



Introduction
Canine hookworms are important nematodes of dogs worldwide and are the causative agents of zoonotic cutaneous larva migrans (CLM) in humans. Of the three canine-adapted hookworm species in the United States, Ancylostoma caninum, Ancylostoma braziliense and Uncinaria stenocephala, A. caninum is the most common hookworm in dogs (Bowman et al., Reference Bowman, Montgomery, Zajac, Eberhard and Kazacos2010). Ancylostoma ceylanicum, a zoonotic hookworm endemic in other parts of the world has not been reported in the continental United States.
The prevalence of canine hookworms in the United States between 2017 and 2019, assessed by faecal flotation and antigen testing in dogs that presented to veterinarians for wellness visits, was 4.1%, whereas those that presented for other clinical visits were 4.2% (Sweet et al., Reference Sweet, Hegarty, McCrann, Coyne, Kincaid and Szlosek2021). Annual hookworm incidence in dogs across all states increased by 47% between 2015 and 2018 (Drake & Carey, Reference Drake and Carey2019), while some states such as Colorado had an increase of 137% between 2013 to 2017, likely due to dog movement and importation of infected dogs (Drake & Parrish, Reference Drake and Parrish2020). Even in large urban areas of the United States where 68.8% of dog owners reported providing parasite control medication to their pets, 7.1% of dogs randomly sampled in dog parks were infected with hookworms (Stafford et al., Reference Stafford, Kollasch and Duncan2020). Additionally, A. caninum isolates resistant to multiple anthelmintics are an emerging problem in United States’ greyhounds raised in racing kennels (Jimenez Castro et al., Reference Jimenez Castro, Howell, Schaefer, Avramenko, Gilleard and Kaplan2019; Kitchen et al., Reference Kitchen, Ratnappan, Han, Leasure, Grill, Iqbal, Granger, O'Halloran and Hawdon2019).
The transmission of hookworms begins when infective A. caninum third-stage larvae (L3) penetrate the skin of dogs and humans from dog-faeces contaminated environments. The intensity and life cycle of A. caninum infections in dogs depend on host age and immunity. In immunocompetent dogs, infection typically begins by ingestion of L3 larvae or skin penetration by L3 larvae. L3s that penetrate the skin migrate through the bloodstream, to the dogs’ lungs and trachea, where they are then coughed up and swallowed and make their way into the intestinal tract. L3s develop, mature and moult to fourth-stage larvae and later to adult males and females. A proportion of L3s undergo migration within the body to musculature or fat tissue, where they reside as ‘somatic larvae’ in an arrested state of development. Upon endocrine stimulation during the last trimester of pregnancy, these arrested larvae reactivate and can pass vertically to nursing puppies through the mammary glands (Burke & Roberson, Reference Burke and Roberson1985). Additionally, ‘larval leak’ may occur, a phenomenon in which reactivated somatic larvae migrate to the lumen of the intestine when existing populations of adult worms in the intestinal niche are eliminated. Thus, infection may be acquired by dogs from many sources. In dogs, infections result in loss of blood due to the haematophagous nature of the pre-adult and adult stages (Stassens et al., Reference Stassens, Bergum and Gansemans1996). Clinically, blood loss manifests as a spectrum of signs ranging from asymptomatic infections in adult dogs to peracute disease and death in neonates (Hill, Reference Hill1946; Miller, Reference Miller1968). Asymptomatic dogs pose the same level of risk as sources of human infections as symptomatic animals (Savilla et al., Reference Savilla, Joy, May and Somerville2011).
