered structures (lithiophorite, chalcophanite, birnessite and vernadite); and spinel-like structures (hausmannite) (Table 1). Pyrolusite (β-MnO2) and ramsdellite usually occur in low-temperature hydrothermal deposits (Post, Reference Post1999). Nsutite (γ-MnO2) is the intergrowth of pyrolusite and ramsdellite structures (Zwicker et al., Reference Zwicker, Groeneveld Meijer and Jafee1962; Turner and Bruseck, Reference Turner and Buseck1983), which forms metasomatic veins in Mn carbonate rocks. These Mn minerals also transform into each other during geological evolution (Post, Reference Post1999).
Table 1. Classification of manganese (oxyhydr)oxides and structure types.
Recent studies have demonstrated that rocks with a dark Mn-rich varnish are generally found on the sunlit side of landforms, and Mn-poor rocks commonly occur on the shaded side (Nealson, Reference Nealson2015; Schindler and Dorn, Reference Schindler and Dorn2017; Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a). In addition, the growth rate of Mn-rich coatings was found to be much faster in high-UV flux or photic environments (Krinsley et al., Reference Krinsley, Dorn and DiGregorio2009; Koschinsky and Hein, Reference Koschinsky and Heine2017). This phenomenon implies a close relationship between the formation of supergene Mn oxides and solar light (Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a).
Manganese mineral evolution
According to stellar nucleosynthesis theory (the B2FH theory), Mn formed 400 million years after the Big Bang (Burbidge et al., Reference Burbidge, Burbidge, Fowler and Hoyle1957; Burbidge, Reference Burbidge2008). After the formation of the Earth, the sedimentary geochemistry of Mn has been controlled primarily by redox reaction, and is influenced by multiple Earth processes such as changing sea level and climate (Roy, Reference Roy2006). The high oxidation states Mn(III) and Mn(IV) exist in insoluble (oxyhydr)oxides in the oxidising environment and the low oxidation state (Mn(II)) is more easily dissolved in the anoxic waters (Johnson et al., Reference Johnson, Webb, Ma and Fischer2016). The early Archean atmosphere and hydrosphere system were extremely oxygen deficient (Lyons, Reference Lyons, Reinhard and Planavsky2014). The super mantle plume and high-temperature hydrothermal activities from mantle heat flux introduced a large amount of soluble Mn(II) into the anoxic ocean (Kasting, Reference Kasting, Eggler and Raeburn1993). From the Archean to the Proterozoic, the rapid burial of organic matter and the gradual reduction of reducing gases in the oxidised mantle resulted in the decrease of an oxygen sink and the increase of photosynthetic oxygen production (Roy et al., Reference Roy2006). Approximately 2.75 Ga, ‘whiffs’ of oxygen (Kaufman et al., Reference Kaufman, Johnston, Farquhar, Masterson, Lyons, Bates, Anbar, Arnold, Garvin and Buick2007; Frei et al., Reference Frei, Gaucher, Poulton and Canfield2009; Lyons et al., Reference Lyons, Reinhard and Planavsky2014), possibly contributed to the initial widespread formation of Mn oxides (Fig. 1; Hazen et al., Reference Hazen, Downs, Eleish, Fox, Gagné, Golden, Grew, Hummer, Hystad, Krivovichev, Li, Liu, Ma, Morrison, Pan, Pires, Prabhu, Ralph, Runyon and Zhong2019). The main Mn minerals in these Archean deposits are still kutnohorite (CaMn(CO3)2), rhodochrosite (MnCO3) and braunite (MnIII6MnIIO8SiO4) (Johnson et al., Reference Johnson, Webb, Ma and Fischer2016). In the period 2.4–2.06 Ga, the initial increase of O2 in the atmosphere did not bring about the complete oxidation of the global ocean, but instead only oxidised surface waters (Reinhard et al., Reference Reinhard, Raiswell, Scott, Anbar and Lyons2009; Kendall et al., Reference Kendall, Reinhard, Lyons, Kaufman, Poulton and Anbar2010). Manganese oxides in this environment could not be dominant; Mn would have a tendency to be reduced and precipitate carbonates due to the degradation of organics. After 2.06 Ga, many Mn-bearing deposits were found to have increased episodes, in which the source of dissolved Mn(II) was crucial (Roy, Reference Roy2006). Some tectonic activities and glacioeustatic sea-level changes would have enhanced the weathering of silicates and improve the concentration of dissolved Mn(II), giving rise to its precipitation as carbonates in Neoproterozoic, early Palaeozoic and early Jurassic (Beukes, Reference Beukes and Gutzmer1996; Böttcher and Huckried, Reference Böttcher and Huckriede1997; Roy, Reference Roy2006). Large Mn oxide deposits after the Proterozoic (e.g. Cretaceous age) were produced by transgression–regression in a greenhouse climate (Fan et al., Reference Fan, Liu and Ye1992).
The average oxidation state of Mn increased with the enhancement of atmospheric oxygen content, which can be divided into five stages (Holland, Reference Holland2006) (Table 2). In the first stage (4.60–2.45 Ga), the O2 content of the Archaean atmosphere was generally less than ~10–5 present atmospheric level (PAL) and the average oxidation state of Mn was 2–2.5 (e.g. rhodochrosite). In the second stage of the Columbia supercontinent period (2.45–1.85 Ga), the free oxygen content in the atmosphere rose sharply to between 0.02 and 0.04 atm due to the Great Oxidation Event, and the average oxidation state of Mn was 2.2–2.6 (Hazen et al., Reference Hazen, Downs, Eleish, Fox, Gagné, Golden, Grew, Hummer, Hystad, Krivovichev, Li, Liu, Ma, Morrison, Pan, Pires, Prabhu, Ralph, Runyon and Zhong2019). The main Mn minerals at that time, for example in the Koegas subgroup and Hotazel formation in South Africa, were braunite, kutnohorite, rhodochrosite, Mn-rich siderite and Mn-rich calcite (Cairncross and Beukes, Reference Cairncross and Beukes2013). In the third stage during the Rodinia supercontinent period (1.85–0.85 Ga), there was no significant change in atmospheric oxygen content and the average oxidation state of Mn was 2–2.8. Deposits such as Santa Cruz in the Jacadigo Basin, Brazil, were formed where the main Mn minerals are braunite, rhodochrosite, kutnohorite and caryopilite (Piacentini et al., Reference Piacentini, Vasconcelos and Farley2013). In the fourth stage during the Pannotia supercontinent period (0.85–0.54 Ga), the atmospheric oxygen content rose to a value of more than 0.2 atm, which resulted in three ice ages (snowball Earth events; Hoffman and Schrag, Reference Hoffman and Schrag2010) with the Mn average oxidation state of 2.3–2.6. In the fifth stage (0.54 Ga to present), the oxygen content in the atmosphere rose higher to the maximum value of 0.3 atm in the Carboniferous, and then returned to the present value 0.2 atm. During the Carboniferous, the average oxidation state of Mn was 2.5–3 and in deposits such as those in the Sea of Japan, the main Mn minerals are magnesite, siderite, Mn-rich calcite/kutnohorite and Ca-rich rhodochrosite (Matsumoto, Reference Matsumoto, Pisciotto, Ingle, von Breymann and Barron1992). Modern Mn deposits are mainly supergene or marine sedimentary deposits (Roy, Reference Roy2006), where the Mn minerals are mainly tetravalent (Maynard, Reference Maynard2010; Johnson et al., Reference Johnson, Webb, Ma and Fischer2016).
Table 2. Five stages of Mn average oxidation state changing with atmospheric oxygen content.

