


1. Introduction
The III-nitrides (III-N) are interesting semiconductors because of their superior electrical, optical, thermal, and acoustical properties, as well as chemical and physical stability. Phosphors based on III-N host materials were first realized when visible emission was observed from Er-doped GaN Reference Steckl and Birkhahn[1]. Since this initial breakthrough, a number of rare-earth-doped III-N phosphors have been prepared Reference Birkhahn, Garter and Steckl[2] Reference Heikenfeld, Garter, Lee, Birkhahn and Steckl[3] Reference Steckl, Garter, Lee, Heikenfeld and Birkhahn[4] Reference Lee, Heikenfeld, Birkhahn, Garter, Lee and Steckl[5] Reference Heikenfeld and Steckl[6] Reference Lozykowski, Jadwisienczak and Brown[7] Reference Lozykowski, Jadwisienczak and Brown[8] Reference Jadwisienczak, Lozykowski, Perjeru, Chen, Kordesch and Brown[9] Reference Lozykowski, Jadwisienczak and Brown[10]; these phosphors cover the entire visible spectrum. Most of these phosphors have employed single-crystal host matrices, and these crystalline phosphors have performed admirably. Unfortunately, high quality, defect-free films are apparently required to realize the full performance potential of the crystalline films. Additionally, most of these materials require growth and/or post-processing at elevated temperatures. The requirements for high quality crystalline materials and for high temperature luminescence activation represent undesirable constraints that severely limit scale-up potential, cost efficiency, and device design options.
A recent theoretical paper asserted that an amorphous III-N semiconductor could serve as a useful electronic material Reference Stumm and Drabold[11]. This revelation prompted activity in growing amorphous III-N materials and many doped amorphous III-N phosphors have subsequently been reported that emit in the visible range Reference Dimitrova, Van Patten, Richardson and Kordesch[12] Reference Caldwell, Richardson and Kordesch[13] Reference Chen, Gurumurugan, Kordesch, Jadwisienczak and Lozykowski[14] Reference Caldwell, Martin, Dimitrova, Van Patten, Kordesch and Richardson[15] Reference Dimitrova, Van Patten, Richardson and Kordesch[16] Reference Martin, Spalding, Dimitrova, Van Patten, Caldwell, Kordesch and Richardson[17]. The development of amorphous materials suitable for device fabrication ameliorates some of the materials constraints alluded to above. Unfortunately, the new amorphous phosphors still require a thermal activation step that precludes device growth on low melting point substrates such as polymers. In this paper we report an alternative to thermal activation that promises to enable the integration of plastic constituents into luminescent devices based on rare-earth-doped III-N phosphors.
2. Experimental
Thin films of pure AlN:Eu3+ were prepared at room temperature by rf magnetron sputtering of an Al target of 99.999% purity with two slugs (3/16 in diameter) of Eu metal in a pure nitrogen atmosphere. The background vacuum in the chamber was less than 1 × 10−6 Torr. All the films were deposited onto 1 in. × 1 in., or less, p-doped silicon (111) substrates. The deposition pressure and the sputtering power during the film growth were 6 × 10−4 Torr and 199 W. The growth rate was between 0.03 and 0.04 nm/s and the total thickness of the films, as determined from the transmission IR spectrum of the AlN:Eu films, was between 200 and 290 nm. The Eu concentration in the films was estimated to be ~ 10%, based upon the slug size and experience with similar sputtering targets. Any long-range order of the pure as-deposited films was determined by x-ray diffraction (XRD). No diffraction peaks were observed, indicating that the as-deposited films were amorphous. Luminescent activation of these films was accomplished by heating at 923 K for 30 minutes in a tube furnace under flowing N2. In general, the unactivated, oxygen-free films do not show any luminescence. After the thermal treatment, XRD showed that the films were still amorphous.
Oxygen-contaminated films were grown in the same manner as the pure films except oxygen gas was introduced along with nitrogen gas during rf sputtering. By regulating the partial pressure of oxygen in the chamber, we were able to control plasma composition. The composition of the plasma is referred to by its percent oxygen composition:
where p O and p N are the partial pressures in the chamber of oxygen and nitrogen, respectively.
CL data were collected by placing the sample into a vacuum chamber pumped to 10−5 Torr and excited with 2.85 kV electrons. The electron beam at the sample had a diameter of ~5 mm, and the sample was placed ~55 degrees from normal to the electron beam. The beam current was ~ 65 μA.
Excited-state spectra and lifetimes were collected using a nitrogen-pumped dye laser (Photon Technology, Inc.) to excite at 395 nm. This wavelength is too long to excite the semiconductor matrix, but allows direct excitation of the Eu3+ centers. The excitation beam was sent through a bandpass filter (400 nm center, 10 nm FWHM), and emission was collected through a double monochromator using a gated photomultiplier tube. A longpass filter (425 nm cut-on) was used to improve rejection of light scattered from the sample surface. The instrument response function had a full-width-at-half-maximum of approximately 5 µs. For one set of experiments, a neutral density filter (O.D. = 1.0) was placed in the excitation beampath to test for fluence dependent dynamics. No fluence dependence was observed. PL spectra were collected by setting the detector gate to include the entire luminescence decay.
3. Results and Discussion
3.1 Cathodoluminescence
The CL spectra of Eu-doped amorphous AlN thin films grown with varying degrees of oxygen contamination are shown in Figure 1. The sample spectra are nearly identical and are dominated by transitions from the 5D0 excited state of Eu3+ to the 7FJ (J = 0→6) manifold. The largest peaks (613 and 592 nm) correspond to transitions to the 7F2 and 7F1 levels, respectively. The emission peaks, which are normally narrow in crystalline materials, are broadened considerably in all samples and fine structure is evident in some (the 5D0 → 7F1 transition has at least four peaks). The fine structure originates from the lifting of the degeneracy of each term due to local symmetry of the Eu3+ ion. Each of these lines, in turn, exhibits significant inhomogeneous broadening due to the amorphous nature of the sample.

