


INTRODUCTION
Pitt-Hopkins syndrome (PTHS) is a rare genetic disorder with no cure caused by insufficient expression of the TCF4 gene found on chromosome 18q21.2 (Amiel et al., Reference Amiel, Rio, de Pontual, Redon, Malan, Boddaert and Colleaux2007). Since the TCF4 gene was identified in 2007, the genetic etiology of PTHS has been associated with an autosomal dominant de novo molecular variant leading to haploinsufficiency of the TCF4 gene (Zweier et al., Reference Zweier, Peippo, Hoyer, Sousa, Bottani, Clayton-Smith and Rauch2007). Transcription Factor 4 (TCF4), the protein product of the TCF4 gene, is believed to be involved in brain development and neuronal differentiation (Forrest, Hill, Quantock, Martin-Rendon, & Blake, Reference Forrest, Hill, Quantock, Martin-Rendon and Blake2014). While the exact prevalence of PTHS is unknown, Rosenfeld et al. (Reference Rosenfeld, Leppig, Ballif, Thiese, Erdie-Lalena, Bawle and Shaffer2009) estimated that the population frequency of PTHS due to 18q21 microdeletion is 1:34,000 to 1:41,000; however, approximately 500 individuals have been identified with the disorder worldwide (Sweatt, Reference Sweatt2013).
PTHS was first described in 1978 in two individuals sharing similar characteristics, including dysmorphic facial features, clubbed fingers, abnormal breathing, and developmental delay (Pitt & Hopkins, Reference Pitt and Hopkins1978). Since then, details regarding the clinical phenotype of PTHS have continued to be described in the literature (Amiel et al., Reference Amiel, Rio, de Pontual, Redon, Malan, Boddaert and Colleaux2007; de Winter et al., Reference de Winter, Baas, Bijlsma, van Heukelingen, Routledge and Hennekam2016; Marangi et al., Reference Marangi, Ricciardi, Orteschi, Lattante, Murdolo, Dallapiccola and Zollino2011; Zweier et al., Reference Zweier, Sticht, Bijlsma, Clayton-Smith, Boonen, Fryer and Rauch2008). Characteristic craniofacial features (e.g., bitemporal narrowing, upslanting palpebral fissures, wide nasal bridge, prominent ears, and broad mouth) and other physical features (e.g., fetal finger pads, palmar creases, and postnatal growth restriction and microcephaly) are frequently seen (de Winter et al., Reference de Winter, Baas, Bijlsma, van Heukelingen, Routledge and Hennekam2016). Additionally, common medical comorbidities, including constipation (in severe cases, Hirschsprung’s disease and intestinal malrotation; 70%); early onset myopia (54%), and strabismus (49%); unique breathing patterns of hyperventilation followed by apnea (48%); and/or recurrent seizures (38%) have been reported in individuals with PTHS (Amiel et al., Reference Amiel, Rio, de Pontual, Redon, Malan, Boddaert and Colleaux2007; Brockschmidt et al., Reference Brockschmidt, Filippi, Charbel Issa, Nelles, Urbach, Eter and Weber2011; Goodspeed et al., Reference Goodspeed, Newom, Morris, Powell, Evans and Golla2018; Hasi et al., Reference Hasi, Soileau, Sebold, Hill, Hale, O’Donnell and Cody2011; Ouvrier, Reference Ouvrier2008; Zweier et al., Reference Zweier, Peippo, Hoyer, Sousa, Bottani, Clayton-Smith and Rauch2007).
Neuroimaging often demonstrates non-specific abnormalities including dysplasia of the corpus callosum, bulging of the caudate nuclei, enlarged asymmetric ventricles, and cerebral atrophy, but no pathognomonic finding has been described (Andrieux et al., Reference Andrieux, Lepretre, Cuisset, Goldenberg, Delobel, Manouvrier-Hanu and Holder-Espinasse2008; de Pontual et al., Reference de Pontual, Mathieu, Golzio, Rio, Malan, Boddaert and Amiel2009; Giurgea et al., Reference Giurgea, Missirian, Cacciagli, Whalen, Fredriksen, Gaillon and Moncla2008; Peippo et al., Reference Peippo, Simola, Valanne, Larsen, Kahkonen, Auranen and Ignatius2006; Stavropoulous, MacGregor, & Yoon, Reference Stavropoulous, MacGregor and Yoon2010; Zweier et al., Reference Zweier, Sticht, Bijlsma, Clayton-Smith, Boonen, Fryer and Rauch2008). Neurodevelopmentally, most cases of PTHS are characterized by severe intellectual disability (ID), global developmental delay, and comorbid autism spectrum disorder (ASD; Cody et al., Reference Cody, Sebold, Heard, Carter, Soileau, Hasi-Zogaj and Hale2015; Sweatt, Reference Sweatt2013; Van Balkom, Vuijk, Franssens, Hoek, & Hennekam, Reference Van Balkom, Vuijk, Franssens, Hoek and Hennekam2012). Speech is often absent, and motor delays (independent ambulation after 5 years of age or not at all), ataxia, and motor incoordination occur in the majority of cases.