Mn4CaO5 and water oxidation reactions
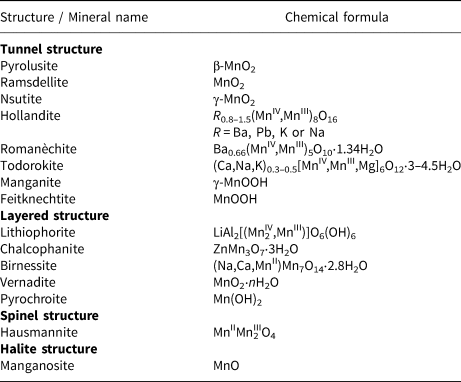
Water oxidation is one of the most important reactions in Nature (Hocking et al., Reference Hocking, Brimblecombe, Chang, Singh, Cheah, Glover, Casey and Spiccia2011). The origin of water oxidation can be traced back to the late Archean, in which cyanobacteria have served as the earliest important producers of O2 by photosynthesis and may produce trace amounts of oxygen, accumulating in some local environments (Lalonde et al., Reference Lalonde and Konhauser2015; Lyons et al., Reference Lyons, Reinhard and Planavsky2014). Interestingly, studies have shown that Mn oxide, a by-product of photosynthesis, already existed before the emergence of cyanobacteria (Johnson et al., Reference Johnson, Webb, Thomas, Ono, Kirschvink and Fischer2013). That is, the photocatalytic oxygen production by layered Mn oxides is possibly the embryonic form of photosynthesis, due to the similar fine structures of the oxygen-evolving centre (OEC) and Ca-birnessite (Fig. 2) (Russell, Reference Russell2002; Sauer and Yachandra, Reference Sauer and Yachandra2002). In PSII, the photocatalytic oxygen production reaction occurs when Mn clusters, Mn4CaO5, are converted into a birnessite-like structure (Hocking et al., Reference Hocking, Brimblecombe, Chang, Singh, Cheah, Glover, Casey and Spiccia2011). The Mn clusters have a cubic alkane structure, which is composed of highly distorted Mn4Ca-oxo and overhanging Mn4CaO5. During the process of photosynthesis (i.e. S-state cycle), the structure of Mn clusters and the oxidation state of Mn are changed (Fig. 3). Birnessite is a layered mineral with a repeated single-layer of MnO2. Each MnO2 layer is composed of [MnO6] octahedra with shared edges, and the interlayer between MnO2 layers contains alkaline cations and water molecules (Robinson et al., Reference Robinson, Go, Mui, Gardner, Zhang, Mastrogiovanni, Garfunkel, Li, Greenblatt and Dismukes2013). The photocatalytic decomposition of water by birnessite (Fig. 4) is similar to the S-state cycle of PSII described above, in which the processes both consist of the first oxidation of Mn(III), the formation of O–O bonds and the final reduction of Mn(IV) (Yang et al., Reference Yang, An, Zhou and Li2015; Table 3).

Fig. 2. Structure comparison of the active centres in (a) PSII and (b) Ca-birnessite. Reprinted with permission from Yang et al. (Reference Yang, An, Zhou and Li2015). Copyright 2015 American Chemical Society.
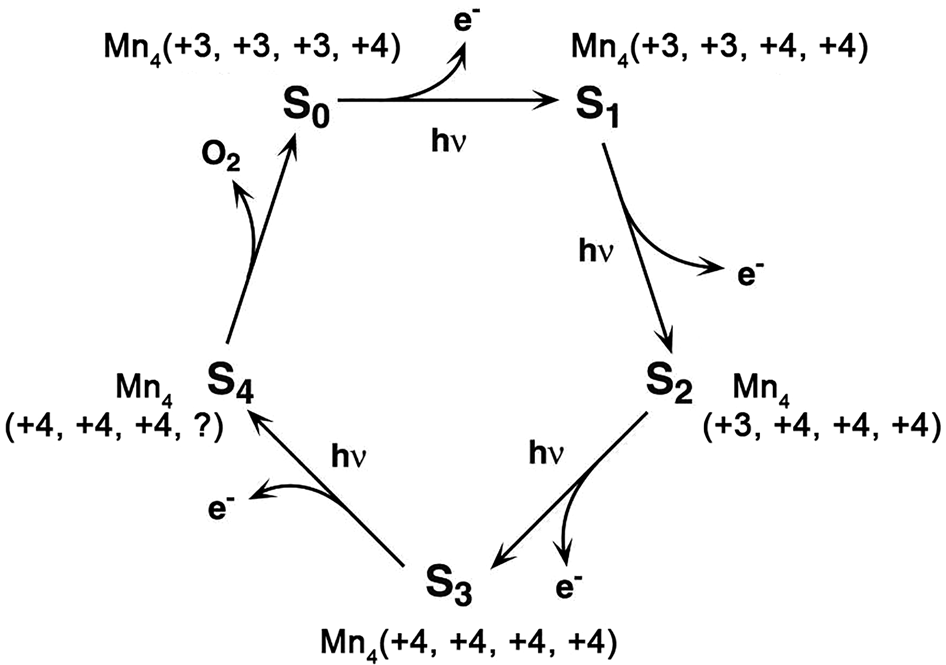
Fig. 3. The Kok cycle of S-state transitions in photosynthetic water oxidation (Reprinted with permission from Sauer and Yachandra, Reference Sauer and Yachandra2002, Copyright (2002) National Academy of Sciences).
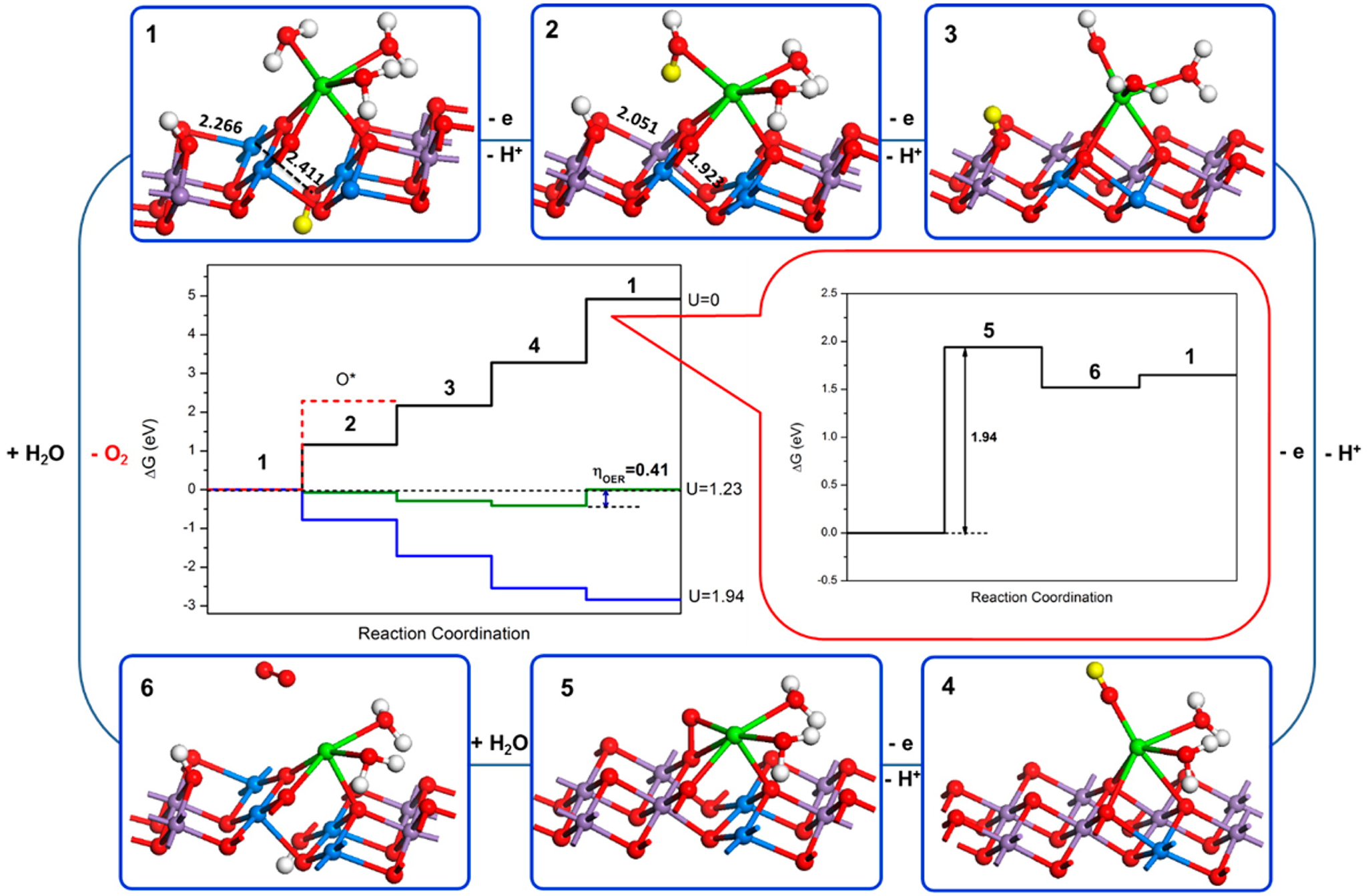
Fig. 4. Scheme of water oxidation on the surface of Ca-birnessite. The purple and blue atoms represent Mn (IV) and Mn (III), respectively. The yellow proton represents the proton to remove in the next step (Reprinted with permission from Yang et al. (Reference Yang, An, Zhou and Li2015). Copyright 2015 American Chemical Society).
