



Introduction
Metallic nuclear fuels such as U–Zr and U–Pu–Zr alloys have several advantages over ceramic fuels, such as superior heat conductivity, large prompt negative temperature coefficient of reactivity, and better safety margins [Reference Sofu1]. Historically, U–Zr alloy fuels were used in several types of test and research reactors including the Experimental Breeder Reactor-II. The research interests in the U–Zr binary alloy also come from the candidacy of U–Pu–Zr ternary alloys for future fast spectrum reactors [Reference Janney, Papesch, Burkes, Cole, Fielding, Frank, Hartmann, Hyde, Keiser, Kennedy, Maddison, Mariani, Middlemas, O'Holleran, Sencer and Squires2, Reference Crawford, Porter and Hayes3, Reference Delage, Carmack, Lee, Mizuno, Pelletier and Somers4]. Within the scope of U–Zr binary system whose equilibrium phase diagram is presented in Fig. 1 [Reference Sheldon and Peterson5], most of the research efforts have been placed in the uranium-rich compositions driven by the interests in metallic fission-reactor fuels. Recent experimental and theoretical approaches by Basak, Ahn, Irukuvarghula, Xiong, and Bagchi have pushed the compositional boundary toward the zirconium-rich region up to U–50 wt.% Zr [Reference Xiong, Xie, Shen and Morgan6, Reference Ahn, Irukuvarghula and McDeavitt7, Reference Basak, Prasad, Kamath and Prabhu8, Reference Basak, Prabhu and Krishnan9, Reference Basak10, Reference Irukuvarghula, Ahn and McDeavitt11]. The motivation for studying these zirconium-rich compositions is twofold. Firstly, uranium-lean (as much as U–50 wt.% Zr) compositions are found in the center region of high burnup U–10 wt.% Zr fuel rods. Secondly, uranium-lean and zirconium-rich U–Zr alloys are considered for advanced nuclear reactors such as the Korean subcritical Hybrid Power Extraction Reactor (HYPER) [Reference Park, Shin, Han, Song, Choi and Park12]. U–Zr alloy microstructure and metallurgical behavior are critical for fuel performance codes, fabrication, and core design.
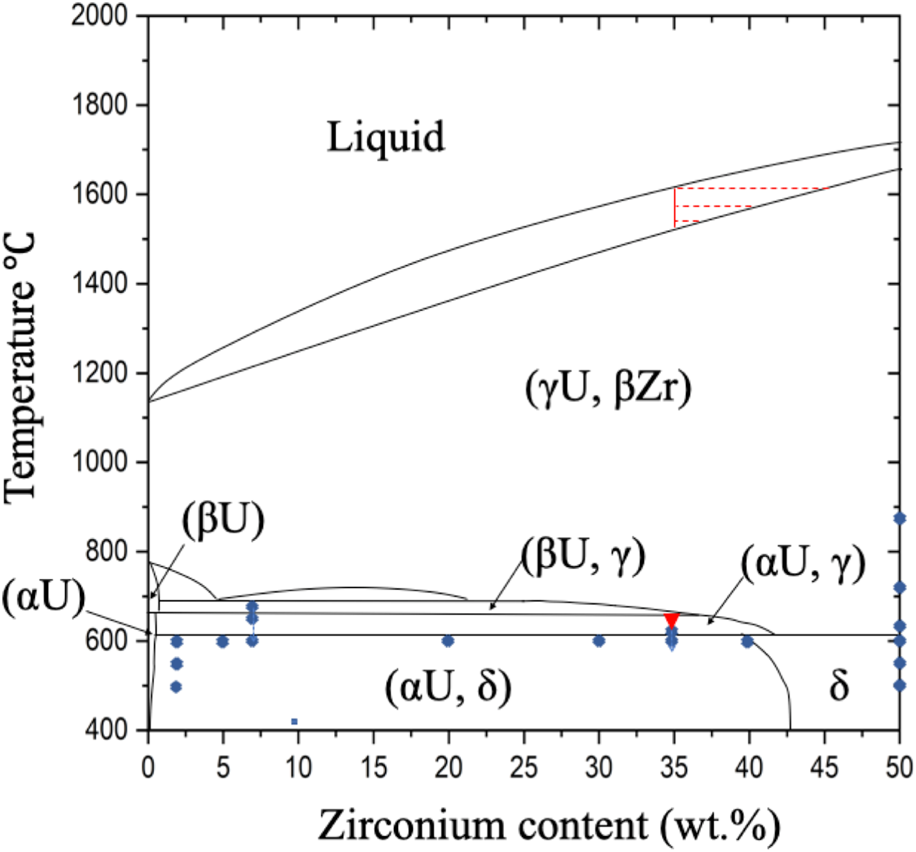
Figure 1: U–Zr binary phase diagram replotted from Ref. Reference Delage, Carmack, Lee, Mizuno, Pelletier and Somers4. The blue dots represent prior microstructure study temperatures and compositions [Reference Basak, Prasad, Kamath and Prabhu8, Reference Basak, Prabhu and Krishnan9, Reference Park, Shin, Han, Song, Choi and Park12]. The red triangle represents this study's annealing conditions.