Stereotypic hand movements characteristic of ASD, including lateral movements, clapping and flapping, and repeated hand–mouth movements, are also common (Brockschmidt et al., Reference Brockschmidt, Todt, Ryu, Hoischen, Landwehr, Birnbaum and Weber2007; Cody et al., Reference Cody, Sebold, Heard, Carter, Soileau, Hasi-Zogaj and Hale2015; Van Balkom et al., Reference Van Balkom, Vuijk, Franssens, Hoek and Hennekam2012). Emotional and behavioral problems including anxiety, aggression, and self-injury have also been described in a minority of PTHS cases (Goodspeed et al., Reference Goodspeed, Newom, Morris, Powell, Evans and Golla2018; Van Balkom et al., Reference Van Balkom, Vuijk, Franssens, Hoek and Hennekam2012). Additionally, TCF4 mutations have been identified as genetic risk factors for schizophrenia, leading to higher risk of schizophrenia in individuals with PTHS (Blake et al., Reference Blake, Forrest, Chapman, Tinsley, O’Donovan and Owen2010). Although many cases of PTHS have been described in the literature, the relationship between neurocognitive profiles and medical comorbidities has not been systematically explored.
Due to the overlapping phenotype of microcephaly, epilepsy, absent speech, and constipation, PTHS is clinically very similar to several other neurodevelopmental disorders, including Angelman syndrome, Rett syndrome, and Mowat-Wilson syndrome (Marangi & Zollino, Reference Marangi and Zollino2015). Angelman syndrome is the neurodevelopmental disorder that most closely resembles PTHS in phenotype, and 2% of patients initially suspected as having Angelman syndrome were found to have PTHS with a mutation in TCF4 (Takano, Lyons, Moyes, Jones, & Schwartz, Reference Takano, Lyons, Moyes, Jones and Schwartz2010). However, PTHS is associated with distinct facial features not found in Angelman syndrome, including bitemporal narrowing, broad nasal bridge and flared nostrils, and large mouth with prominent M-shaped upper lip (Marangi & Zollino, Reference Marangi and Zollino2015).
Individuals with PTHS also have been previously diagnosed with Rett syndrome; however, Rett syndrome usually presents as a progressive encephalopathy, with regression of language and motor skills following typical early development, which is not typical of PTHS (Armani et al., Reference Armani, Archer, Clarke, Vasudevan, Zweier, Ho and Christodoulou2012). Mowat-Wilson syndrome, while phenotypically similar with regards to severe constipation, also presents with a variety of major malformations not found in PTHS, including heart defects and genitourinary anomalies, in addition to a distinctive pattern of facial anomalies (i.e., uplifted earlobe configuration, hypertelorism, and linear mandible arch; Marangi & Zollino, Reference Marangi and Zollino2015).
Although a variety of pathogenic variants of TCF4 have been reported, no studies have a found a conclusive link between specific genotypes and phenotypes in PTHS (Zweier et al, Reference Zweier, Peippo, Hoyer, Sousa, Bottani, Clayton-Smith and Rauch2007). However, when Van Balkom et al. (Reference Van Balkom, Vuijk, Franssens, Hoek and Hennekam2012) divided previously published case studies based on proven deletion or mutation, differing trends between the two categories were noted. At that time, higher frequencies were found in published cases with proven TCF4 mutations (n=49) versus TCF4 deletions (n=17) in breathing abnormalities (71% vs. 35%), epilepsy (39% vs. 29%), nonverbal presentation (92% vs. 77%), and severe ID (100% vs. 82%).