Table 3. Comparison of photocatalytic decomposition of water by PSII oxygen production and birnessite.

There are two opinions on the evolution of the Mn4CaO5 cluster. The first is that the OEC evolved from Mn-bearing proteins, whereby the transfer of electrons by Mn-bearing catalase may be the intermediate step of aerobic photosynthesis (Blankenship and Hartman, Reference Blankenship and Hartman1998). However, there is no obvious similarity between the PSII core protein and Mn-bearing catalase, and there is no evidence that H2O2 was abundant in the Archean ocean. Olson (Reference Olson1970) proposed a hypothesis that Mn(III)–porphyrin cytochrome could replace Fe-bearing cytochromes in the evolution of OEC and pointed out a series of nitrogen-containing compounds such as NO and NO2– as transition electron donors of intermediate redox potential (Olson, Reference Olson1970). However, from today's perspective of OEC, this may be incorrect because Mn is not bound in cytochrome but exists in Mn4CaO5 clusters, and whether the transition state of nitrogen compounds existed at that time is still questionable (Sauer and Yachandra, Reference Sauer and Yachandra2002).
The other viewpoint is that OEC is derived from the integration of Mn oxides, because OEC is similar to some layered Mn oxides (e.g. birnessite) in the short-range structure (Russell, Reference Russell2002; Sauer and Yachandra, Reference Sauer and Yachandra2002; Russell et al., Reference Russell, Allen, James, Allen, Gantt, Golbeck and Osmond2008). However, there is no integration process of Mn oxides in the formation of OEC (Tamura and Cheniae, Reference Tamura and Cheniae1987; Johnson et al., Reference Johnson, Webb, Thomas, Ono, Kirschvink and Fischer2013). If that occurred, the OEC-like function of natural Mn oxides would have made a pioneering contribution to atmospheric oxygen before the Great Oxidation Event of primitive Earth. Moreover, such a model for abiotic oxygen production has been probably working on Mars, where Mn oxides have always been abundant and exposed to the Sun (as described for the Earth above) (e.g. Lanza et al., Reference Lanza, Wiens, Arvidson, Clark, Fischer, Gellert, Grotzinger, Hurowitz, McLennan, Morris, Rice, Bell, Berger, Blaney, Bridges, Calef, Campbell, Clegg, Cousin, Edgett, Fabre, Fisk, Forni, Frydenvang, Hardy, Hardgrove, Johnson, Lasue, Le Mouélic, Malin, Mangold, Martìn-Torres, Maurice, McBride, Ming, Newsom, Ollila, Sautter, Schröder, Thompson, Treiman, VanBommel, Vaniman and Zorzano2016).
Vast distribution of Mn oxides on modern Earth
Manganese oxides are widespread in modern natural settings especially in both oceanic and terrestrial environments (Post, Reference Post1999; McKeown and Post, Reference McKeown and Post2001; Dorn, Reference Dorn, Nash and McLaren2007; Nash et al., Reference Nash, McLaren, Nash and McLaren2007; Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a). They are the predominant components of Mn nodules that develop across the ocean floor and on the bottom of fresh-water lakes (Bauman, Reference Bauman1976; Jiang et al., Reference Jiang, Lin, Yao, Zhai and Guo2007). These Mn oxides are generally birnessite, todorokite and vernadite (Burns and Burns, Reference Burns, Burns and Glasby1977; Manceau et al., Reference Manceau, Lanson and Takahashi2014). The formation of Mn-enriched nodules is thought to arise from upward diffusion of Mn through the underlying reducing sediments and further accumulated by the catalytic oxidation of Mn(II) adsorbed on preliminarily formed fine-grained MnO2 (Lei and Bostrom, Reference Lei and Bostrom1995). In addition, some Mn-oxidising bacteria (such as Pseudomonas putida and Bacillus species) have been found in marine nodules, which are considered responsible for the biogenic origin of Mn oxides (Crerar and Barnes, Reference Crerar and Barnes1974; Tebo et al., Reference Tebo, Bargar, Clement, Dick, Murray, Parker, Verity and Webb2004; Webb et al., Reference Webb, Tebo and Bargat2005; Villalobos et al., Reference Villalobos, Lanson, Manceau, Toner and Sposito2006).
In terrestrial settings, Mn is the second most abundant heavy metal element in the Earth's crust, of which Mn(IV/III) are the most prevalent forms (Tebo et al., Reference Tebo, Bargar, Clement, Dick, Murray, Parker, Verity and Webb2004). In terrestrial weathering environments especially, the most representative Mn deposits are rock varnish, which attaches typically to the uppermost surface of rocks from arid to semiarid regions such as in the Gobi Desert (McKeown and Post, Reference McKeown and Post2001; Nash et al., Reference Nash, McLaren, Nash and McLaren2007; Xu et al., Reference Xu, Ding, Li, Lu, Li and Wang2018). The Mn enrichment of rock varnish has been reported to be ~50–200 times of that in upper crust (Thiagarajan and Lee, Reference Thiagarajan and Lee2004; Dorn, Reference Dorn, Nash and McLaren2007; Goldsmith et al., Reference Goldsmith, Stein and Enzel2014; Xu et al., Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019), and the dominant Mn oxide phases are birnessite, todorokite and romanèchite (McKeown and Post, Reference McKeown and Post2001; Garvie et al., Reference Garvie, Burt and Buseck2008; Xu et al., Reference Xu, Ding, Li, Lu, Li and Wang2018; Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a). Several formation mechanisms for rock varnish have been proposed. Thiagarajan and Lee (Reference Thiagarajan and Lee2004) and Goldsmith et al. (Reference Goldsmith, Stein and Enzel2014) proposed direct aqueous atmospheric deposition as one method, whereas Northup et al. (Reference Northup, Snider, Spilde, Porter, Van De Kamp, Boston, Nyberg and Bargar2010) suggested that biogenic Mn oxidation by fungi and bacteria will also induce Mn nucleation and precipitation. Recently, Xu et al. (Reference Xu, Ding, Li, Lu, Li and Wang2018, Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019) put forward a new theory that light-enhanced photocatalytic oxidation of Mn by semiconducting minerals such as Fe oxides co-present in rock varnish is responsible for its formation.
Widespread Mn coatings on the Earth's surface
The Mn (oxyhydr)oxide mineral coatings are found to overlay vast expanses of natural rock/soil surfaces in various natural environments (Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a). These Mn coatings are usually tens to hundreds of micrometres in thickness. In arid and semiarid regions, they are typically dark brown in colour and well separated from the substrate rocks by a distinct interface. Manganese coatings in relatively humid areas, such as karst regions with limestone deposits, are dark-grey in colour and porous in structure. The soil coatings (i.e. soil cutans) are generally red to dark brown, wrapping around quartz, feldspar particles and clay mineral aggregates. Lu et al. (Reference Lu, Li, Ding and Wang2019b) and Xu et al. (Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019) discovered that the Mn coatings were generally developed on the upper rock surfaces that are exposed to abundant solar irradiation. The geographical distribution of Mn coatings exactly coincides with intense solar irradiation regions around the Earth (Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a). All these lines of evidence suggest that sunlight has the greatest effect on the formation of Mn coatings on the Earth's surface. In laboratory studies, the photo-generated reactive oxygen species (such as 1O2 and OH⋅) in rock varnish have been detected, providing direct evidence for the model proposed by Xu et al. (Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019).
Birnessite is the primary Mn oxide found in Mn coatings, whereas tunnelled Mn oxides (such as todorokite, romanèchite and hollandite) possibly occur in desert Mn coatings or soil cutans, and Mn(II)-carbonate has been found in karst Mn coatings (McKeown and Post, Reference McKeown and Post2001; Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a; Xu et al., Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019). Moreover, the fine structural features of Mn coatings can vary in different environmental settings. According to our recent investigations by X-ray absorption spectroscopy, the average Mn oxidation state was as low as 3.21 in soil cutans, whereas it could reach up to 3.62 in desert Mn coatings (unpublished work). In karst Mn coatings, nanocrystalline vernadite was most common (Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a), which possessed random layer stacking and abundant vacant sites in its structure (Post, Reference Post1999; Villalobos et al., Reference Villalobos, Toner, Bargar and Sposito2003; Villalobos et al., Reference Villalobos, Lanson, Manceau, Toner and Sposito2006; Sherman and Peacock, Reference Sherman and Peacock2010). Among semiconducting minerals, birnessite in Mn coatings has been revealed to exhibit highly responsive and stable photon-to-electron conversion and is expected to actively participate in the inorganic and organic redox chemistry on the Earth's surface (Sherman, Reference Sherman2005; Ling et al., Reference Ling, Post, Heaney, Kubicki and Santelli2017; Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a).