A few reports of A. caninum reaching patency in humans exist, albeit in a minor fraction of infections in tropical regions (George et al., Reference George, Levecke, Kattula, Velusamy, Roy, Geldhof, Sarkar and Kang2016; Furtado et al., Reference Furtado, Dias, Rodrigues, Silva, Oliveira and Rabelo2020). In a larger proportion of people, larval skin penetration by A. caninum produces epidermal migration of larvae which manifests as ephemeral papular, pustular, or follicular lesions (Diakou et al., Reference Diakou, Di Cesare and Morelli2019). CLM in humans is often associated with travel and/or close human–animal bonds, especially in areas where anthelmintics are not used in dogs (Heukelbach et al., Reference Heukelbach, Mencke and Feldmeier2002; Chris & Keystone, Reference Chris and Keystone2016). Variability in host and parasite factors have been hypothesized to play a role in the incubation period (Siriez et al., Reference Siriez, Angoulvant, Buffet, Cleophax and Bourrat2010). Parasite factors such as strain variability have been suggested to be related to pathogenicity in the related hookworm Necator americanus (Clements & Addis Alene, Reference Clements and Addis Alene2022). Understanding the genetic variability in parasite populations is crucial to elucidating genetic drift in populations, estimating the level of gene flow between and/or within populations, determining if cryptic species exist and explaining phylogeography (Ouborg et al., Reference Ouborg, Pertoldi, Loeschcke, Bijlsma and Hedrick2010; Cavallero et al., Reference Cavallero, Snabel, Pacella, Perrone and D'Amelio2013; Zhao et al., Reference Zhao, Lu and Li2022).
The mitochondrial cytochrome oxidase (cox1) is a more commonly used barcoding gene to understand population structure than nuclear loci such as internal transcribed spacer 1 and internal transcribed spacer 2 (Gasser et al., Reference Gasser, Cantacessi and Campbell2009). There is relatively little data available on the population structure and molecular epidemiology of A. caninum globally. In previous studies, seven haplotypes were identified from 38 adult A. caninum from Australia (Hu et al., Reference Hu, Chilton, Zhu and Gasser2002), 18 haplotypes were identified from 62 United States mid-Atlantic samples from Pennsylvania and North Carolina (Moser et al., Reference Moser, Carbone, Arasu and Gibson2007) and 30 haplotypes were identified from 160 adult worms from Brazil (Miranda et al., Reference Miranda, Tennessen, Blouin and Rabelo2008). United States samples from the other states have not been studied. Additionally, sequences from the previous United States study (Moser et al., Reference Moser, Carbone, Arasu and Gibson2007) are not available in GenBank or other publicly available databases for comparative studies.
The aim of the present study was to understand the haplotype distribution, diversity and phylogenetics of A. caninum populations isolated from naturally infected dogs in the United States using the partial mitochondrial cox1 gene and to compare it with those reported globally. We hypothesized that geographical isolation would drive haplotypic differentiation between A. caninum populations in the United States and other global regions. Based on the evidence of association of genetic structuring in parasites with host movement (Blouin et al., Reference Blouin, Yowell, Courtney and Dame1995), we hypothesized that A. caninum isolates within the United States would have population structure.
Materials and methods
Parasites
Faecal samples from naturally infected pet dogs were submitted with owner and veterinarian consent to the Kansas State Veterinary Diagnostic Laboratory between December 2020 and April 2022 for diagnostic parasitology testing. Ancylostoma caninum eggs were isolated from faecal samples using a double centrifugal faecal flotation technique. Briefly, approximately 5 g of the faecal sample was mixed with water, strained, sedimented with centrifugation and the supernatant was discarded. The sediment was then mixed with Sheather's sugar solution (Sp. gr. 1.275) and a centrifugal floatation was performed with a coverslip covering the tubes. All parasite stages observed on the coverslip were recorded. If Ancylostoma spp. eggs were present, the eggs were collected for inclusion in the study.
Three adult A. caninum samples were opportunistically obtained from samples submitted for diagnostic identification to Iowa State University College of Veterinary Medicine by veterinarians. All samples were anonymized for genetic studies, except for the state of origin and reported dog breeds, which were recorded as metadata. Samples for which the dog breed was not provided were coded as ‘unknown’. Samples used in the final genetic analysis were obtained from: Kansas (51 samples); New York (four samples); Iowa (three samples); Florida (one sample); and Massachusetts (one sample).