Accurate phase equilibria determination uncertainties remain [Reference Xiong, Xie, Shen and Morgan6]. This is especially true for the Zr-rich (αU, δ), (αU, γ), and (βU, γ) boundaries. As an example, Ahn pointed out several discrepancies between experimental measurements and the existing phase diagram while studying the solid phase transformation behavior in the (βU, γ) phase using differential scanning calorimetry and thermogravimetric analysis [Reference Ahn, Irukuvarghula and McDeavitt7]. One of the many solid phase boundary determination challenges include intrinsic sluggish phase transformation kinetics preventing U–Zr alloy phase equilibrium. One U–35 wt.% Zr alloy sluggish kinetics example includes Basak [Reference Basak10] finding zirconium supersaturation in α-U precipitates after 500-h soaking at 630 °C and 72-h annealing at 600 °C.
In this study, we investigated a U-35 wt.% Zr alloy annealed at 650 °C which is in the fuel operation temperature range of sodium-cooled faster reactor. Early-stage microstructure and chemical evolution from the as-cast conditions were assessed with short 1-h and 24-h annealing times. Besides providing scientific insights, this study also addresses the research need in understanding the homogenization process for cast metallic fuels [Reference Morais, Lopes and Schön13, Reference Morais, Lopes and Schön14, Reference Prabhakaran, Gardner, Joshi, Burkes and Lavender15, Reference Hu, Wang, Joshi and Lavender16]. Microstructural evolution, microchemical evolution, and phase transformation from the as-cast condition with shorter annealing times than previously done by other researchers are investigated.
Results and discussion
Quality sample surface finish was achieved by electrolytic polishing to allow reliable microstructure characterization. Samples electrolytically polished at room temperature and −20 °C, respectively, shown in Figs. 2(a) and 2(b) highlight the effects of electrolytic temperature in scanning electron microscopy (SEM) images. Three contrast initially seen in backscatter electron (BSE) micrographs were bright precipitates, dark features, and a gray matrix. Bright precipitates and gray matrix in BSE micrographs along with the phase diagram in Fig. 1 qualitatively suggest that α-U phase precipitated out of a δ-UZr2 or γ phase matrix. The bright contrast of the α-U precipitated in BSE micrographs come from electrons being backscattered by the high Z–U atoms. Pits left behind from etched precipitates display as dark features in Fig. 2(a) after electrolytic polishing at room temperature. With all other conditions held the same, room temperature electrolytic polishing etched about half the α-U precipitates, whereas cold (−20 °C) electrolytic polishing preserved α-U precipitates as seen in Fig. 2(b). Additionally, cold electrolytic polishing achieved minimal scratching and pitting while removing residual oxidation. The rest of this study uses cold electrolytic polished samples for consistency. More sample polishing details are elaborated in the methods section.

Figure 2: SEM-BSE micrographs of samples electrolytic polished at (a) room temperature and (b) −20 °C.