Electronic structure of Mn oxides
Theoretical calculations have been applied widely to analyse the electronic structure of birnessite (Kwon et al., Reference Kwon, Refson and Sposito2009; Lucht and Mendoza-Cortes, Reference Lucht and Mendoza-Cortes2015; Peng et al., Reference Peng, McKendry, Ding, Thenuwara, Kang, Shumlas, Strongin, Zdilla and Perdew2017). According to these results, the valence band and conduction band of birnessite consist mainly of O(2p) and Mn(3d) orbitals, respectively, whose electronic structure can be interpreted by crystal field theory (Fig. 5). Due to the repulsion between O ions and the 3d orbitals of Mn in the [MnO6] octahedron, Mn 3d orbitals would be split into t2g (e.g. dxy, dyz and dzx) symmetry and eg (e.g.  $d_{x^2-y^2}$ and
$d_{x^2-y^2}$ and  $d_{z^2}$) symmetry (Sherman, Reference Sherman2005; Kwon et al., Reference Kwon, Refson and Sposito2009). Considering that 3d electron configuration of Mn(IV) ions is
$d_{z^2}$) symmetry (Sherman, Reference Sherman2005; Kwon et al., Reference Kwon, Refson and Sposito2009). Considering that 3d electron configuration of Mn(IV) ions is  $t_{2g}^3 e_g^0 $, the valence band maximum (VBM) of birnessite is occupied by the t2g triplet and its conduction band minimum (CBM) is dominated by the eg doublet (Xu and Schoonen, Reference Xu and Schoonen2000). Mn (or O) vacancies and introduced transition metal cations change the band structure of birnessite and are discussed in detail below.
$t_{2g}^3 e_g^0 $, the valence band maximum (VBM) of birnessite is occupied by the t2g triplet and its conduction band minimum (CBM) is dominated by the eg doublet (Xu and Schoonen, Reference Xu and Schoonen2000). Mn (or O) vacancies and introduced transition metal cations change the band structure of birnessite and are discussed in detail below.

Fig. 5. The density of states (DOS) of (a) non-vacancy birnessite with its O 2p and Mn 3d contribution to the total DOS. Total DOS of birnessite with vacancies (filled areas) in a (b) 4 × 4 × 1 supercell and (c) 2 × 2 × 1 supercell, with the corresponding total DOS of non-vacancy birnessite (from Kwon et al., Reference Kwon, Refson and Sposito2009).
Effect of Mn (or O) vacancies
Manganese (or O) vacancies in birnessite can affect the electronic structure and reduce the bandgap (Kwon et al., Reference Kwon, Refson and Sposito2008, Reference Kwon, Refson and Sposito2009). For Mn vacancies, the surrounding O around the Mn vacancy usually associates with four H atoms to balance the charge. By calculating the energy, the structure is stable when four extra H atoms lie out of the ab plane in proximity to each other (Ruetschi, Reference Ruetschi1984; Kwon et al., Reference Kwon, Refson and Sposito2009). Attributed to the Mn 3d and O 2p orbitals around the Mn-vacancy site, the bandgap decreases due to some newly-introduced states emerging above its VBM. Further, when increasing the Mn vacancies from 3.3% to 12.5%, the bandgap of birnessite shows a reducing trend (Fig. 5). The bandgap of non-vacancy birnessite is indirect, whereas the bandgap of birnessite with Mn vacancy is direct at Γ (Fig. 6a,b). There is also a difference in the CBM and VBM partial charge of non-vacancy and Mn-vacancy birnessite (Fig. 6c,d). In non-vacancy birnessite, most of the electron and hole states are overlapping, whereas they are well-separated in birnessite with Mn vacancies (Kwon et al., Reference Kwon, Refson and Sposito2009). The electronic transitions from valence band to conduction band upon illumination would leave holes at VBM and excited electrons at CBM. The separation between the VBM and the CBM would promote photoinduced charge carries efficiently.

Fig. 6. MnO2 band structure of (a) non-vacancy and (b) Mn-vacancy views of charge distributions of valence band maximum (VBM) hole states [orange or light grey] and conduction band minimum (CBM) electron states [blue or grey] in (c) non-vacancy and (d) Mn-vacancy (from Kwon et al., Reference Kwon, Refson and Sposito2008).
Oxygen vacancies, another common defect in birnessite, has been proposed to have dual roles in the alteration of electronic structure. They can generate an extra electron in each [MnO6] octahedron and convert Mn(IV)  $\lpar t_{2g}^3 e_g^0 \rpar $ into unstable high-spin Mn(III)
$\lpar t_{2g}^3 e_g^0 \rpar $ into unstable high-spin Mn(III)  $\;\lpar t_{2g}^3 e_g^1 \rpar $. To maintain a low-energy state in the strong Jahn–Teller effect, the extra electron must occupy another split Mn(III) d-orbital, i.e.
$\;\lpar t_{2g}^3 e_g^1 \rpar $. To maintain a low-energy state in the strong Jahn–Teller effect, the extra electron must occupy another split Mn(III) d-orbital, i.e.  $e_g^1 \,\, \lpar d_{z^2}\rpar $ within the bandgap (Fig. 7) (Lucht and Mendoza-Cortes, Reference Lucht and Mendoza-Cortes2015; Peng et al., Reference Peng, McKendry, Ding, Thenuwara, Kang, Shumlas, Strongin, Zdilla and Perdew2017). Such a configuration can remarkably decrease the bandgap of O-defected birnessite compared with the non-vacancy one. In contrast, O vacancies also commonly exist in other metal oxides and work as active sites in reactions (Wang et al., Reference Wang, Li, Jiang, Zhou, Zhang and Yu2017; Cao et al., Reference Cao, Zhang, Liu and Zheng2019). In Nature, multiple surfaces may be exposed and therefore perform different levels of photoactivity. Theoretical calculations have compared the function of O vacancies exposed on the (001) and (100) crystal face of birnessite. The formation energy of O vacancies is 1.45 eV for the (001) face and 1.43 eV for the (100) face, indicating the latter is more available than the former. The density of states (DOS) in Fig. 8 also shows O vacancies reduce the bandgap and the (100) crystal face exhibits half-metallicity characteristics with near-zero bandgap. These results suggest that O vacancies can effectively change the surface state, which is consistent with the experimental studies (Yang et al., Reference Yang, Zhu, You, Yan, Ma, Lu, Gao, Hao and Li2018).
$e_g^1 \,\, \lpar d_{z^2}\rpar $ within the bandgap (Fig. 7) (Lucht and Mendoza-Cortes, Reference Lucht and Mendoza-Cortes2015; Peng et al., Reference Peng, McKendry, Ding, Thenuwara, Kang, Shumlas, Strongin, Zdilla and Perdew2017). Such a configuration can remarkably decrease the bandgap of O-defected birnessite compared with the non-vacancy one. In contrast, O vacancies also commonly exist in other metal oxides and work as active sites in reactions (Wang et al., Reference Wang, Li, Jiang, Zhou, Zhang and Yu2017; Cao et al., Reference Cao, Zhang, Liu and Zheng2019). In Nature, multiple surfaces may be exposed and therefore perform different levels of photoactivity. Theoretical calculations have compared the function of O vacancies exposed on the (001) and (100) crystal face of birnessite. The formation energy of O vacancies is 1.45 eV for the (001) face and 1.43 eV for the (100) face, indicating the latter is more available than the former. The density of states (DOS) in Fig. 8 also shows O vacancies reduce the bandgap and the (100) crystal face exhibits half-metallicity characteristics with near-zero bandgap. These results suggest that O vacancies can effectively change the surface state, which is consistent with the experimental studies (Yang et al., Reference Yang, Zhu, You, Yan, Ma, Lu, Gao, Hao and Li2018).