DNA extraction, amplification and sequencing of cox1
Eggs collected from coverslips (n = 63 samples) were washed with 1X phosphate-buffered saline and stored at 4°C until DNA extraction. Adults (n = 3 samples) were stored in 70% ethanol at room temperature until DNA extraction. DNA was extracted from whole adult worms or eggs suspended in a 200 μl volume using the Qiagen DNeasy Blood and Tissue kit (Valencia, CA) according to the manufacturer's instructions. Total genomic DNA was eluted in 100–200 μl of water and stored at −20°C.
An approximately 400 base pairs (bp) fragment of the cox1 gene was amplified using primers JB3 (5′-TTTTTTGGGCATCCTGAGGTTTAT-3′) and JB4.5 (5′-TAAAGAAAGAACATAATGAAAATG-3′) (Hu et al., Reference Hu, Chilton, Zhu and Gasser2002). Polymerase chain reaction (PCR) was carried out in a 25 μl volume with 2 μl of DNA, 1 × PCR buffer, 3 mm MgCl2, 200 μM of each dNTP, 200 nM of each primer and 0.3 units of Taq polymerase (GoTaq Flexi, Promega, Madison, WI). PCR conditions were 94°C for 5 min followed by 30 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 30 s, with the final extension of 72°C for 5 min. PCR reactions were analysed by agarose gel electrophoresis to confirm amplification. Amplicons were enzyme purified (Applied Biosystems, Thermo Fisher Scientific, Vilnius, Lithuania) and sequenced using Sanger sequencing technology (Eurofins Genomics, Louisville, KY). Sequences were analysed and contigs assembled with GeneStudio ver. 2.2.0.
Sequence analysis
Nucleotide Basic Local Alignment Search Tool searches were used to confirm species identity. Ancylostoma caninum nucleotide sequences from (Hu et al., Reference Hu, Chilton, Zhu and Gasser2002) and others from the same portion of the cox1 gene were obtained from GenBank for inclusion in the present study (table 1). For phylogenetic analyses, cox1 sequences of A. tubaeforme, A. duodenale and A. ceylanicum were also obtained from GenBank for inclusion. The cox1 sequences of A. braziliense were not available in the nucleotide database of GenBank for inclusion (accessed 19 December 2022).
Table 1. Summary of haplotype analysis of mitochondrial cytochrome oxidase (cox1) gene sequences obtained from the present study and sequences derived from GenBank for comparative analysis.

Multiple sequence alignment was performed with MAFFT (Katoh & Standley, Reference Katoh and Standley2013), trimmed to 380 bp in MegaX (Kumar et al., Reference Kumar, Stecher, Li, Knyaz and Tamura2018) for haplotypic and network analyses, and with Block Mapping and Gathering with Entropy (Criscuolo & Gribaldo, Reference Criscuolo and Gribaldo2010) for phylogenetic analysis. Haplotype diversity, nucleotide diversity and nucleotide difference analyses were performed in DnaSp ver. 6 (Rozas et al., Reference Rozas, Ferrer-Mata, Carlos Sanchez-DelBarrio, Guirao-Rico, Librado, Ramos-Onsins and Sanchez-Gracia2017). Geographical distribution of haplotypes was visualized by mapping using Datawrapper (Datawrapper, 2021) and Microsoft Excel ver. 16.63. A heatmap of breed distribution against haplotypes in the present study was created using plotly (Plotly Technologies, 2015). Median joining networks were generated in Population Analysis with Reticulate Trees (Leigh & Bryant, Reference Leigh and Bryant2015). Maximum likelihood phylogenetic analysis was performed using PhyML 3.0 (Guindon et al., Reference Guindon, Dufayard, Lefort, Anisimova, Hordijk and Gascuel2010) after the best substitution model was determined using Bayesian information criterion criteria in Smart Model Selection (Lefort et al., Reference Lefort, Longueville and Gascuel2017). The tree was visualized with Interactive Tree Of Life ver. 6 (Letunic & Bork, Reference Letunic and Bork2021).
Data availability
Unique haplotypic sequences generated in the present study have been deposited in the GenBank nucleotide database under the accession numbers ON980650–ON980674.