These findings suggest that genetic variability might play a role in neuropsychological and medical outcomes, although larger studies with systematic analysis are needed to further explore this hypothesis. However, these frequencies must also be interpreted with caution, as individual neuropsychological outcomes are not only influenced by gene expression, but also by interactions with social and learning environments (Van Balkom et al., Reference Van Balkom, Vuijk, Franssens, Hoek and Hennekam2012).
Additionally, emerging evidence from in vitro studies by Sepp, Pruunsild, and Timmusk (Reference Sepp, Pruunsild and Timmusk2012) reveals that not all deletions and truncating mutations in TCF4 result in complete loss of function in all TCF4 protein products. Functional analyses in this study of PTHS-associated reading frame elongating and missense mutations demonstrated that resulting protein products showed variable mechanisms (i.e., protein destabilization, alteration of dimerization preferences, and loss of DNA-binding and transactivation ability) and extent of impairments (i.e., from subtle deficiencies to dominant-negative effects). This range of impairment was found to be dependent on protein dimer context (i.e., homo- or hetero-dimer) and whether the mutation occurred within the basic helix-loop-helix domain (i.e., outside missense mutations caused no deficiencies). This work represents in vitro functional analyses of known human pathogenic variants of TCF4 and highlights the complexity of this gene, which can be transcribed into many different transcripts of varying lengths. The authors emphasize that size and location of the variant may determine functionality of the resultant protein products. While this research has not been translated to human subjects, it offers a context for hypotheses regarding genotype and phenotype correlations in PTHS.
Although there is no cure for PTHS, research is continuing to identify the molecular pathways affected by TCF4 mutations, while clinical management includes evaluation and intervention for common medical comorbidities and neuropsychological difficulties. Individuals with PTHS are classically thought to be severely delayed, and formal neuropsychological assessments of patients with PTHS are lacking in the literature. In a sample of 10 children (median age=10 years), Van Balkom et al. (Reference Van Balkom, Vuijk, Franssens, Hoek and Hennekam2012) reported maximum developmental age-equivalents of 15 months on the mental scale and 19 months on the motor scale, as assessed by the Bayley Scales of Infant Development – Second Edition. However, there may be a wider range of cognitive abilities in PTHS than reported in much of the scientific literature. As most patients diagnosed with PTHS are nonverbal and have severe ID irrespective of pathogenic variant, the present case study is to the authors’ knowledge the first case of formal neuropsychological assessment in an adolescent with PTHS to date and expands the existing understanding of PTHS by presenting a unique phenotype in an adolescent male with higher cognitive abilities and fewer medical comorbidities.
METHODS
Case History
The presenting patient was a 13-year-old, right-handed, Caucasian male diagnosed with PTHS at 12 years of age. He was the product of a full-term gestation and uncomplicated pregnancy and delivery. Early motor developmental milestones were generally acquired within normal limits, as he was noted to sit (8 months), stand (13 months), and walk (14 months) within the typical timeframe. Early language developmental milestones were reportedly delayed, as he had three words at age 2 years, first used two-word phrases at age 4 years, and first used sentences at age 6 years. He received speech therapy beginning at age 2.5 years. The patient had medical history significant for ASD (diagnosed at age 3 years), celiac disease (diagnosed at age 5 years), and attention deficit/hyperactivity disorder (AD/HD) (diagnosed at age 7 years). Vision was noted to be normal, with no history of strabismus. He had a long-standing history of constipation (treated with Miralax), with moderate stool burden regularly shown on abdominal X-rays. He also had history of sleep disturbance (treated with melatonin), including occasional parasomnias with sleepwalking and sleeptalking.
Notably, his medical history was not significant for several common medical comorbidities found in PTHS, including apneic spells and seizures. Academically, the patient was enrolled in 7th grade with an Individualized Education Plan under the primary classification of ID (not formally diagnosed), secondary classification of Speech Impairment, and tertiary classification of Other Health Impairment (AD/HD), and he received his education in a self-contained classroom with special education services (speech therapy and assistive technology). Regarding psychological history, he struggled with frustration tolerance, social communication, restricted/repetitive behaviors, attention/impulsivity, adaptive functioning, and academic progress. Extended family history was significant for ID, learning disabilities, language delays, AD/HD, depression, and anxiety.