Fig. 7. For Mn3+ (right), the Jahn–Teller effect causes elongation of the Mn–O bond compared to Mn4+ (left). Because of the loss of degeneracy, the orbitals for the Mn3+ are offset to lower the energy of the partially filled orbitals. Reprinted with permission from Lucht and Mendoza-Cortes (Reference Lucht and Mendoza-Cortes2015). Copyright 2015 American Chemical Society.

Fig. 8. (a,b) Top view of pure birnessite-type MnO2 and corresponding DOS; (c,d) O vacancy configurations and corresponding DOS of (001) MnO2; and (e,f) (100) MnO2 (from Yang et al., Reference Yang, Zhu, You, Yan, Ma, Lu, Gao, Hao and Li2018).
Effect of metal cations
Cation exchange capacity is a significant property of layered minerals, which adsorb cations reversibly in order to compensate for the negative charge of layers (Mukhopadhyay, Reference Mukhopadhyay2011). Transition metal cations are commonly observed in natural birnessite because of their similar crystal field environments with Mn ions. Generally, cations coordinate in two kinds of sites. One is cations incorporating into the birnessite sheet (INC), resulting in surrounding ions distorting and the second is cations forming triple-corner-sharing (TCS) in the interlayer of birnessite, resulting in [MnO6] layers with, either increased interlayer spacing, or expanded average bonding distances between Mn and O atoms. The electronic structure of birnessite will be changed in both cases. When Fe ions occupy a vacanct Mn site, the bandgap of birnessite reduces because of Fe 3d orbits mixing with Mn 3d and O 2p in VBM and CBM (Lucht and Mendoza-Cortes, Reference Lucht and Mendoza-Cortes2015; Liu et al., Reference Liu, Gu, Luo, Fan, Liao, Tian, Niu, Fu, Wang, Wu, Lv and Mei2018a). For Cu cations, the influence of INC and TCS sites on the electronic structure has been discussed by Li et al. (Reference Li, Liu, Xu, Liu, Li, Ding, Chen, Yin, Lin, Wang and Lu2019a). The bandgap will shrink in both situations. Specifically, the bandgap of Cu ions in hexagonal birnessite reduces more in INC configuration compared with Cu ions in TCS, whereas for triclinic birnessite, the influence of Cu ions on the bandgap is greater in TCS than in INC.
Moreover, the electronic structure of transition metal cations will affect the coordination trend (i.e. in the TCS and INC model). For example, through simulating the INC and TCS model of Co, Ni, Cu and Zn cations in birnessite, the energy difference between forming INC and TCS species is −214, −23, +4 and +34 kJ/mol, respectively. For INC models, with the increasing content of cations, it exerts greater stress on the surrounding ions by Jahn–Teller distortion through the interaction between the 3d orbit of metal cations and ligands O. Qualitatively, with increasing electronic number of Co, Ni, Cu and Zn, the unoccupied 3d orbital reduces and thus decreases the Jahn–Teller distortion. In Zn ions, strong interaction would not happen due to fully occupied 3d orbits, and the INC site for Zn is unstable in birnessite (Kwon et al., Reference Kwon, Refson and Sposito2013). The DOS of Zn in the INC site also shows the weak interaction because the 3d electronic is located mainly in a deep valence band without spin-exchange.
Photo-reactivity of natural semiconducting Mn oxides
Photocatalytic self-reduction of natural Mn oxides
In aquatic environments, the vertical profile of Mn in a multitude of oceanic and lacustrine water columns shows a gradual decrease in the concentration of aqueous Mn with depth (Klinkhammer and Bender, Reference Klinkhammer and Bender1980; Landing and Bruland, Reference Landing and Bruland1987; Sunda and Huntsman, Reference Sunda and Huntsman1990; Davison, Reference Davison1993; Statham et al., Reference Statham, Yeats and Landing1998). Theoretically, aqueous Mn should be oxidised to Mn(III/IV) oxides by superoxide radicals generated by sunlight irradiation or organisms in the photic zone, resulting in lower concentrations. However, contrary to these thermodynamic estimates, aqueous Mn predominates in the photic zone (Sunda et al., Reference Sunda, Huntsman and Harvey1983; Tebo et al., Reference Tebo, Bargar, Clement, Dick, Murray, Parker, Verity and Webb2004; Morgan, Reference Morgan2005; Zhang et al., Reference Zhang, Liu, Tan, Suib, Qiu and Liu2018). This phenomenon is related to the photocatalytic self-reduction of natural Mn oxides, which can be caused either by a ligand-to-metal charge transfer process involving organics, or by the reduction of photoelectrons (Sunda et al., Reference Sunda, Huntsman and Harvey1983; Sunda and Huntsman, Reference Sunda and Huntsman1990, Reference Sunda and Huntsman1994). Narsito et al. (Reference Narsito, Santosa and Lastuti2008) showed that the charge transfer process involves three steps: (1) humic acid is adsorbed on the surface of MnO2 to form an intermediate complex; (2) one delocalised π electron is excited and transferred; (3) one electron transfers from an oxygen centre to the manganese centre of an intermediate complex, causing the reductive dissociation of Mn(IV) → Mn(II) (Fig. 9a).

Fig. 9. (a) (Upper) Excitation of the humic acids aromatic system in the intermediate HA-MnO2 complex through π–π* transition. (Lower) Reductive dissociation of exited intermediate of the HA-MnO2 complex, producing water soluble Mn(II) of MnO22– (from Narsito et al., Reference Narsito, Santosa and Lastuti2008). (b) Proposed model for the evolution of metal redox chemistry during δ-MnO2 photoreduction (from Marafatto et al., Reference Marafatto, Strader, Gonzalez-Holguera, Schwartzberg, Gilbert and Pena2015).
Manganese oxides have a suitable band gap of <3.1 eV and can absorb and convert photons to produce photoelectron-hole pairs under the excitation of visible light (Sherman, Reference Sherman2005). The photoelectrons/holes can change the valence states and existing forms of elements. It means the sunlight irradiation can induce photoelectrons in the Mn 3d orbitals in conduction bands and holes in O 2p orbitals in the valence band. If the electrochemical potential of the valence band is above one half-reaction then the hole in the valence band could have oxidation potential (Sherman, Reference Sherman2005; Pinaud et al., Reference Pinaud, Chen, Abram and Jaramillo2011). If the conduction-band potential is more negative than one half-reaction then the electron excited into the conduction band would cause reduction. Sherman (Reference Sherman2005) calculates that the conduction band for pyrolusite and birnessite are lower than the redox potential 1.23 V (SHE, Standard Hydrogen Electrode) of MnO2/Mn(II); that is, they could theoretically adsorb sunlight to generate photoelectrons for self-photoreduction. Hence, a photoelectron could be excited to the conduction band to reduce Mn(IV) to Mn(III) under sunlight irradiation (Luther, Reference Luther2005; Sakai et al., Reference Sakai, Ebina, Takada and Sasaki2005)
The valence band is occupied mainly by O 2p and eg of Mn 3d orbits. One electron in the O 2p orbital can transfer to the empty eg orbital of Mn(IV) under sunlight irradiation to form intermediate Mn(III) (Luther, Reference Luther2005; Sakai et al., Reference Sakai, Ebina, Takada and Sasaki2005). Mn(III) is in a tetragonally distorted rather than octahedral geometry, resulting in higher kinetic lability (ligand exchange) and reactivity (Marafatto et al., Reference Marafatto, Strader, Gonzalez-Holguera, Schwartzberg, Gilbert and Pena2015; Chan et al., Reference Chan, Kitchaev, Weker, Schnedermann, Lim, Ceder, Tumas, Toney and Nocera2018). The eg of Mn(III) therefore are prone to accept the photo-electron and thus the Mn(III) is further reduced to Mn(II) (Luther, Reference Luther2005). Hence, the photocatalytic self-reduction of Mn oxides is a two-step one-electron transfer process. The conduction band states are localised Mn 3d orbitals that contribute to electron transfer, consequently, promoting the reduction of Mn(IV) to Mn(III). Marafatto et al. (Reference Marafatto, Strader, Gonzalez-Holguera, Schwartzberg, Gilbert and Pena2015) has proved the two-step one-electron transfer process by light-initiated time-resolved X-ray absorption spectroscopy (LITR–XAS). This involves the layered Mn(IV) oxide obtaining a photo-electron and being reduced to layered Mn(III) oxide; the Jahn–Teller distorted Mn(III) migrating into the interlayer; and then the interlayer staking increasing (Fig. 9b). In addition, the formation of valence band holes accompanying the photocatalytic self-reduction and the generation of Mn(III) can oxidise water or dissolved organic matter (Chan et al., Reference Chan, Kitchaev, Weker, Schnedermann, Lim, Ceder, Tumas, Toney and Nocera2018). In addition, in the Raman spectra of the oxygen evolution reaction, Mn(III)-induced local strain on the oxide sublattice is observed, which further proves the one-electron transfer mechanism (Chan et al., Reference Chan, Kitchaev, Weker, Schnedermann, Lim, Ceder, Tumas, Toney and Nocera2018).