Results
Analysis of cox1 sequences reveals high haplotypic variability
An approximately 400 bp region of the cox1 gene was amplified from 57 of 63 A. caninum-positive faecal floats and from three of three A. caninum adult worms. The sequences obtained from the 60 samples were found to be distributed across 25 unique haplotypes (table 1, fig. 1a), with haplotype diversity of 0.904 (variance 0.00085 and standard deviation 0.029) and nucleotide diversity of 0.00886 (standard deviation 0.00086). Haplotype designations were provided to the sequences based on frequency of occurrence. The most frequent haplotype (frequency 0.28) was designated US Hap 1, the next most frequent was designated US Hap 2, etc. As shown in fig. 1a, samples from Kansas (n = 51) represented 22 haplotypes (haplotypic diversity: 0.886 ± 0.04). Sequences from Iowa (n = 3) belonged to the same haplotypes as sequences from Kansas (US Hap 3, US Hap 6 and US Hap 8; haplotypic diversity: 1.0 ± 0.27). Three of the sequences from New York represented two unique haplotypes (US Hap 11 and US Hap 14), while the other belonged to US Hap 1 (haplotypic diversity: 0.83 ± 0.22). Two sequences from Massachusetts and Florida (n = 1 each) belonged to a unique haplotype (US Hap 7).

Fig. 1. (a) Haplotype distribution of mitochondrial cytochrome oxidase (cox1) gene sequences from the present study. Haplotype diversity is represented as pie charts with analysis performed according to the state of origin of samples. Hd indicates the haplotypic diversity in each state.(b) Multiple sequence alignment of translated protein coding region of cox1 haplotypes. Consensus amino acid at each location is shown above the multiple sequence alignment. Each line of the alignment represents a haplotype, shown on the left (with GenBank accession number). Percentage of consensus amino acid at each location is shown at the bottom.
Global haplotype analysis was performed using 60 sequences from the present study, in addition to 11 sequences derived from the GenBank nucleotide database. Sequences from a study in Brazil (Miranda et al., Reference Miranda, Tennessen, Blouin and Rabelo2008) were derived from a different portion of the cox1 gene with no overlap with the sequences in Hu et al. (Reference Hu, Chilton, Zhu and Gasser2002) and the present study. Sequences from a previous United States study (Moser et al., Reference Moser, Carbone, Arasu and Gibson2007) were not available in GenBank or any other database for inclusion. Haplotype analysis revealed that the 71 sequences in the final dataset represented 35 haplotypes, with a haplotype diversity of 0.931 (variance: 0.00048 and standard deviation: 0.022) and nucleotide diversity of 0.01624 (standard deviation: 0.00303). Of 380 bp analysed, 59 sites were determined to be polymorphic (variable) with 63 mutations and 43 sites were determined to be parsimony informative. Most of the mutations were silent (synonymous) (fig. 1b), with two missense (non-synonymous) mutations occurring at amino acid 40 (US Haplotypes 23 and 24) and at 113 (US Haplotype 21).
The occurrence of haplotypes in the different dog breeds included in the present study is shown (table 1). A diverse set of haplotypes were recorded from greyhounds and unknown breeds, which were both overrepresented in the sample set. The most frequently occurring haplotype, US Hap 1, was not restricted to any breed. Unique haplotypes (US Hap 12–25) were overrepresented in greyhounds due to the sampling bias associated with opportunistic sampling.