Neuropsychological Assessment
The patient was referred to the University of Texas Southwestern/Children’s Health Center for Autism and Developmental Disabilities by his neurologist (coauthor K.G.) for a neuropsychological evaluation (coauthors A.T. and V.B.E.) following diagnosis to document current neurocognitive functioning secondary to his medical condition to assist with planning treatment interventions. He was assessed over the course of a single day and was accompanied by his mother. Relevant information was obtained through a review of available medical and educational records, as well as a clinical interview with his mother.
This information was supplemented by detailed examination of his neurobehavioral repertoire and his performance during a neuropsychological test battery that included the following: Stanford Binet Intelligence Scales, Fifth Edition (SB-5); Conners’ Continuous Performance Test, Third Edition (CPT-3); Drexel Tower of London, Second Edition (TOL-2); Wide Range Assessment of Memory and Learning, Second Edition (WRAML-2); Beery Buktenica Developmental Test of Visual-Motor Integration, Sixth Edition (Beery VMI-6); Grooved Pegboard Test (GPT); and Wechsler Individual Achievement Test, Third Edition (WIAT-III). His mother also completed the Behavior Rating Inventory of Executive Function (BRIEF); Behavior Assessment System for Children, Second Edition (BASC-2); Social Communication Questionnaire (SCQ); Social Responsiveness Scales, Second Edition (SRS-2); and Vineland Adaptive Behavior Scales, Second Edition (Vineland-2). The patient’s mother consented to this case report, and data included in this manuscript was obtained in compliance with the Helsinki Declaration.
RESULTS
Neurological Findings
The patient received a late diagnosis of PTHS at age 12 years following whole exome sequencing that identified a de novo pathogenic variant of a single base-pair deletion (c.1933delG) in exon 19 of 20 of the TCF4 gene, resulting in a frameshift mutation, as seen in Figure 1. This finding was in the context of a previously unremarkable chromosomal microarray at age 9 years. The patient’s most recent electroencephalography (EEG) at age 12 years revealed no focal, lateralized, or epileptiform abnormalities. During the maximally alert state on the EEG, a medium-amplitude posterior dominant rhythm and low-amplitude frontocentral beta frequencies were observed to be symmetrical and well regulated.
The patient’s most recent magnetic resonance imaging (MRI) at age 9 years is shown in Figure 2. Results revealed a dysmorphic corpus callosum that was slightly shorter in anteroposterior dimension and thicker than normal, consistent with corpus callosal abnormalities described in the PTHS literature. The retrocerebellar cerebrospinal fluid spaces were also noted to be prominent, but insignificant, and no additional parenchymal abnormalities were found. Facial features consistent with PTHS were observed, including midface flattening, mild hypotelorism, bulbous nasal tip, and Cupid’s bow lip. Appearance was also significant for slightly prominent finger pads and toe pads. No significant findings were found on recent neurological review of systems including cranial nerve, sensory, cerebellar, reflexes, and gait examination. Examination of motor systems revealed hypotonia, but normal strength. Finally, a recent mental status examination revealed adequate eye contact, normal orientation, good cooperation, and ability to speak in full sentences despite poor articulation and immature content/syntax.
Fig. 1 Representative drawing of the TCF4 gene to demonstrate location of the patient’s mutation (arrow). Numbered boxes represent exons. Exons 1 and 20 (white boxes) are non-coding regions.
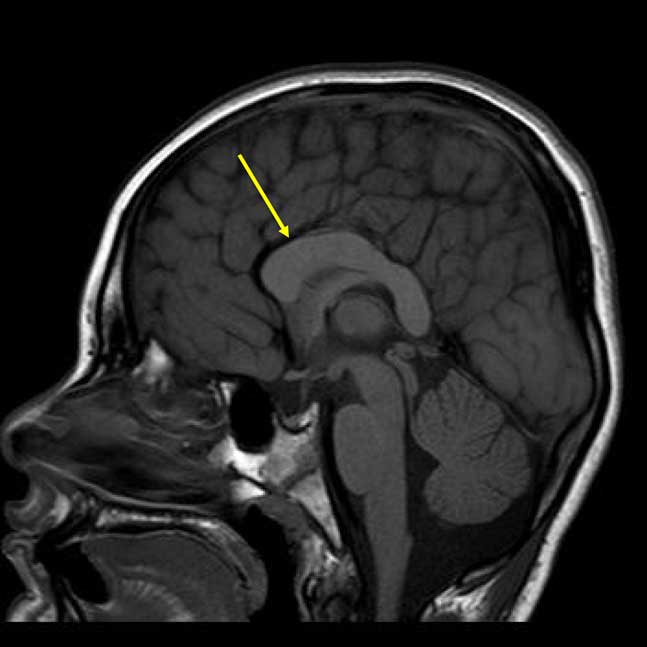
Fig. 2 Sagittal T1-weighted magnetic resonance imaging demonstrating the dysplastic corpus callosum (arrow).
