



Introduction
Tracheophilus cymbius (Diesing, 1850) Skrjabin, 1913, belongs to the family Cyclocoelidae (Stossich, 1902), and is parasitic in the trachea of ducks and other waterfowl, often causing mild clinical symptoms, including cough, malnutrition, weight loss and dyspnoea, but sometimes even causing death (Bisseru, Reference Bisseru1957; Tang & Tang, Reference Tang and Tang1978; Scott et al., Reference Scott, Rau and McLaughlin1982). Tracheophilus cymbius have been reported in Asia, Europe, America and Africa (Bisseru, Reference Bisseru1957; Gu et al., Reference Gu, Pan and Qiu1973; Tang & Tang, Reference Tang and Tang1978; Scott et al., Reference Scott, Rau and McLaughlin1982; Wang et al., Reference Wang, Qiu and Wang2004). Tracheophilus cymbius-infected domestic ducks and wild aquatic birds have also been widely recorded in China, including Fujian, Hebei, Ningxia, Jiangxi and Heilongjiang Provinces (Gu et al., Reference Gu, Pan and Qiu1973; Tang & Tang, Reference Tang and Tang1978; Wang et al., Reference Wang, Qiu and Wang2004). The prevalence of T. cymbius in domestic ducks ranges from 5.8% to 30% in Fujian Province, and from 2.6% to 37.8% in wild aquatic birds in Bai-Yang-Dian Lake, Hebei Province (Gu et al., Reference Gu, Pan and Qiu1973; Tang & Tang, Reference Tang and Tang1978). Tracheophilus cymbius is particularly common and prevalent in domestic ducks in Heilongjiang Province, causing severe clinical symptomology. Our research shows 5.4% (12/222) T. cymbius prevalence in Heilongjiang Province in 2003, and 22.3% (21/94) in 2017, with an infection intensity of 1–8 (Wang et al., unpublished data), causing significant economic loss to the duck industry. Therefore, effective control and prevention of this trematode is important to the poultry industry.
Traditional approaches for the identification and differentiation of parasites based on morphological features have long been used worldwide. However, morphological approaches have limitations in identifying and distinguishing closely related species (McManus & Bowles, Reference McManus and Bowles1996). Although the family Cyclocoelidae was first named over a century ago, the status of the family and several cyclocoelid species remains controversial and uncertain. This is particularly the case with the important tracheal fluke T. cymbius Skrjabin (1913), considering that Tracheophilus Skrjabin and Typhlocoelum Stossich were classified into two different genera based on testes shape (Skrjabin, Reference Skrjabin1913; Bisseru, Reference Bisseru1957). However, Joyeux & Baer (Reference Joyeux and Baer1927) recognized Tracheophilus Skrjabin and Typhlutimum Witenberg as synonyms of Typhlocoelum Stossich, 1902, and the two species were combined into the same genus, namely Typhlocoelum cymbius (Diesing, 1850) (syn. Tracheophilus sisowi Skrjabin, 1913, Typhlocoelum sisowi Skrjabin, 1913) and Typhlocoelum cucumerinum (Rudolphi, 1809) (Joyeux & Baer, Reference Joyeux and Baer1927; Bisseru, Reference Bisseru1957). Tang & Tang (Reference Tang and Tang1978) reported that T. cymbius and T. sisowi should be considered the same species based on life cycle, in spite of the two trematodes having one key difference in morphological features (T. cymbius with the posterior vitelline united, versus T. sisowi with a separated posterior vitelline) (Tang & Tang, Reference Tang and Tang1978). Scott et al. (Reference Scott, Rau and McLaughlin1982) used Typhlocoelum cucumerinum sisowi (Skrjabin, 1913) and Typhlocoelum cucumerinum (Rudolphi, 1809) metacercariae, identified by morphological observation, to infect wild waterfowl, and the results suggested that the two parasites are not separate species, and rather should be considered two separate sub-species (Scott et al., Reference Scott, Rau and McLaughlin1982).
Mitochondrial (mt) genome and nuclear ribosomal DNA (rDNA) sequences can effectively identify species of the parasite (Wang et al., Reference Wang, Wang and Zhao2011; Gao et al., Reference Gao, Zhang and Yang2017). However, only a partial Typhlocoelum sp. 28S ribosomal RNA (rRNA) sequence (KT956960) is currently available in GenBank, and no complete mt genome data for any parasite belonging to the family Cyclocoelidae is available. This is a crucial limitation into investigations of the systematics and phylogeny of this family.
Thus, the purpose of our study was to determine the complete T. cymbius mt genome, and to analyse the phylogenetic relationships of T. cymbius with other trematodes based on a dataset consisting of the concatenation of 12 protein-coding gene nucleotide sequences. The results will help resolve issues of taxonomy, population genetics and systematics within and beyond the fluke family Cyclocoelidae.
Materials and methods
Parasite and DNA extraction
Adult T. cymbius flukes were collected from the trachea of naturally infected ducks in Daqing, Heilongjiang Province, China. Specimens were washed in physiological saline, and then morphologically identified to the species level (Tang & Tang, Reference Tang and Tang1978). Next, the flukes were fixed in 70% ethanol and stored at −20°C until further use. Total genomic DNA was extracted from individual worms with a TIANamp Genomic DNA Kit (Tiangen, Beijing, China), according to the manufacturer's protocol.