Haplotype network analysis supports the presence of three clusters
A median joining network of the haplotype dataset (n = 35 sequences; each representing one haplotype) was constructed. Haplotypic nomenclature, geographical origin and number of sequences for each haplotype was added from previous studies when available, as summarized in table 1. Three distinct clusters could be visualized, with all haplotypes from the United States and four of the seven haplotypes from Australia forming a cluster (cluster A). Three haplotypes from Australia formed a distinct cluster (cluster B), which had an average nucleotide difference of 27.6 from cluster A. Fixation index (Fst) between haplotypes in clusters A and B was 0.80. Fst between all United States haplotypes and all Australian haplotypes (in both clusters A and B) was 0.249. A single GenBank sequence from China was distinct (designated cluster C) with an average nucleotide difference of 16.9 from cluster A. Clusters B and C had an average difference of 32.7 nucleotides. Fst between cluster C and other clusters could not be calculated because cluster C comprised a single sequence. Additionally, there is some evidence for a sub-cluster within cluster A formed by the haplotypes US Hap12, US Hap 18, Aus Hap 7 and Jap Hap1. Fst and average number of nucleotide differences between the sub-cluster population and all the other haplotypes of cluster A is 0.335 and 8.8, respectively. Taken together, these data suggest that there is moderate genetic structuring of global A. caninum populations.
Phylogenetic analysis supports the existence of genetic clusters
A maximum likelihood tree of unique haplotypes was constructed using the generalized time reversible GTR model for nucleotide substitutions with discrete gamma model parameters (gamma classes: four, shape parameter: 0.057) and bootstrap branch support (fig. 2). The Cooperia oncophora cox1 sequence (GenBank Accession: GQ888713) was used as the outgroup. All sequences of A. caninum formed a monophyletic clade with high statistical support (91%). Sequences from the United States in the study were in cluster A, which appeared monophyletic but with low statistical support (32%). Three haplotypic sequences from Australia formed a distinct cluster (cluster B) with high statistical support (96%), but this cluster was monophyletic with the United States isolates with 64% statistical support. The single cox1 sequence from China formed a distinct cluster (cluster C) but had low statistical support (<50%). The identity of A. caninum sequences from the present study as being distinct from the sequences of other Ancylostoma spp. was further highly supported by the monophyly of A. tubaeforme (100%), A. duodenale (84%) and A. ceylanicum (99%).