Behavioral Observations
Behaviorally, the patient presented as a polite child with a pleasant disposition consistent with PTHS. He appeared well-groomed and dressed appropriately for his age and setting. During the interview, the patient was able to provide correct answers in response to standard personal information questions including name, age, birthday, home address, school, grade level, teacher’s name, and family members. He transitioned from the interview to one-on-one work with the examiner without difficulty. Rapport was slowly established but easily maintained. Affect was largely appropriate for context. He was generally able to sustain his attention and regulate his behavior throughout the evaluation with hourly breaks. However, he was often impulsive in his responses and used a trial-and-error approach in completing tasks. Additional prompting and increased structure was needed to reduce impulsivity.
The patient was able to speak in complete sentences with occasional articulation difficulties. His expressive language for conversation was limited to answering specific questions by the examiner, and he struggled to engage in conversational exchange. The patient was able to comprehend simple task instructions, but he struggled with multistep instructions. As such, he often required simplification and repetition of instructions and was helped by the use of direct commands over questions.
The patient ambulated independently and presented with a mildly clumsy gait. He preferred his right hand for fine-motor tasks and used a tripod pencil grip resulting in legible print. The patient presented with difficulties consistent with ASD, including inconsistent nonverbal communication (e.g., limited use of eye contact, gestures, and facial expressions for social interaction), poor social reciprocity, repetitive motor mannerisms (e.g., body rocking), and sensory-seeking behaviors (e.g., visual inspection of objects). Overall, the patient displayed good effort and persistence with behavioral reinforcement (i.e., visual checklists, rewards, and social praise) used for motivation and encouragement. While accommodations made during testing (e.g., increased structure, repetition/simplification of instructions) may have impacted standardization of the assessments, the findings were thought to provide a valid reflection of his current functioning.
Neurocognitive Testing
The results of neurocognitive testing are presented in Table 1. The patient’s overall cognitive ability was documented to be in the Impaired range consistent with a 6-year, 3-month-old level. He demonstrated relative strengths in verbal fluid reasoning and nonverbal visual-spatial processing (i.e., Borderline range). The patient demonstrated impairments in aspects of attention and executive functioning, including vigilance, processing speed, planning, and problem-solving; however, he was able to adapt to a novel problem-solving task. He performed in the Borderline range on both verbal and visual memory tasks. During a story memory task, he was easily overwhelmed by the amount of information, but on a list-learning task requiring practice and repetition, he demonstrated improved learning with repetition and retention of information over time. On both verbal tasks, he was also able to recall the information that he had learned after a period of encoding and consolidation. His performance on visual memory tasks was marked by visual overload for simultaneous processing of information on a design memory task, but was improved when provided thematically related visual cues. Performance on visual memory tasks improved considerably (i.e., greater than one standard deviation) with multiple-choice items for recognition.
Table 1 Neurocognitive test results
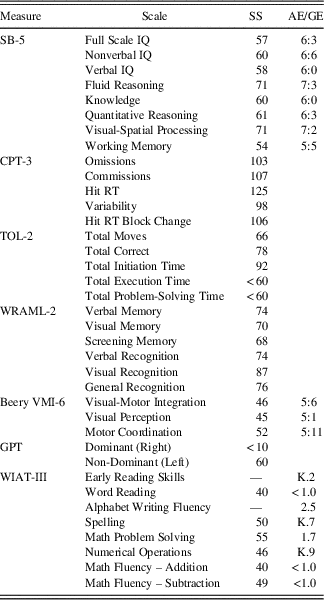
SS=standard score; AE=age equivalent; GE=grade equivalent; IQ=intelligence quotient; RT=reaction time.