Earlier studies on metallic U–Zr sample preparation have not accounted for electrolytic polishing temperature's significant impact on sample preparation quality [Reference Kelly, Thoma, Field, Cerreta and Hackenberg17]. An electropolishing potential of 3–4 V versus traditional 20–30 V [Reference Eckelmeyer, Romig and Weirick18] is suggested by Kelly [Reference Kelly, Thoma, Field, Cerreta and Hackenberg17] to prevent etching or pitting around inclusions. Suggested electrolytic polishing removed sample oxide layers and scratches typically found after sample storage and mechanical polishing. However, fine α-U precipitate removal observed after room temperature electrolytic polishing suggests preferential α-U etching. The low sample dissolution rate associated with lower electrolytic polishing potential voltage led to nonsaturated sample surface uranium cation concentration. Under this scenario, the electrochemical reaction is chemical reaction dominate rather than mass transfer dominate [Reference Fujita, Enda and Morisue19]. A higher reaction rate of α-U phase over the matrix phase is expected because of a higher concentration of the more-active element uranium in α-U. Surface cation supersaturation and mass-transfer controlled mechanisms are favored by reducing the environment temperature from room temperature to −20 °C where the saturation concentration and diffusion coefficient of uranium cations are lower. The resulted diffusion-limited electrolytic polishing [Reference Yang, Wang, Tawfiq, Wei, Zhou and Chen20] minimizes the reaction rate difference between α-U precipitates and matrix. Fine α-U precipitate preservation achieved is presented in Fig. 2(b).
An X-ray diffraction (XRD) pattern for the 24-h annealed sample presented in Fig. 3 identifies and labels characteristic peaks for the α, γ, and δ phases. The residual profile in the bottom of Fig. 3 illustrates good fitting with Rietveld refinement for all the three phases. The weighted profile residual between the calculated and experimental pattern is 15.2%, with an expected profile residual of 11.1%, determines an acceptable goodness of fit of 1.9 [Reference Asmi and Low21]. The three-phase weight fractions estimated following the Hill and Howard method [Reference Hill and Howard22] are listed in Table I.

Figure 3: XRD patterns and refinement for the 24-h annealed sample.
TABLE I: Rietveld refinement results for the XRD data on the sample annealed at 650 °C for 24 h.

It is known that Rietveld refinement of XRD patterns provides a semiquantitative analysis of the phase fractions. The small fraction of α-U phase precipitates qualitatively agrees to the phase diagram shown in Fig. 1 and the microstructural analysis presented later in this article. Most of the matrix phase being δ rather than γ suggests phase transformation took place during sample cooling after annealing at 650 °C. During the phase transformation from γ to δ, a metastable ω phase has been reported by Irukuvarghula when the sample was quenched from elevated temperatures [Reference Irukuvarghula, Ahn and McDeavitt11]. The ω phase has the same chemical composition with the δ phase but with a disordered hexagonal structure. To distinguish ω from δ phase via XRD is challenging because of their structural and chemical similarities. In this study, the samples were cooled through nature convention inside of a glovebox with a relatively slow cooling rate. Thus, it is reasonable to believe that more ordered δ phase formed through atomic diffusion compared to Irukuvarghula's study [Reference Irukuvarghula, Ahn and McDeavitt11]. Recent studies using transmission electron microscopy (TEM) also evidenced the formation of δ phase in a more rapid fashion than traditionally believed [Reference Janney, Papesch, Burkes, Cole, Fielding, Frank, Hartmann, Hyde, Keiser, Kennedy, Maddison, Mariani, Middlemas, O'Holleran, Sencer and Squires2].
Figure 4 presents the optical and SEM images for the samples annealed for 1 h in Figs. 4(a) and 4(b) and 24-h in Figs. 4(c) and 4(d). As seen in Fig. 4(a), the 1-h annealed sample displays a black dendritic microstructure characteristic of the solidification process. The comparison between the optical micrograph [Fig. 4(a)] and the BSE image [Fig. 4(b)] shows that the dendrites are precipitate free while high-density α-U precipitate clusters between dendrite branches. Most α-U precipitates have an elongated cross-sectional shape, with some of them showing a polygonal shape as seen in Fig. 4(b) inset. The 3-dimensional morphology of these precipitates is thus implied to be either needle-like or disc-like. After 24 h of annealing, the microstructure was homogenized as presented in Figs. 4(c) and 4(d). The dendritic microstructure disappeared with the α-U precipitates coarsened considerably.

Figure 4: Microstructure of the 1-h annealed sample in (a) optical micrograph and (b) SEM-BSE micrograph; and 24-h annealed sample in (c) optical micrograph and (d) SEM-BSE micrograph.