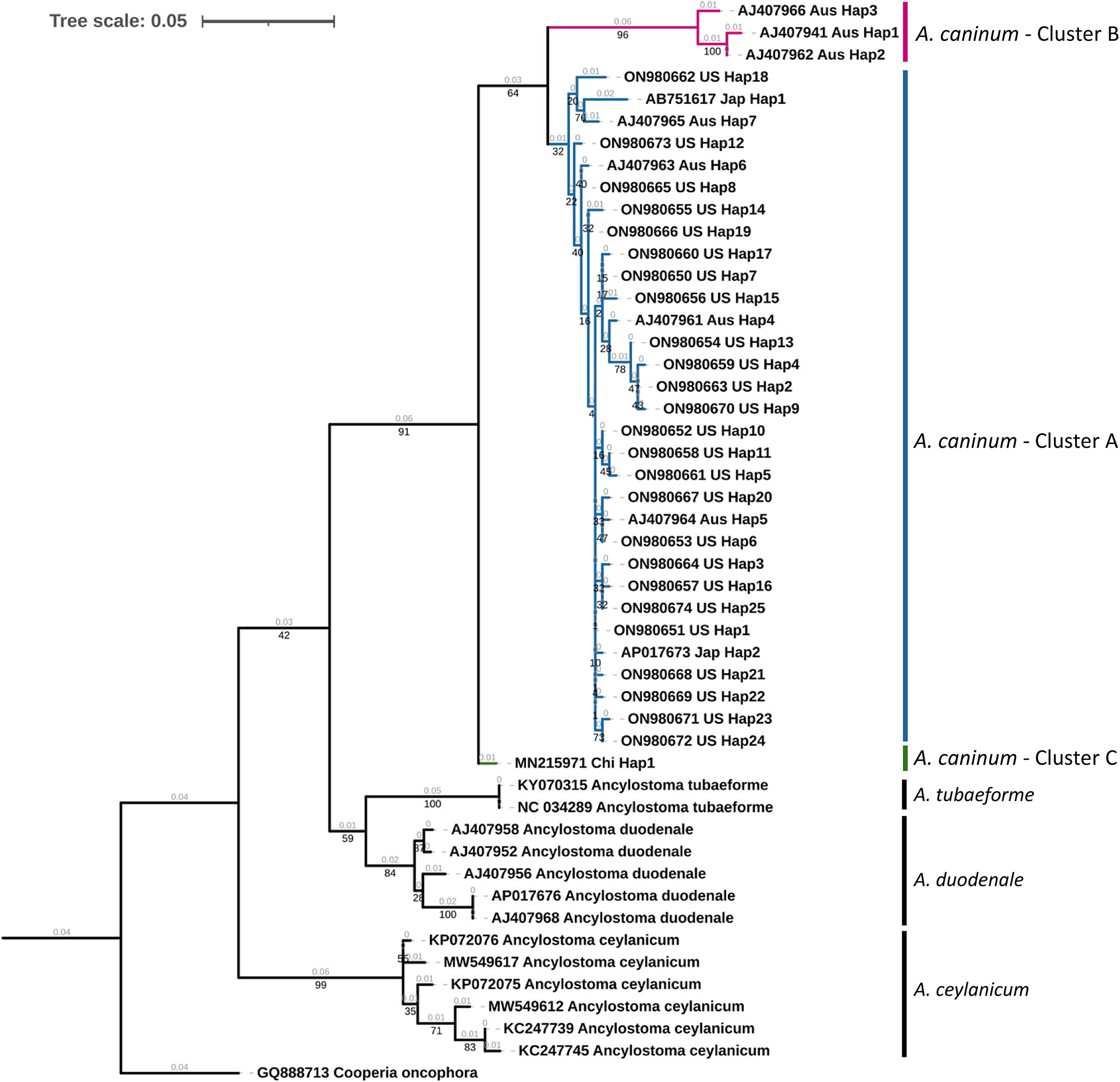
Fig. 2. Phylogenetic tree of Ancylostoma spp. A maximum likelihood tree of haplotypes generated from Ancylostoma caninum partial mitochondrial cytochrome oxidase (cox1) gene sequences in the present study and Ancylostoma spp. sequences derived from GenBank. Accession numbers, country code and haplotypic designation are given for each haplotype. Cooperia oncophora cox1, trimmed from the complete mitochondrial genome record (Accession number: GQ888713), was used as the outgroup. Cluster designations based on the haplotype network (fig. 3) are provided. Bootstrap values are represented in black under branches. Computed branch lengths are represented in grey above the branches.
Discussion
Ancylostoma caninum is one of the most common nematode parasites of dogs in the United States and has the potential to cause significant disease in young dogs. As one of the causative agents of human CLM, its prevalence in dogs, incidence, genetic variation and viability in the environment, among other factors are important from a One Health perspective. Human CLM can be reduced by proper anthelmintic administration to dogs, by removing dog faeces immediately after defecation after donning appropriate personal protective equipment and instituting hygiene practices. Understanding the molecular epidemiology in dogs is a crucial starting point for the control of the infection in humans since prevalence and differences in pathogenicity in CLM at the population level is unknown (Heukelbach et al., Reference Heukelbach, Mencke and Feldmeier2002). Yet, there is an important need for this information due to the reported increase in the annual incidence of infection in dogs (Drake & Carey, Reference Drake and Carey2019), which increases the risk of human infection. We hypothesized that due to dog movements/importation, A. caninum isolates within the United States would have population structure. To test these hypotheses, we examined A. caninum in dog isolates from the United States and characterized prevalent cox1 haplotypes.