The patient exhibited impaired performance on tasks of visual-motor integration, visual-spatial skills, and fine-motor coordination. Assessment of the patient’s academic functioning revealed impaired reading, writing, and math skills consistent with his cognitive functioning. However, he was able to identify and write letters, sound out letters of words phonemically, and spell his name and multiple sight words (e.g., “cat” and “be”). He was also able to identify numbers, understand simple math concepts (e.g., most, less, same, longest), recognize shapes, and solve single-digit addition and subtraction problems. Additionally, he was able to demonstrate early reading skills (i.e., rhyming, two-letter phonemes) and solve word problems when accompanying visual information was presented. Additional language testing was not conducted due to time constraints of the evaluation.
Parent-Rated Questionnaires
The results of parent-rated questionnaires are presented in Table 2. Parent ratings revealed clinically significant elevations in difficulties with behavioral regulation, metacognition, externalizing problems, and behavioral symptoms, consistent with his previously diagnosed AD/HD. The patient’s mother also rated clinically significant difficulties with social communication/interaction and restricted interests/repetitive behaviors on the SRS-2, consistent with his previously diagnosed ASD. Additionally, the raw score on the parent-rated SCQ was 20, which was above the cutoff for ASD set at 15. Finally, adaptive skills were revealed to be largely impaired, including functional communication, activities of daily living, and socialization. However, gross motor skills were rated to be age-appropriate and noted as a strength in comparison to the majority of patients with PTHS.
Table 2 Parent-rated questionnaire results

* at risk.
** clinically significant.
SS=standard score.
DISCUSSION
To the authors’ knowledge, this unique individual is the first adolescent with PTHS to complete formal neuropsychological assessment. Evaluation of the patient’s intellectual, attention/executive, memory, visual-motor/fine-motor, academic, adaptive, and emotional/behavioral functioning revealed global impairment across all areas of functioning consistent with a formal diagnosis of mild to moderate ID. Additionally, results from caregiver interview, behavioral observations, and direct standardized assessment revealed ongoing difficulties consistent with ASD and AD/HD. However, he demonstrated abilities beyond what has been detailed in the literature (Van Balkom et al., Reference Van Balkom, Vuijk, Franssens, Hoek and Hennekam2012) regarding individuals with PTHS including use of full sentences, capacity to learn and solve novel problems, basic academic functioning, and independent ambulation.
Additionally, he presented with fewer medical comorbidities, as his vision was reportedly normal, and he had never demonstrated any spells of abnormal breathing or seizure-like activity. Thus, this unique case report demonstrates that there may be a wider range of clinical phenotype and neuropsychological functioning in PTHS than previously reported in the scientific literature, suggesting that more individuals may be affected by TCF4 mutations who exhibit milder symptoms.
Due to the patient’s higher cognitive abilities and fewer medical comorbidities, he did not receive a diagnosis of PTHS until age 12. The reason he has attained higher cognitive abilities compared to previously reported cases remains unclear. Many individuals with PTHS due to TCF4 deletions display similar severity of medical and cognitive phenotypes (Giurgea et al., Reference Giurgea, Missirian, Cacciagli, Whalen, Fredriksen, Gaillon and Moncla2008) compared to individuals with PTHS due to sequence variants of TCF4, although this conclusion has not held true in all genotype–phenotype linkage analyses (de Winter et al., Reference de Winter, Baas, Bijlsma, van Heukelingen, Routledge and Hennekam2016; Rosenfeld et al., Reference Rosenfeld, Leppig, Ballif, Thiese, Erdie-Lalena, Bawle and Shaffer2009; Whalen et al., Reference Whalen, Heron, Gaillon, Moldovan, Rossi, Devillard and Giurgea2012).
Engelen et al. (Reference Engelen, Moog, Weber, Haagen, van Uum and Hamers2003) identified a young male patient with an interstitial deletion of chromosome region 18q21.1 → 18q21.3 by karyotype before the discovery of TCF4 as the disease-associated gene with PTHS. Similar to the present case study, the child displayed mild cognitive delay, emerging language, independent ambulation, and mild facial anomalies; however, this patient was never given the diagnosis of PTHS, nor did he have the characteristic facial features of PTHS.
Kalscheuer et al. (Reference Kalscheuer, Feenstra, Van Ravenswaaij-Arts, Smeets, Menzel, Ullmann and Ropers2008) reported a case with a de novo balanced translocation involving TCF4 and CHD6 (chromodomain helicase DNA binding protein 6) in an adolescent female patient, a gene which has not been implicated in PTHS. Similar to the present case study, this adolescent female presented with mild to moderate ID, basic academic functioning, minor facial anomalies, ability to speak in full sentences, independent ambulation, and absence of epilepsy or breathing abnormalities. She was not given the diagnosis of PTHS, although disruption of the TCF4 gene by translocation was presumed to be the cause of her ID.