Photocatalytic oxidation of water by Mn oxides
Although there are several Mn oxide minerals in natural environments, such as pyrolusite, todorokite, tithiophorite and hausmannite (Boppana and Jiao, Reference Boppana and Jiao2011; Meng et al., Reference Meng, Zhao, Chen, Wang, Li and Qiu2014; Huang et al., Reference Huang, Dai, Singewald, Liu, Saxena and Zhang2019), only the birnessite local structure bears a striking similarity to the OEC in PSII (Fig. 10a,b) (Hocking et al., Reference Hocking, Brimblecombe, Chang, Singh, Cheah, Glover, Casey and Spiccia2011; Robinson et al., Reference Robinson, Go, Mui, Gardner, Zhang, Mastrogiovanni, Garfunkel, Li, Greenblatt and Dismukes2013; Lucht and Mendoza-Cortes, Reference Lucht and Mendoza-Cortes2015; Thenuwara et al., Reference Thenuwara, Shumlas, Attanayake, Cerkez, McKendry, Frazer, Borguet, Kang, Zdilla, Sun and Strongin2015; Yang et al., Reference Yang, An, Zhou and Li2015). Consequently, the photo-oxidation activity of birnessite has drawn increasing attention, especially for water oxidation (Hocking et al., Reference Hocking, Brimblecombe, Chang, Singh, Cheah, Glover, Casey and Spiccia2011; Wiechen et al., Reference Wiechen, Zaharieva, Dau and Kurz2012; Thenuwara, et al., Reference Thenuwara, Shumlas, Attanayake, Cerkez, McKendry, Frazer, Borguet, Kang, Zdilla, Sun and Strongin2015; McKendryet al., Reference McKendry, Thenuwara, Shumlas, Peng, Aulin, Chinnam, Borguet, Strongin and Zdilla2018; Elmacı et al., Reference Elmacı, Özgenç, Kurz and Zumreoglu-Karan2020).

Fig. 10. (a) Structural comparison for Mn4CaO5 in natural vs. birnessite (Reprinted with permission from Yang et al. (Reference Yang, An, Zhou and Li2015). Copyright 2015 American Chemical Society). (b) Comparison of the extended X-ray absorption fine structure (EXAFS) spectra for several Mn oxides: i: [Mn4O4L6](ClO4); ii: [(bipy)2Mn(O)2Mn(bipy)2](ClO4)3; iii: Pyrolusite (1 × 1 tunnel); iv: Hollandite (2 × 2 tunnel); v: Todorokite (3 × 3 tunnel); vi: K-birnessite; vii: natural birnessite (disordered layer). (c) Comparison of X-ray absorption near edge structure (XANES) spectra during various states of catalytic cycling (from Hocking, et al., Reference Hocking, Brimblecombe, Chang, Singh, Cheah, Glover, Casey and Spiccia2011).
The birnessite structure type is responsible for the high photocatalytic activity in the water oxidation process (Fig. 10). Hocking et al. (Reference Hocking, Brimblecombe, Chang, Singh, Cheah, Glover, Casey and Spiccia2011) found the tetranuclear Mn cluster [Mn4O4L6] (L = diarylphosphinate), an efficient water-oxidation catalyst that converts to a birnessite-like structure during the water oxidation. This photoreactive process is similar to biological photosynthesis (Wiechen et al., Reference Wiechen, Zaharieva, Dau and Kurz2012) in thylakoids. Like Ca-cubane, birnessite also occurs in 5 states (S0, S1, S2, S3 and S4) during photocatalytic water oxidation (Fig. 10c) (Hocking et al., Reference Hocking, Brimblecombe, Chang, Singh, Cheah, Glover, Casey and Spiccia2011). The valence states of the Mn ions are S0 (+3, + 3, +3, +4), S1 (+3, +3, +4, +4), S2 (+3, +4, +4, +4), S3 (+4, +4, +4, +4) and S4 (+4, +4, +4, ?) (Fig. 3) (Sauer and Yachandra, Reference Sauer and Yachandra2002; Cox et al., Reference Cox, Pantazis, Neese and Lubitz2013; Zhang et al., Reference Zhang, Chen, Dong, Shen, Dau and Zhao2015). The photocatalytic water oxidation by birnessite indicates the oxygen evolution on Earth is closely related to Mn oxides, which may have long been present through geological time prior to the emergence of biologic photosynthesis and Great Oxidation Event (Fig. 11).
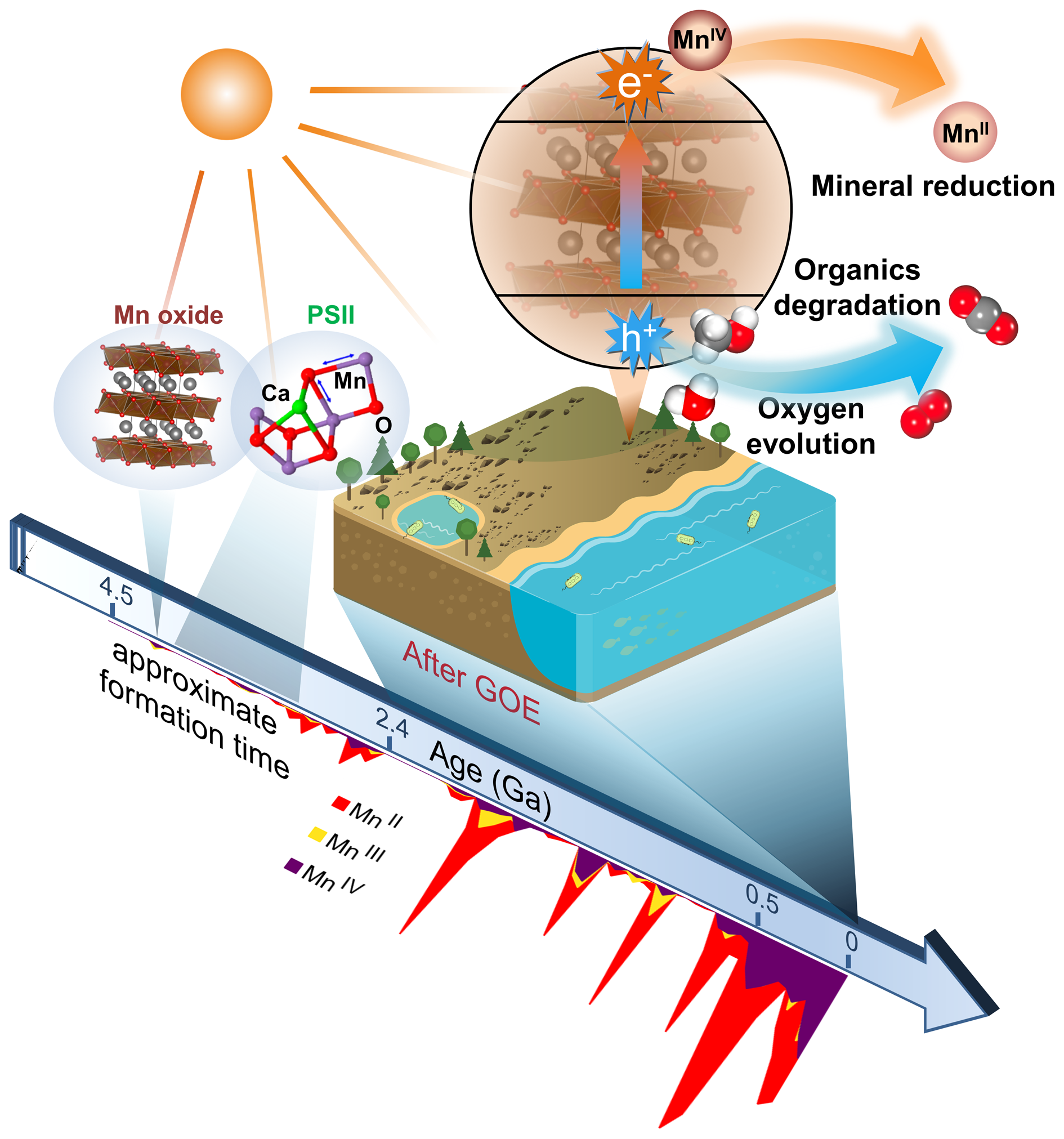
Fig. 11. Schematic model showing biogeochemical reactions dominated by photocatalytic Mn oxides on the sunlit Earth's surface in a long period of geological history. The evolution average oxidation state of Mn-bearing minerals (after Hazen et al., Reference Hazen, Downs, Eleish, Fox, Gagné, Golden, Grew, Hummer, Hystad, Krivovichev, Li, Liu, Ma, Morrison, Pan, Pires, Prabhu, Ralph, Runyon and Zhong2019) is shown for indicating the formation time of Mn oxides.