Amplification and sequence analysis
The entire mt genome of a single T. cymbius specimen was amplified in seven overlapping fragments using primers (supplementary table S1) designed from relatively conserved regions of the mt genome nucleotide sequences of closely related species. Polymerase chain reaction (PCR) cycling conditions used to amplify the T. cymbius mtDNA genome were based on a previous report (Li et al., Reference Li, Qiu and Zeng2019). PCR products were sent to Sangon Biotech Company (Shanghai, China) for sequencing in both directions using the same primers.
Sequences were assembled and aligned manually against the complete Fasciola gigantica (NC024025) and Fasciola hepatica (NC002546) mt genome sequences to identify gene boundaries using the program DNAStar v. 5.0 (Burland, Reference Burland2000). Each protein-coding gene was translated into amino acid sequences using the trematode mt genetic code in MEGA X (Kumar et al., Reference Kumar, Stecher and Li2018). The secondary structures of the 22 predicted tRNA genes were estimated using the online program ARWEN (http://130.235.46.10/ARWEN/), and/or by visual identification combined with manual proofreading. The rRNA genes were identified by comparison with the mt genomes of closely related species.
Using the individual protein-coding genes of T. cymbius, Echinochasmus japonicus, F. hepatica and Echinostoma miyagawai, all members of Echinostomata, comparisons were made based on complete mt genome size, gene arrangement, A + T content, AT/GC-skew and nucleotide and amino acid sequence differences. Nucleotide and amino acid sequences differences were calculated using DNAStar v. 5.0 (Burland, Reference Burland2000). The AT-skew and GC-skew values in both coding and non-coding regions (NCRs) were calculated using the following equations: AT-skew = (A − T)/(A + T), and GC-skew = (G − C)/(G + C).
Phylogenetic analyses
Nucleotide sequences from the 12 protein-coding genes in the T. cymbius mt genome were concatenated and aligned with those from published trematodes mt genomes using MEGA X (Kumar et al., Reference Kumar, Stecher and Li2018). We used the Gblocks online server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) to exclude ambiguously aligned regions from the multiple nucleotide sequence alignment, specifying the ‘less stringent’ selection option (Castresana, Reference Castresana2000). We used 38 trematode mt genomes available in GenBank, in addition to our T. cymbius data, to create our phylogenetic datasets (see supplementary table S2). Gyrodactylus salaris (NC008815) was included as an outgroup. Phylogenetic trees were reconstructed using the Bayesian inference (BI) and maximum likelihood (ML) methods on the concatenated nucleotides sequences of the 12 protein-coding genes.
BI was performed using the mixed model in MrBayes v. 3.1.1 (Ronquist & Huelsenbeck, Reference Ronquist and Huelsenbeck2003) with 1,000,000 metropolis-coupled Markov chain Monte Carlo generations (Ronquist & Huelsenbeck, Reference Ronquist and Huelsenbeck2003). The first 250 trees were omitted as burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities. We used PAUP v. 4.0 Beta 10 (Swofford, Reference Swofford2002), with 100 random addition searches and tree-bisection–reconnection branch swapping, for the nucleotide ML analysis. ML bootstrap reliability estimates were calculated from 100 bootstrap replicates with ten random additions per replicate, also using PAUP (Swofford, Reference Swofford2002). Phylograms were drawn using Tree View v. 1.65 (Page, Reference Page1996) for all inferences.
Results and discussion
Tracheophilus cymbius mtDNA features
The whole T. cymbius mt genome (GenBank accession code MK355447) is a typical circular mtDNA molecule 13,760 bp in size (supplementary fig. S1). The mt genome contains 36 genes: 12 protein-coding genes (cox 1–3, nad 1–6, nad 4L, atp 6 and cyt b), 22 tRNA genes, two rRNA genes (rrn L and rrn S) plus one NCR (table 1 and supplementary fig. S1). All genes are transcribed in the anticlockwise direction. The T. cymbius mt genome gene arrangement is identical to those of F. hepatica, Clinostomum complanatum and some Opisthorchiidae species (Opisthorchis felineus, Metorchis orientalis and Clonorchis sinensis) (Le et al., Reference Le, Blair and McManus2001; Cai et al., Reference Cai, Liu and Song2012; Chen et al., Reference Chen, Feng and Chen2016; Na et al., Reference Na, Gao and Liu2016). The entire T. cymbius mt genome nucleotide composition is biased toward A and T, with an overall A + T content of 62.82% (table 2). There is a very low base C content (9.75%) in the T. cymbius mt genome. Furthermore, a 40 bp overlap between the nad 4L and nad 4 genes exists in T. cymbius mtDNA, which is consistent with most flukes, but longer than those of Schistosoma haematobium (28 bp) and Schistosoma japonicm (37 bp), and shorter than that of Schistosoma mekongi (64 bp) (Le et al., Reference Le, Blair and McManus2001, Reference Le, Nguyen and Nguyen2016; Littlewood et al., Reference Littlewood, Lockyer and Webster2006; Cai et al., Reference Cai, Liu and Song2012; Liu et al., Reference Liu, Zhang and Liu2016).