Figure 1. CL spectra of AlN:Eu processed as follows: (a) no O2 and no heat treatment; (b) no O2 but heated to 923 K; (c) grown under 1.6% O2; (d) grown under 3.8% O2; and (e) grown under 20% O2.
While the spectra of the samples are qualitatively similar, their luminescence intensities vary widely depending on oxygen concentration. Figure 2 is a photograph taken of four samples butted up against one another being excited with 2.85 kV electrons. As shown in Figure 2, there is very little visible CL emission from as-grown, oxygen-free samples. When these oxygen-free samples are heated in a tube furnace at 923 K for 30 minutes under a flow of nitrogen, the luminescence intensity increases by a factor of approximately 20 (see figure 1). For comparison, samples grown under 1.6% oxygen exhibit approximately a 60-fold CL enhancement without any thermal treatment. Further enhancement is obtained by increasing oxygen from 1.6% to 3.8%. Samples grown under 3.8% oxygen are approximately 550 times brighter than as-grown, oxygen free samples. Further increases on oxygen content appear to have only a minor impact on CL intensity; samples grown under 20% oxygen show CL enhancement factors of approximately 620.

Figure 2. Photograph of CL emission from AlN:Eu3+ samples doped with varying amounts of oxygen.
Figure 3 compares the integrated intensities from CL and PL with percentage of oxygen in the plasma. All integrated intensities have been plotted relative to the integrated intensity from a non-activated, oxygen free sample. It is clear that the PL data exhibits an intensity trend qualitatively similar to that of the CL data. Still, significant quantitative differences between the CL and PL intensities can be discerned. In particular, the dependence of intensity on oxygen content is much weaker for PL than for CL. A discussion of these differences and their implications can be found in a subsequent section.

Figure 3. The integrated CL(x) and PL(?) intensities from AlN:Eu3+ samples versus oxygen content of the plasma, normalized to the emission intensity from an oxygen-free sample.
3.2 Photoluminescence
A PL spectrum from the sample grown under 20% oxygen is shown in Figure 4. The PL spectrum of this sample, like the others, was identical to the CL spectrum obtained. This spectral similarity strongly suggests that the emitting states are the same following CL and PL excitation. Consequently, this conclusion implies that the excited state decay kinetics observed in PL and CL experiments should also be identical. Photoluminescence dynamics of these samples are presented and discussed below.
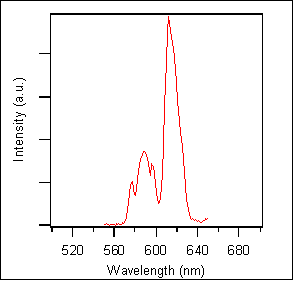
Figure 4. PL emission spectrum from AlN:Eu3+ grown under 20% oxygen.
3.3 Excited-state decays
Figure 5 shows PL emission decays for the four AlN:Eu3+ samples with varying degrees of oxygen contamination. All four decays can be well-characterized as double exponential decays with one very fast component (indistinguishable from the instrument response function) and one slow component with a lifetime of 0.4 ms. Since the emitting states in PL and CL experiments are believed to be identical, the kinetics measured in these experiments should be identical to those observed in time-resolved CL experiments, which are not available in our laboratories. Significantly, the slow component lifetime (0.4 ms) is identical to the reported lifetime of the Eu3+ 5D0 state in ion-implanted, single-crystal samples of AlN grown via MOCVD Reference Jadwisienczak, Lozykowski, Berishev, Bensaoula and Brown[18]. Jadwesienczak et al. Reference Jadwisienczak, Lozykowski, Berishev, Bensaoula and Brown[18] assign this slow component as the radiative lifetime of the 5D0 state. The kinetic similarities between amorphous and crystalline AlN:Eu3+ samples support the notion that amorphous III-nitride phosphors can be designed with low emission quenching.