Interestingly, on the Earth's surface, rare earth elements (REEs) are slightly enriched in mineral coatings compared with the underlying bedrock (Thiagarajan and Lee, Reference Thiagarajan and Lee2004; Goldsmith et al., Reference Goldsmith, Stein and Enzel2014; Xu et al., Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019). Particularly, the enrichment of cerium (Ce) in mineral coatings can reach 12 times, and Ce is concentrated in Mn-rich regions (Kalintsev et al., Reference Kalintsev, Brugger, Etschmann and Ram2021). The occurrence of Ce is either in the form of interlayer cations in birnessite, or as Ce(OH)x nanoclusters adsorbed on corners of birnessite layers. The ratio of Ce to Mn can reach 1:20 (wt.%) in samples (Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a,Reference Lu, Li, Ding and Wangb). Similar Ce enrichment has been also reported in rock varnish of the arid desert over western America and the Middle East (Thiagarajan and Lee, Reference Thiagarajan and Lee2004; Goldsmith et al., Reference Goldsmith, Stein and Enzel2014). Interlayer cations can also affect the photoactivity of Mn oxides (e.g. Wiechen et al., Reference Wiechen, Zaharieva, Dau and Kurz2012; Lucht and Mendoza-Cortes, Reference Lucht and Mendoza-Cortes2015; Remsing et al., Reference Remsing, McKendry, Strongin, Kein and Zdilla2015; Rong et al., Reference Rong, Zhao, Chen, Xu, Zhao, Yang and Li2016). The different intercalated cations could tune the Jahn−Teller effect and adjust the hybridisation of electron orbits of birnessite, which in turn affects the electronic structure and band gap. By substituting the intercalated cations, it could tune the light capture ability and modify the intrinsic water-splitting catalytic property of birnessite. More specifically, Ce possesses a unique 4f electronic structure and properties of excellent oxygen adsorption and oxygen release, which are potentially advantageous for photocatalytic applications (Hernandez-Alonso et al., Reference Hernandez-Alonso, Hungria, Martinez-Arias, Fernandez-Garcia, Coronado, Conesa and Soria2004). The redox cycle of Ce(IV)–Ce(III) and oxygen vacancies in the structure enables it to have a strong oxygen storage capacity, which was beneficial to photocatalytic oxidation activity. For example, Ce1−xMnxO2 (x = 0, 0.1, 0.2, 0.3 and 1.0) samples have been used as photocatalysts for increasing the degradation efficiency of the textile diazo dye Naphthol Blue Black (Borker and Salker, Reference Borker and Salker2006). More importantly, Gobi et al. (Reference Gobi, Ramaraj and Kaneko1993) reported that Mn Schiff base complexes in the presence of Ce(IV) could oxidise water. Hence, the enrichment and occurrence of Ce in mineral coatings can improve the understanding of the environmental functions of semiconducting minerals on the Earth's surface under sunlight.
It is noteworthy that the average abundances of Mn in the core, mantle and crust have low values, whereas Mn is extraordinarily concentrated in ultrathin layers of mineral coatings (Lu et al., Reference Lu, Li, Ding, Xu, Li, Ren, Liang, Liu, Hong, Chen, Chu, Liu, Li, Wang, Ding, Wang, Lai, Liu, Dick, Liu and Hochella2019a). We conclude that the solar light response and photocurrent production of widespread semiconducting mineral coatings are capable of playing important roles in biogeochemical processes on the Earth's surface (Sunda et al., Reference Sunda, Huntsman and Harvey1983; Matsunaga et al., Reference Matsunaga, Ohyama, Kuma, Kudo and Suzuki1995; Sherman, Reference Sherman2005; Lu et al., Reference Lu, Li, Ding and Wang2019b), as shown schematically in Fig. 11. The photoelectrons from the conduction band of birnessite, similar to other semiconductors, can provide a driving force for the reduction of elements such as Mn, regenerate NADPH from NADP+ (Guo et al., Reference Guo, Suastegui, Sakimoto, Moody, Xiao, Nocera and Joshi2018), and provide a new form of energy for bacterial metabolisms (Lu et al., Reference Lu, Li, Jin, Wang, Wu, Zeng, Li and Dong2012; Sakimoto et al., Reference Sakimoto, Wong and Yang2016). The inexhaustible electron donors (e.g. water, organics and reduced ions) in Nature can complete those electron transport chains and bring about continuous redox chemistry, in which the semiconducting mineral coatings act as the vital bridge. Overall, the solar harvesting and photoelectric converting properties of these coatings broaden the pathways for utilising solar energy from the well-known organic world to the mineral semiconductor world (Lu et al., Reference Lu, Li, Ding and Wang2019b). As water, organic/inorganic matter and energy exchange on and penetrate through, these thin mineral coatings, it acts more like a functional ‘membrane’ covering the Earth's surface; therefore, we can describe it as a ‘mineral membrane’.
Environmental functions of Mn oxides controlled by Mn redox cycling
Reductive dissolution of Mn oxides mediated by organic matter
In soil and sediment, Mn oxides are capable of reacting with surrounding organic matter such as citrate, oxalate, humic and fulvic acids (Stone, Reference Stone1987; Wang and Stone, Reference Wang and Stone2006a,Reference Lu, Li, Ding and Wangb; Wang et al., Reference Wang, Yang and Zhu2018; Flynn and Catalano, Reference Flynn and Catalano2019). During redox reactions Mn(IV/III) will be reduced to low-valent Mn(III/II), some of which can detach from the structure and release off as soluble Mn(II) (Fig. 11). The mechanisms for the redox reactions between Mn oxides and organics are intrinsically attributed to the adsorption, complexation and reduction processes (Wang and Stone, Reference Wang and Stone2006b). Adsorption refers to the formation of surface complexes, Mn(IV/III)–organic anions, which are essential to the reactions on the (hydr)oxide surfaces. Complexation induces the release of surface-located Mn(III) and the formation of Mn(III)-organic complexes in solution, which cause the ligand-assisted dissolution. Reduction processes involve the conversion of Mn(IV) and Mn(III) to dissolved Mn(II), i.e. reductive dissolution.
The reduction efficiency of Mn oxides is affected closely by pH condition. At more acidic pH, Mn reduction will proceed more completely and rapidly, giving rise to both the accumulation of structural Mn(III/II) and the massive dissolution of Mn oxides into Mn(II) (Stone and Morgan, Reference Stone and Morgan1984; Waite et al., Reference Waite, Wrigley and Szymczak1988; Wang and Stone, Reference Wang and Stone2006b; Wang et al., Reference Wang, Yang and Zhu2018; Flynn and Catalano, Reference Flynn and Catalano2019). The H+-enhanced Mn reduction could be rationalised by the H+-consuming property of the birnessite–organics reaction (Wang et al., Reference Wang, Yang and Zhu2018). Also, the protonation of the birnessite surface is beneficial for the surface adsorption of organic anions and thus facilitates further reactions (Stone and Morgan, Reference Stone and Morgan1984). In addition, Mn(II) desorption from edge sites into solutions will be promoted under acidic conditions (Wang and Stone, Reference Wang and Stone2006a).