Such complex U–Zr dendritic microstructure as seen in Fig. 4 has not been reported by previous studies whose focuses have been on the near-equilibrium conditions [Reference Basak10]. The complex microstructure formed through a two-step mechanism. Firstly, microsegregation took place during solidification. As shown in Fig. 1, the melting temperature of U–Zr system increases with Zr concentration, so the solidification initialized with a higher Zr concentration in the solid phase than the liquid. Compositional gradients were observed with Zr-rich dendrite cores and Zr-lean dendrite boundaries. Dendrite Zr compositions as high as 45 wt.% Zr are expected, a composition so rich that it is in the single γ/δ-UZr2 phase indicated by red dashed lines in Fig. 1. Molten U-rich alloy pockets trapped between dendrite branches become uranium rich. Secondly, when the sample cooled down, the decomposition of the high-temperature γ phase primarily occurred within the interdendritic region with uranium supersaturation, leading to a high local density of α-U precipitates seen in Figs. 2(b) and 4(b). The dendrites remain precipitate free because they remain in a single-phase solid solution. α-U precipitates heavily decorate grain boundaries because grain boundaries are the last molten region during solidification.
Recently, dendrite formation in cast alloys of uranium–zirconium [Reference Zhang, Wang, Zeng, Wang, Jia, Sheng and Zhang23], uranium–zirconium–niobium [Reference Morais, Lopes and Schön13, Reference Morais, Lopes and Schön14], and uranium–molybdenum [Reference Prabhakaran, Gardner, Joshi, Burkes and Lavender15, Reference Hu, Wang, Joshi and Lavender16, Reference Chakraborty, Choudhuri, Somayajulu, Agarwal and Khan24, Reference Chakraborty, Choudhuri, Banerjee, Agarwal, Khan and Kumar25, Reference Jana, Sweet, Neal, Schemer-Kohrn, Lavender and Joshi26] has been reported to prevent microstructural homogenizition after thermomechanical treatments such as hot rolling [Reference Morais, Lopes and Schön13]. The microstructure evolution reported herein shows that the dendrites can be annealed out in U–35 wt.% Zr system at 650 °C for about 24 h, suggesting that annealing within the temperature range of (αU, γ) may be an effective strategy to homogenize the as-cast dendritic microstructures in U–Zr alloys.
Figure 5 presents BSE micrographs obtained at medium magnifications (3000–4000×) in (a and b), with their containing α-U phase identified by machine learning and presented in (c and d). Trainable Weka Segmentation [Reference Arganda-Carreras, Kaynig, Rueden, Eliceiri, Schindelin, Cardona and Sebastian Seung27] machine learning was used and is elaborated more in the Methods section. Machine learning micrograph magnification played a significant role in precipitate recognition and was optimized. Machine learning classification improved with high magnification; however, sample heterogeneity makes use of a small area associated with high magnification impractical for capturing bulk properties. Low-magnification micrograph machine learning errors caused by finer precipitates missed are because of low image resolution. High-magnification micrographs decrease the machine learning phase identification errors with the same image resolution because of precipitates having more pixels; however, missed larger features impact results significantly. The area fractions of α-U phase in U–35 wt.% Zr 1-h and 24-h annealed samples are 12 ± 2 % and 13 ± 1%, respectively. Precipitate sizes were assessed by measuring more than 200 precipitates for each sample. The average lengths were 0.3 ± 0.2 µm and 1.1 ± 0.7 µm for 1-h and 24-h annealed samples, respectively. Area fraction cannot determine volume fraction [28]; however, α-U precipitates coarsened with annealing without significantly changing the area fraction was found by means of machine learning–based microstructure analysis.

Figure 5: Original SEM-BES micrographs of U–35 wt.% Zr annealed for (a) 1 h and (b) 24 h. Machine learning–based precipitate recognition of U–35 wt.% Zr annealed for (c) 1 h and (d) 24 h. Trainable Weka Segmentation [Reference Arganda-Carreras, Kaynig, Rueden, Eliceiri, Schindelin, Cardona and Sebastian Seung27] classified α-U precipitates as black and γ/δ-UZr2 as white.