Figure 5. Semilogarithmic PL decays from AlN:Eu3+ samples doped with varying amounts of oxygen. The slow component of each decay has a rate constant of (0.4 ms)−1. The decays have been vertically offset for clarity.
The oxygen-free sample (solid line) is dominated (>99%) by the fast decay component and is essentially indistinguishable from the instrument response function. The samples grown under 1.6% and 3.8% oxygen (dotted line and circles, respectively) both contain major contributions from the fast decay component (approximately 83%), but with an appreciable slow component (17%). These two samples exhibit nearly identical excited state dynamics, as expected from analysis of their nearly identical spectral intensities. The sample grown under 20% oxygen (crosses) shows an increase in the relative intensity of the slow component to approximately 28% of the total decay intensity.
The role of oxygen in increasing the PL emission yield cannot be determined based exclusively on these data. However, the nature of the kinetic data, the relatively small quantities of oxygen required to effect substantial changes, and the highly oxophilic nature of the lanthanides suggest a possible enhancement mechanism. From the kinetic data it can be seen that the oxygen "toggles" the coupling between Eu centers and non-radiative trap states in a discrete, binary manner. If the coupling to non-radiative trap states (via exciton migration through the matrix, for example) were varied continuously, then multi-exponential decay kinetics with a broad distribution of lifetimes would be expected. The presence of even a small quantity of oxygen in the plasma seems to turn on emission from a substantial number of luminescent centers. Further increases in oxygen doping provide only limited additional gain. The fact that the small oxygen quantity can cause such a substantial change in Eu photophysics suggests that the oxygen may actually be concentrated near the luminescent centers in these samples. Eu's strong affinity for oxygen could explain a highly inhomogeneous oxygen distribution within the sample.
3.4 CL / PL intensity discrepancy
Since the emitting states are the same for PL and CL, differences between PL and CL intensity trends imply that oxygen affects the PL and CL excitation processes differently. In particular, the presence of oxygen apparently enhances the yield of the emitting state in CL to a greater extent than it does in PL. Oxygen-induced CL enhancements are approximately 600-fold, while PL enhancements are approximately 25-fold. Interestingly, this means that the CL excitation enhancement relative to PL is approximately 25-fold (600 ÷ 25). As in the previous section, the role of oxygen in causing this observed enhancement is subject to conjecture; however, we can follow the arguments proposed above to arrive at a plausible model. If the oxygen distribution is indeed non-uniform and is concentrated most heavily around the Eu centers, then electron beam excitation could be enhanced due to a larger electron impact cross section of oxide relative to nitride.
Differences between the spectra in Figure 1 are subtle. There is no way to confirm the mechanism proposed above through analysis of these data. Indeed, the absence of striking differences in these spectra may seem to argue against our model. Incorporation of oxide ions into the local coordination sphere of Eu might be expected to yield significant changes in the emission spectra. In fact, Eu2O3 emission spectra are not radically different from those presented here. We have collected spectra from Eu2O3 powder (99.99%) and compared those spectra with the ones presented here. The oxide emission peak wavelengths are nearly identical to the peak wavelengths in these spectra. The primary difference is that the oxide peaks are much narrower (only a few nanometers FWHM), owing to the homogeneity of that sample. Oxide spectra have also been reported in the literature. Recently published spectra of Eu:Y2O3 Reference Kumar, Sankar, Cho, Craciun and Singh[19] also closely resemble our spectra (only narrower peaks). Especially noteworthy in the previously published Eu:Y2O3 spectra is the splitting of the 5D0 → 7F1 transition into four peaks as seen in our spectra. Based on these considerations, we submit that any expected spectral shifts in these samples may indeed be small enough to be obscured by the tremendous inhomogeneous broadening.
4. Conclusion
We have demonstrated that it is possible to activate visible emission from an amorphous Eu-doped III-nitride semiconductor by intentionally incorporating oxygen. This activated phosphor gives rise to strong emission in the orange-yellow (~613 nm) region. The incorporation of as little as a few percent oxygen in the sputtering chamber during growth results in luminescence activation. The CL intensities of samples prepared in this way are several times greater than otherwise similar films activated thermally at 950K.
ACKNOWLEDGMENTS
This work is supported by BMDO URISP grant N00014-96-1782 entitled “Growth, Doping and Contacts from Wide Band Gap Semiconductors” and grant N00014-99-1-0975 entitled “Band-Gap Engineering of the Amorphous In-Ga-Al Nitride Semiconductor Alloys for Luminescent Devices from the Ultraviolet to the Infrared”. The authors would also like to thank the Material Research Society for partial funding this project under the 2000-2001 Undergraduate Materials Research Initiative.