The dissolved Mn(II) and the structural Mn(III/II) could further modify the fine structures of Mn oxides, or even promote the inter-transformation among different mineral phases (Lefkowitz et al., Reference Lefkowitz, Rouff and Elzinga2013; Hinkle et al., Reference Hinkle, Flynn and Catalano2016). For birnessite especially, the vacancy abundance, layer stacking, in-plane crystallinity within the phyllomanganate sheets and local coordination structure centring the Mn atoms will all be likely to alter (Wang et al., Reference Wang, Yang and Zhu2018; Flynn and Catalano, Reference Flynn and Catalano2019). Notably, these structural responses after the redox reaction with organics are also liable to be affected by pH conditions. In a weakly acid environment of pH 4–6, Mn(III/II) tends to migrate into the inter-layer space and adsorb in the layer above vacancies (Wang et al., Reference Wang, Yang and Zhu2018; Flynn and Catalano, Reference Flynn and Catalano2019), which will strengthen the layer stacking (Marafatto et al., Reference Marafatto, Strader, Gonzalez-Holguera, Schwartzberg, Gilbert and Pena2015; Flynn and Catalano, Reference Flynn and Catalano2019); in contrast, some of the Mn(III) also appears to locate at the layer edge sites, which, together with the abundant presence of dissolved Mn(II) in acidic solutions, might help induce the re-ordering of layer sheets towards triclinic symmetry (Hinkle et al., Reference Hinkle, Flynn and Catalano2016; Flynn and Catalano, Reference Flynn and Catalano2019). Under neutral to alkaline solutions, the generated Mn(III) is more likely to be incorporated into vacant sites and reside in the layer (Wang et al., Reference Wang, Yang and Zhu2018); further, with the effect of Mn(II), additional mineral phases may form, such as manganite, feitknechtite and hausmannite (Lefkowitz et al., Reference Lefkowitz, Rouff and Elzinga2013; Wang et al., Reference Wang, Yang and Zhu2018; Zhang et al., Reference Zhang, Liu, Tan, Suib, Qiu and Liu2018). In addition, Mn oxide minerals, such as birnessite, in the soil can be used as electron acceptors for microorganisms (such as the Dietzia species), accompanied with the oxidation of organic matter and fixation as carbonate minerals (e.g. rhodochrosite, Li et al., Reference Li, Wang, Li, Duan, Jia, Ding, Lu, Wang, Nie and Wu2019b). It thus can be summarised that the coupling effect of birnessite and microorganisms in the soil system under sunlight can convert atmospheric CO2 into organics effectively and, further, into carbonate minerals assisting with carbon sequestration.
Oxidative formation of Mn oxides and heavy-metal sorption
Until recently, the formation mechanism of natural Mn oxides has been controversial, and mainly divided into biogenic and abiotic oxidation. Naturally occurring Mn-oxidising microorganisms (bacteria and fungi) are responsible for the biogenic catalytic oxidation of Mn(II) into Mn(III/IV) oxides (Tebo et al., Reference Tebo, Bargar, Clement, Dick, Murray, Parker, Verity and Webb2004; Webb et al., Reference Webb, Tebo and Bargat2005; Villalobos et al., Reference Villalobos, Lanson, Manceau, Toner and Sposito2006). The key to the catalytic process should be attributed to the properly-modified pH and redox conditions of the local aqueous environment by microorganisms, or the release of metabolic products to oxidise Mn(II) chemically (Richardson et al., Reference Richardson, Aguilar and Nealson1988; Hullo et al., Reference Hullo, Moszer, Danchin and Martin-Verstraete2001; Tebo et al., Reference Tebo, Bargar, Clement, Dick, Murray, Parker, Verity and Webb2004). By contrast, Mn(II) is stable as redox-neutral at pH values up to 8.5 and hence the abiotic homogeneous oxidation of Mn(II) is very slow and requires years for completion (Tebo et al., Reference Tebo, Johnson, McCarthy and Templeton2005; Kim et al., Reference Kim, Bargar, Nealson, Flood, Kirschvink, Raub, Tebo and Villalobos2011). However, some researchers have observed the rapid photo-oxidative generation of Mn oxides by coexisting natural semiconducting iron (hydr)oxides, such as hematite and goethite (Madden and Hochella, Reference Madden and Hochella2005; Xu et al., Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019). Also, the photo-stimulated production of reactive oxygen species in natural Mn coatings, like 1O2 and OH⋅ with strong oxidising capability, can contribute to Mn oxide formation (Xu et al., Reference Xu, Li, Li, Lu, Qiao, Liu, Ding and Wang2019).
During the oxidative formation of layered Mn oxides, i.e. birnessite, some heavy metal ions are likely to be adsorbed or incorporated into the structure, such as Cu2+, Pb2+, Zn2+ and Ni2+ (Sherman and Peacock, Reference Sherman and Peacock2010; Zhu et al., Reference Zhu, Ginder-Vogel and Sparks2010; Wang et al., Reference Wang, Feng, Villalobos, Tan and Liu2012; Kwon et al., Reference Kwon, Refson and Sposito2013; Yin et al., Reference Yin, Li, Wang, Ginder-Vogel, Qiu, Feng, Zheng and Liu2014; Qin et al., Reference Qin, Xiang, Liu, Xiong, Koopal, Zheng, Ginder-Vogel, Wang, Feng, Tan and Yin2017; Liu et al., Reference Liu, Li, Chen, Ding, Zhang, Liu, Yin, Chu, Wang and Lu2018b). As revealed in these studies, the sorption sites and binding structures of the metal ions can be varied: most commonly, they are inclined to migrate into interlayer space and bind above the vacancy, forming a triple-corner-sharing (TCS) complex; sometimes they tend to be incorporated into the layer vacancy as INC species; in addition, the metal ions can be adsorbed at layer edges to form multinuclear species by double corner-sharing or edge-sharing complexing (DCS or DES).
The sorption pathways of heavy metals largely affect their preferential binding sites. When the metal cations are simultaneously co-precipitated with birnessite, they will predominantly adsorb above the layer vacancies; when the metal cations are post-adsorbed to birnessite, they tend to be incorporated into the vacancies or bind at layer edges (Li et al., Reference Li, Liu, Xu, Liu, Li, Ding, Chen, Yin, Lin, Wang and Lu2019a). In contrast, the metal sorption mechanism is sensitively impacted by the fine structure of birnessite. A large interlayer space will allow the layer accommodation of metal cations to bind at vacancies as TCS and INC species (Zhu et al., Reference Zhu, Ginder-Vogel and Sparks2010; Wang et al., Reference Wang, Feng, Villalobos, Tan and Liu2012; Liu et al., Reference Liu, Li, Chen, Ding, Zhang, Liu, Yin, Chu, Wang and Lu2018b). These species will potentially induce the structural transformation towards tunnelled Mn oxides (Manceau et al., Reference Manceau, Lanson and Takahashi2014). In other cases when the birnessite is accumulated structurally with Mn(III), the metal ions will be repelled to the edge sites (Liu et al., Reference Liu, Li, Chen, Ding, Zhang, Liu, Yin, Chu, Wang and Lu2018b; Li et al., Reference Li, Liu, Xu, Liu, Li, Ding, Chen, Yin, Lin, Wang and Lu2019a). The correspondingly formed DES and DCS complexes are structurally unsteady and readily released off from the layer, especially under extreme pH conditions (Qin et al., Reference Qin, Xiang, Liu, Xiong, Koopal, Zheng, Ginder-Vogel, Wang, Feng, Tan and Yin2017).
Concluding remarks
Manganese oxides distributed widely among various natural settings on Earth are diversified in mineral phase, crystal structure and Mn oxidation state. The most common Mn oxides on the Earth's surface occur in rock/soil surface Mn coatings, which are predominantly present as birnessite, which has a layered structure and can contain interlayer metal cations. Natural birnessite displays a marked photoelectric response to visible light and acts as a semiconducting material. The electronic structure is sensitive to the fine structure such as Mn/O vacancies and adsorbed metal ions. With excellent photocatalytic property, birnessite actively participates in the photoredox reactions on Earth, during which birnessite itself is reduced while organics and even water is oxidised. Notably, the water oxidation and oxygen-evolving centre in the biological photosynthesis system, Mn4CaO5, features a birnessite-type structure. The oxidative formation of birnessite occurs mainly through bio-catalysis by microorganisms such as Pseudomonas putida and Bacillus species; in other cases, the photo-oxidation is mediated by coexisting semiconducting minerals or reactive oxygen species. Manganese oxides act as heavy-metal scavenger in the environment, while also acting to degrade organics as well as oxidise water. Together they are all involved in the biogeochemical cycle driven by Mn photocatalytic reduction and oxidation in Nature.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (91851208, 91951114, 41820104003, 41872042), the National Key Research and Development Program of China (2019YFC1805901) and the DDE-IUGS Big Science Program.