Mitochondrial cox1 has been used as a barcoding marker in multiple parasite studies (Busi et al., Reference Busi, Snábel, Varcasia, Garippa, Perrone, De Liberato and D'Amelio2007; Small et al., Reference Small, Tisch and Zimmerman2014; Jesudoss Chelladurai et al., Reference Jesudoss Chelladurai, Murphy, Snobl, Bader, West, Thompson and Brewer2017; Zhao et al., Reference Zhao, Lu and Li2022). Ideally, molecular epidemiology studies should use published protocols if any exist (such as using the same primer sets) and must deposit sequence data generated in an easily accessible public database. This approach has resulted in a plethora of phylogeographical epidemiological data being generated for important zoonotic parasites such as Ascaris spp. and Echinococcus spp. (Cavallero et al., Reference Cavallero, Snabel, Pacella, Perrone and D'Amelio2013; Spotin et al., Reference Spotin, Boufana, Ahmadpour, Casulli, Mahami-Oskouei, Rouhani, Javadi-Mamaghani, Shahrivar and Khoshakhlagh2018). In the case of A. caninum, however, genetic data are scant due to unavailability of data from previous studies and this is complicated by using different primer sets by different research groups. The complete cox1 gene is 1577 bp long in the A. caninum reference mitochondrial genome (GenBank Accession: NC_012309). The primer set used by Hu et al. (Reference Hu, Chilton, Zhu and Gasser2002) flanks nucleotides 754 to 1146 of the cox1, while the primer set used by Miranda et al. (Reference Miranda, Tennessen, Blouin and Rabelo2008) flanks nucleotides 162 to 628 and the primer set used by Mulinge et al. (Reference Mulinge, Zeyhle and Mpario2021) flanks nucleotides 368 to 830. In the present study, the primers used by (Hu et al., Reference Hu, Chilton, Zhu and Gasser2002) and (Moser et al., Reference Moser, Carbone, Arasu and Gibson2007) were used for PCR amplification.
All haplotypes identified in the present study was exclusive to the United States. Our analysis revealed 35 unique A. caninum haplotypes currently reported globally and 25 unique haplotypes within the United States. To date, there has been a paucity of publicly available molecular data from A. caninum populations in the United States. Unique haplotypes were also found in a previous United States study (Moser et al., Reference Moser, Carbone, Arasu and Gibson2007), but the absence of publicly available data from that study makes direct comparisons impossible. Overall haplotypic diversity among United States samples in the present study (0.931; 60 samples) was higher than diversity at state level – 0.886 in Kansas (51 samples) and 0.833 in New York (four samples; fig. 1a). The higher diversity (1.0) reported from Iowa is attributable to the small random sample obtained for inclusion in the study (three samples). State level haplotype diversities from the present study were comparable to A. caninum diversity previously reported from Australia (0.80; 38 samples) (Hu et al., Reference Hu, Chilton, Zhu and Gasser2002), from North Carolina, United States (0.828; 54 samples) (Moser et al., Reference Moser, Carbone, Arasu and Gibson2007) and Brazil (0.88; 164 samples) (Miranda et al., Reference Miranda, Tennessen, Blouin and Rabelo2008). These diversities are significantly higher than the A. caninum diversity reported from Massachusetts, United States (0.25; eight samples; (Moser et al., Reference Moser, Carbone, Arasu and Gibson2007) and diversity of other soil-transmitted zoonotic nematodes such as Ascaris from United States (Iowa) populations (0.596; 100 samples; (Jesudoss Chelladurai et al., Reference Jesudoss Chelladurai, Murphy, Snobl, Bader, West, Thompson and Brewer2017). Additionally, high numbers of A. caninum haplotypes were recorded in greyhounds, a breed overrepresented in reports of anthelmintic resistance (table 1).
Median joining haplotype network analysis demonstrated a complex pattern (fig. 3). Ancylostoma caninum occurred in three clusters, which agrees with Moser et al. (Reference Moser, Carbone, Arasu and Gibson2007). United States’ haplotypes were found only in cluster A, while Australian haplotypes were found in clusters A and B. Cluster B currently only has haplotypes from Australia and cluster C has only one haplotype from China. Combined with the Fst values obtained, it can be concluded that there is moderate population structuring in A. caninum. The maximum likelihood tree provided high statistical support for haplotypes in cluster B (fig. 2). Due to the moderately high statistical support for the supercluster containing clusters A and B (64%), it is evident that the sequence in cluster C is distinct from clusters A and B. Given the high divergence of sequences in the clades, an alternate explanation could be that clades B and C are cryptic species.