In combination with emerging evidence from Sepp et al. (Reference Sepp, Pruunsild and Timmusk2012), comparison with this case study suggests the location of the present patient’s single base-pair deletion in the penultimate exon of the gene may have yielded a more functional protein product, leaving a greater number of unaffected transcripts intact. However, studies investigating genotype and phenotype correlations in this population remain limited, and additional research is needed to elucidate the connection between genetic abnormality, neurological phenotype, and neuropsychological presentation.
Individuals with PTHS require evaluation and management of medical comorbidities (i.e., constipation, myopia, strabismus, apneic spells, seizures, sleep disturbances) in addition to neuropsychological difficulties (i.e., ID, ASD, developmental delays). Successful evaluation and management of such a broad range of symptoms requires a coordinated multidisciplinary team. Because information regarding the range of neuropsychological functioning in various genetic conditions is limited, the ability of neuropsychologists to differentiate between diagnoses of PTHS and other phenotypically similar genetic conditions based on neuropsychological profiles remains limited. However, emerging evidence that a wider range of neuropsychological functioning may be present in these genetic conditions may allow neuropsychologists to contribute to a broader understanding of the link between neurocognitive phenotypes and genetic conditions.
The study of neurocognitive profiles of individuals with monogenic disorders may also provide insight into the underlying neurobiology and connectivity of common neurodevelopmental disorders. Additionally, neuropsychologists play an essential role in assessing the strengths and weaknesses of these individuals and providing recommendations for their care. In the present case, the patient was receiving limited school-based special education services to address significant impairments in cognitive, academic, and behavioral/emotional functioning. However, the neuropsychological evaluation demonstrated increased success for the patient when he was provided with heightened structure and clear expectations, repetition and simplification of instructions, multiple-choice items for recognition, pairing of visual and verbal stimuli, and behavioral reinforcement.
As a result of the evaluation, treatment recommendations included medical service coordination; referrals for speech, occupational, and behavioral therapies; suggestions for educational programming, home-based behavioral management, social skills development, safety planning, and guardianship considerations; and resources for parental support, advocacy, and education. Thus, this case demonstrates the importance of having a treatment team that includes a neuropsychologist to improve quality of life for individuals with PTHS and their families.
PTHS is likely underdiagnosed, and clinicians should use comprehensive genetic testing methods with patients with developmental delays to investigate the possible presence of an underlying genetic etiology. In the present case, chromosomal microarray results were unremarkable, whereas whole exome sequencing identified the TCF4 deletion. While routine cytogenetic testing (including chromosomal microarray) is indicated in the evaluation of any child with global developmental delay lacking a clear etiology, chromosomal microarray often does not establish a diagnosis of PTHS, and higher-tier genetic testing with whole exome sequencing or commercially available panels should be considered (Goodspeed et al., Reference Goodspeed, Newom, Morris, Powell, Evans and Golla2018; Michelson et al., Reference Michelson, Shevell, Sherr, Moeschler, Gropman and Ashwal2011). In the appropriate setting, whole exome sequencing has been shown to have a higher diagnostic yield when used as second-tier testing in place of less specific testing and reduced overall cost (Green, Rubin, & Olson, Reference Green, Rubin and Olson2017; Nolan & Carlson, Reference Nolan and Carlson2016).
Future research is needed to investigate the medical and neuropsychological profiles in this population to advance understanding of the spectrum of this syndrome, identify clinical biomarkers, and develop novel interventions. Failure to recognize this spectrum can result in late identification of an accurate diagnosis and less comprehensive intervention, but with continued research and awareness of PTHS, a more thorough understanding of the spectrum of PTHS may be developed.
For more information about PTHS, visit the Web site for the Pitt Hopkins Research Foundation, whose mission is to support research dedicated to finding treatments for PTHS and to support the Pitt Hopkins community with resource recommendations, parental support, and the latest medical information (https://pitthopkins.org/).
ACKNOWLEDGMENTS
The authors have no conflicts of interest or sources of financial support to disclose. The authors acknowledge the family who allowed us to provide medical care to their beloved child and consented to this case report to contribute to advances in the understanding of PTHS.