Sample elemental analysis was performed with energy dispersive X-ray spectroscopy (EDS). Sample EDS line scans for 1-h and 24-h annealed samples are presented in Fig. 6. Uranium Mα (3.171 kV), Lα (13.614 keV), zirconium Lα (2.044 keV), and Kα (15.775 keV) characteristic X-rays were excited and used for elemental quantification. α-U clusters had more average uranium content than the dendrite branches evident in Fig. 6(a) α-U precipitate clusters have a U-rich center with U-lean compositions on the fringe. A wider range of Zr composition was observed in the 1-h annealed sample as seen in Fig. 6(a) as compared with the 24-h annealed sample presented in Fig. 6(b). Matrix compositions are found to be U–28 ± 3 wt.% Zr and U–29 ± 2 wt.% Zr for 1-h and 24-h annealed samples, respectively. Improved local chemical homogeneity with annealing was indicated in the 24-h annealed sample by the smaller Zr composition standard deviation in the matrix phase.

Figure 6: (a) SEM-BSE micrograph and (b) EDS line scan results of the 1-h annealed sample. (c) SEM micrograph and (d) EDS line scan results of the 24-h annealed sample. The arrows in SEM micrographs indicate line scan distance and direction. Gray semitransparent boxes in line scan results indicate α-U regions in the line scans.
Although the nominal composition of the samples is U–35 Zr wt.%, the measured Zr contents in the EDS line scans presented in Fig. 6 are systematically lower than 35 wt.%. Besides the experimental error from EDS qualification, there are two possible explanations to the lower measured Zr content. Firstly, the impurity-stabilized α-Zr precipitates formed during the sample casting deplete Zr from the rest of the materials. Impurity-stabilized α-Zr is inevitable and often observed in uranium–zirconium metallic alloys [Reference Hirschhorn, Tonks, Aitkaliyeva and Adkins29]. In this study, the α-Zr inclusions are observed as the black features in BSE images in Figs. 5(a) and 5(c). A recent study by Hirschhorn analyzing the chemical composition of U–Zr alloys using mass spectroscopy and EDS showed that the Zr content measured by EDS is lower by >4 at.% because of the impurity-stabilized Zr inclusions not being counted by EDS [Reference Hirschhorn, Tonks, Aitkaliyeva and Adkins29]. Secondly, the small samples received were sectioned from a pin alloy of the U-35 wt.% Zr composition before being remelted. The sectioned samples were not subjected to chemical analysis to determine its composition before this work. Although nuclear fuel pins fabricated using the same method at Idaho National Laboratory generally show very good agreement between the nominal and real chemical compositions, it is possible that the small section of the pin alloy has a composition that is slightly off the alloy composition because of the inhomogeneity in the pin alloy. Nevertheless, the limited difference in chemical composition between the measured and nominal values is tolerable when comparisons were made between samples of the same material with different annealing times to show the short-term microstructural and microchemical evolution.
The chemical composition of the α-U precipitates was measured using EDS technique and presented in Fig. 7. The accuracy in composition determination as influenced by the incident electron energy and particle size is investigated by separately controlling these two variables. Figure 7(a) shows the wt.% of Zr determined by EDS with different electron energies on a α-U particle of 786 nm in width. The measured Zr content decreases with the electron energy. To further illustrate the effect of electron energy, electron scattering Monte Carlo simulation [Reference Joy30] was conducted to present the theoretical activation volumes of 10 and 20 keV electrons in α-U phase in Figs. 7(c) and 7(d). Figures 7(e) and 7(f) provide two examples of EDS patterns obtained from the same particle of submicron width with 10 and 20 keV electron energies, respectively. The Zr characteristic X-ray is negligible for 10 keV electrons but significant for 20 keV electrons. Considering the dependency of activation volume on electron energies, it is suggested that EDS composition measurement of a small particle with a lower electron energy tends to provide more accuracy because of the reduced influence from the surrounding matrix. For this study, an electron energy ≤10 keV is recommended.

Figure 7: (a) The Zr content decided by EDS point scan as a function of electron energy in an α-U precipitate of 786 nm in width. (b) The mean Zr content as a function of α-U particle width using ≤10 keV electron beams. (c, d) Monte–Carlo simulated activation volumes in α-U phase of 10 and 20 keV electrons, respectively. (e, f) EDS spectra of a submicron α-U particle obtained with 10 and 20 keV electron beams, respectively.