Fig. 3. Median joining network of mitochondrial cytochrome oxidase (cox1) gene haplotypes from the present study (designated US Hap 1–25) and GenBank sequences (denoted by country, haplotype designation and accession numbers). Haplotype circles are coloured to represent unique geographical sources of the sequence and are scaled to represent the number of sequences belonging to each haplotype (the present study and Hu et al. Reference Hu, Chilton, Zhu and Gasser2002). Nucleotide differences are denoted by hatch marks across the connecting lines with each mark representing a single nucleotide difference. Unlabelled dark circles represent inferred, unsampled nodes. Three clusters observable in the network are represented by grey boxes.
Haplotype differentiation and spread can be attributed to many factors, such as hybridization, introgression and retention of ancestral polymorphisms (Detwiler & Criscione, Reference Detwiler and Criscione2010). Dog movement and dog importation also play a role in the spread of zoonotic parasites (Wright et al., Reference Wright, Jongejan and Marcondes2020; von Rentzell et al., Reference von Rentzell, van Haaften, Morris and Protopopova2022). Rates of mutation that led to the formation of novel haplotypes is still relatively unknown in zoonotic helminths (Jesudoss Chelladurai et al., Reference Jesudoss Chelladurai, Murphy, Snobl, Bader, West, Thompson and Brewer2017).
Some limitations of the present study include a geographical sampling bias associated with the selection of A. caninum positive faecal samples sent to a state veterinary diagnostic laboratory. Since samples were opportunistically collected, clinical history was not considered as a factor in the analysis and often not provided by submitters. The use of Sanger sequencing allowed the capture of only the predominant cox1 haplotype in each sample. Additionally, the use of cox1 primers from Hu et al. (Reference Hu, Chilton, Zhu and Gasser2002) disallowed us from making comparisons with the dataset from Miranda et al. (Reference Miranda, Tennessen, Blouin and Rabelo2008). Furthermore, the lack of A. caninum gene sequences from across the world in GenBank and/or other public nucleotide databases led to a low global representation of A. caninum populations in the present study. These limitations can be overcome in future studies by prospective selection of dogs with adequate clinical history for inclusion in studies and by using deep amplicon sequencing to detect intra-sample variations in mitochondrial sequences.
Molecular tools can be useful for increasing our understanding of A. caninum, and therefore, CLM epidemiology. The usefulness of cox1 sequences amplified from eggs in describing infection epidemiology has been previously demonstrated with human hookworms (Monteiro et al., Reference Monteiro, Jaeger, Nunes, Calegar, Reis, Bacelar, Santos, Bóia and Carvalho-Costa2019). In the present study, we provide molecular characterization of A. caninum from dogs in the United States. Sequences from the present study are available in GenBank for future comparative analyses. Our results demonstrated that dogs harbour haplotypes of A. caninum unique to the United States. Future studies of A. caninum isolates from endemic regions across the world are essential to understand the genetic diversity of this parasite.
Acknowledgements
The authors thank Chance Kopsa, Dr Brian Herrin, Dr Kamilyah Miller and Dr. Cameron Sutherland for help with holding and storing of hookworm positive samples after diagnostic work was completed at the Kansas State Veterinary Diagnostic Laboratory.
Author contributions
Conceptualization: J.R.J.; Data curation: T.A.Q. and J.R.J.; Formal analysis: T.A.Q. and J.R.J.; Funding acquisition: J.R.J.; Investigation: T.A.Q., W.L.J., D.R., V.S., K.A.M., K.M., M.T.B. and J.R.J.; Project administration: J.R.J.; Resources: M.T.B. and J.R.J.; Supervision: J.R.J.; Visualization: T.A.Q. and J.R.J.; Writing – original draft: J.R.J.; Writing – review and editing: T.A.Q., W.L.J., D.R., V.S., K.A.M., K.M., M.T.B. and J.R.J.
Financial support
This work was supported by start-up funds provided to J.R.J.J.C. by the College of Veterinary Medicine, Kansas State University.
Competing interests
None.
Ethical standards
All protocols were approved by the Institutional Biosafety Committee at Kansas State University (IBC-1637-VCS).





 Open access
Open access