Figure 7(b) shows the wt.% of Zr in α-U particles of different sizes determined by EDS with ≤10 keV electrons. A trend of decreasing Zr content with increasing particle size is observed. It is believed that measurements from larger particles better reflect the true composition of the α-U phase because the activation volume of X-rays is more likely to be within the particle domains. Based on the trends shown in Figs. 7(a) and 7(b), the composition of the α-U phase was estimated by averaging multiple EDS point scans collected from particles with >1 µm using ≤10 keV electron energy. The Zr content in α-U precipitates in the 24-h annealed sample is determined accordingly to be 1.5 ± 0.6 wt.%.
The EDS-decided composition for α-U precipitates in this study qualitatively agrees with the equilibrium value (∼0.5 wt.%) at 650 °C. Comparing with the results from Basak [Reference Basak10], who observed α-U precipitates having as much as 11.6 wt.% of supersaturated Zr in U–35 wt.% Zr alloy after thermal annealing at 630 °C for 500 h followed by 240 h at 600 °C, the result herein indicates that a thermodynamic equilibrium can be approached, if not reached, in U–35 wt.% Zr within 24 h at 650 °C. Basak ascribed the supersaturation of Zr in α-U phase to the sluggish reaction kinetics associated with local thermodynamic equilibrium conditions within the (αU, δ) temperature range [Reference Basak10]. The sluggish kinetics has also been suggested by the absence of peaks from δ phase in XRD patterns in some 15–20 wt.% Zr alloys, as reviewed in Ref. Reference Janney, Papesch, Burkes, Cole, Fielding, Frank, Hartmann, Hyde, Keiser, Kennedy, Maddison, Mariani, Middlemas, O'Holleran, Sencer and Squires2. However, recent investigations using TEM showed that the nucleation of δ phase is actually fast [Reference Janney, Papesch, Burkes, Cole, Fielding, Frank, Hartmann, Hyde, Keiser, Kennedy, Maddison, Mariani, Middlemas, O'Holleran, Sencer and Squires2]. Without TEM analysis, this study suggests a nonsluggish reaction at 650 °C in reaching the equilibrium composition of the α-U phase. The significant coarsening of α-U precipitates during the course of annealing is an evidence of sufficient diffusion.
Conclusion
In this study, we investigated the crystallography, microstructure, and microchemistry for U–35 wt.% Zr alloys annealed at 650 °C for 1 h and 24 h. A modified electrolytic chemical polishing method was applied to the samples to preserve the fine α-U precipitates and reduce the pitting effects, while maintaining the surface smoothness, and remove sample surface oxidation. Two-phase microstructure of α-U and the matrix phase in both the 1-h and 24-h annealed samples was revealed by XRD, SEM, and EDS analysis. The uranium-rich precipitate phase was identified as α-U, whereas the matrix was identified to be δ and γ phases using Rietveld refinement on XRD patterns. Dendritic microstructure in the 1-h annealed sample remnant from as-cast was observed. The dendrites were slightly Zr rich without α-U precipitation, and the interdendritic regions were U rich with high density of α-U precipitates. After a short annealing of 24 h at 650 °C, the microstructure was homogenized accompanied by the coarsening of α-U precipitates and disappearance of the dendrites. The 24-h annealed sample showed a more homogeneous matrix chemical composition than the 1-h annealed sample. Careful EDS-based analysis of the α-U precipitates suggested a thermal equilibrium chemical composition of these precipitates after 24 h of annealing at 650 °C.
Methods
U–35 wt.% Zr alloy samples were melt-casted from high-purity depleted uranium and zirconium using an arc-melting furnace. The materials were flipped three times and remelted to ensure homogeneity. The samples were casted into 10-g buttons in a ceramic crucible and then cooled by natural convection in an argon-filled glove box. After that, the samples were wrapped with a Ta foil, placed inside an argon atmosphere furnace and annealed at 650 °C for 1 h and 24 h separately, and cooled again by natural convection in the glove box. The glove box contains less than 10 ppm of oxygen and water. Before sample characterization, standard metallographic preparations were adopted, and samples were mechanically polished to a mirror-like finish with 0.05-μm alumina suspension.
Surface oxides and fine scratches were removed from the samples by using electrolytic polishing technique. Recommendations of Kelly et al. [Reference Kelly, Thoma, Field, Cerreta and Hackenberg17] were followed with a modified electrolytic polishing temperature of −20 °C instead of room temperature. Electrolytic polishing conditions were 10% H3PO4 (phosphoric acid) methanol solution electrolyte, a 316 stainless steel cathode, the sample as an anode, and 4 V direct current, for 2–5 s. Before and after electrolytic polishing, the sample surface quality was examined using an IM-5000 metallurgical microscope to examine the removal of oxidation and scratches. Oxidation was minimized by analyzing samples within 30 min of being polished. When not used, the samples were stored in a vacuumed desiccator at −20 kPa gauge.
Microstructure characterization was conducted with an FEI QUANTA 600F SEM and an FEI QUANTA 3D SEM using backscattered electron (BSE) imaging and EDS. Elemental composition line scans were done with an EDAX EDS system fitted to the FEI QUANTA 600F SEM. Elemental composition point scans were done with an EDAX EDS system fitted to the FEI QUANTA 3D. The EDS systems were calibrated with standard samples including uranium. The accuracy was checked using a pyrite standard to have a nominal accuracy of ±1.5% absolute maximum. EDS scans were performed with electron beam energies no higher than 10 kV to excite the M uranium and L zirconium characteristic X-rays while maintaining a minimal excitation volume for maximum accuracy. Automatic background subtraction was applied to the EDS spectra followed by the Z-factor, Absorption factor, Fluoresce factor (ZAF) method to quantify the EDS results [Reference Goldstein, Newbury, Michael, Ritchie, Scott and Joy31]. ZAF method uses Z-factor, absorption factor, and fluorescence factor to adjust and correct for atomic number effects, X-ray absorption, and X-ray fluorescence, respectively. Simulating activation volume for EDS was done with EISS-Electron Scattering Monte Carlo Simulation Software [Reference Joy30]. One thousand trajectories were simulated with different beam energies for uranium and zirconium.
XRD was used to obtain crystallographic information of the samples. The XRD spectra were collected using a Bruker-AXS D8 Discover instrument with copper K-α1 X-rays. The scans were done with a step size of 0.01°, step dwelling time of 2 s, and within the 15°–80° 2θ range. The X-ray illuminated area is around 0.6 mm2, which is enough to provide representative results, given the level of microstructural inhomogeneity presented in Fig. 4. The X-ray penetration depth was estimated to be 8 µm for a normal incident beam based on the attenuation of electromagnetic radiation in U–35 wt.% Zr.
Rietveld refinement was conducted to obtain more information from XRD result. Based on the refinement results, the weight fractions of crystalline phases are calculated using the method proposed by Hill and Howard [Reference Asmi and Low21, Reference Hill and Howard22]:
 $${W_k} = {{{S_k}{{\left( {{\rm{ZMV}}} \right)}_k}} \over {\sum\limits_{i = 1}^n {{S_i}{{\left( {{\rm{ZMV}}} \right)}_i}} }}\quad ,$$
$${W_k} = {{{S_k}{{\left( {{\rm{ZMV}}} \right)}_k}} \over {\sum\limits_{i = 1}^n {{S_i}{{\left( {{\rm{ZMV}}} \right)}_i}} }}\quad ,$$where W k is the weight fraction of phase k, S is the Rietveld scale factor, Z is the number of formula units per unit cell, M is the mass of the formula unit, and V is the unit cell volume.
Acknowledgments
This work was supported by Oregon State University startup funding and the U.S. Department of Energy, Office of Nuclear Energy under DOE Idaho Operations Office Contract DE-AC07-05ID14517. Accordingly, the U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript or allow others to do so, for U.S. Government purposes.
Special thanks to Peter Eschbach, Teresa Sawyer for professional and expert training on SEM, and Brady Gibbons and Kyle Grove for assistance on XRD. We acknowledge OSU Electron Microscopy Facility for the access to SEM, and X-ray Diffraction Facility for the access to XRD. MHC thanks Jill Josselyn for the ARCS award. CZ acknowledges Oregon State University Provost's Distinguished Graduate Fellowship for financial support.









 Open access
Open access