Table 1. Mitochondrial genome organization of Tracheophilus cymbius.

aa, amino acid; nt, nucleotide.
Table 2. Comparison of complete mtDNA among Tracheophilus cymbius and other Echinostomata species.
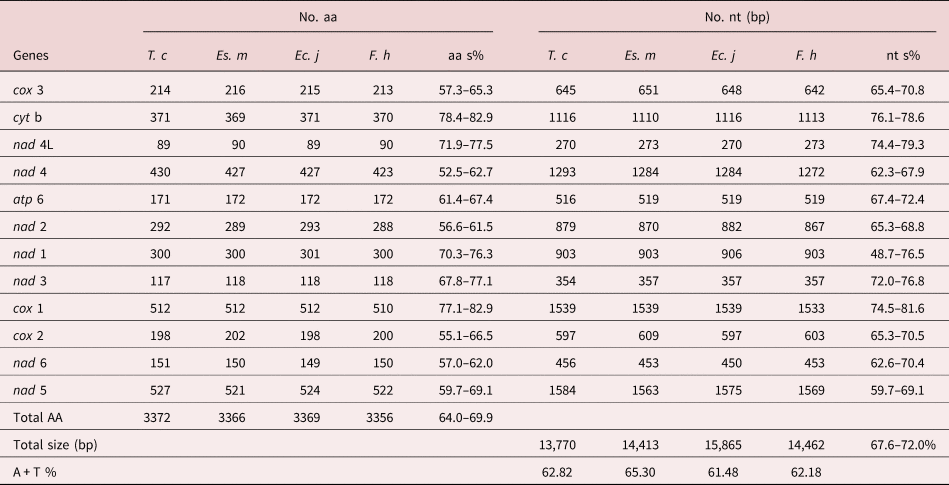
T. c, Tracheophilus cymbius; Es. m, Echinostoma miyagawai; Ec. j, Echinochasmus japonicus; F. h, Fasciola hepatica; nt s, nucleotides similarity; aa s, amino acid similarity.
The lengths of the 12 protein-coding T. cymbius mtDNA genes arrange in the order nad 5 > cox 1 > nad 4 > cyt b > nad 1 > nad 2 > cox 3 > cox 2 > atp 6 > nad 6 > nad 3 > nad 4L. A total of 3372 amino acids are encoded in the T. cymbius mt genome (table 2). Four genes (nad 1, nad 3, nad 4 and nad 5) use GTG as a start codon, eight genes (atp 6, cyt b, cox 1, cox 2, cox 3, nad 2, nad 4L and nad 6) use ATG as a start codon. Additionally, all genes have complete TAG termination codons (table 1). The most frequent codon is TTT (Phe) in the 12 protein coding genes, with a frequency of 12.5%. The least frequent codons are ACC (Thr), CGA (Arg) and AGC (Ser), all with frequencies of 0.12% (supplementary table S3).
The T. cymbius rrn L gene is located between trn T and trn C, and rrn S is located between trn C and cox 2. The lengths of the rrn S and rrn L genes are 760 bp and 985 bp, respectively (table 1). The A + T contents of the rrn L and rrn S are 61.93% and 59.61%, respectively. A total of 22 tRNA genes were identified in the T. cymbius mt genome, ranging from 63 bp to 76 bp in length (table 1). Twenty-one of the tRNA gene products fold into a predicted secondary structure with the conventional cloverleaf shape. The remaining trn S1 transcript has unpaired D-arms replaced by 7-bp loops (supplementary fig. S2). This is different than Echinostoma hortense, Es. miyagawai and F. hepatica, in which the two trn S transcripts both have unpaired D-arms (Le et al., Reference Le, Blair and McManus2001; Liu et al., Reference Liu, Zhang and Liu2016; Li et al., Reference Li, Qiu and Zeng2019). The T. cymbius mt genome NCR is located between trn G and cox 3, with a length of 142 bp (table 1).
Comparative mt genome analysis: T. cymbius and other closely related species
The complete T. cymbius mt genome is shorter than that in Ec. japonicus (15,865 bp), F. hepatica (14,462 bp) and Es. miyagawai (14,413 bp) (table 2). This length discrepancy is mainly due to the NCR lengths: 152 bp in T. cymbius, 931 bp in Es. miyagawai, 2342 bp in Ec. Japonicus and 806 bp in F. hepatica. Full-length mt genome nucleotide sequence identities range from 67.6% to 72.0% among the four Echinostomata trematodes (T. cymbius, Ec. japonicus, F. hepatica and Es. miyagawai) (table 2). Among them, the highest full-length nucleotide sequence identity (69.1%) is between T. cymbius and F. hepatica (70.7% nucleotide identity and 64.8% amino acid identity in the 12 protein-coding gene sequences) (supplementary table S4). The total A + T content (62.82%) is slightly higher than those of Ec. japonicus (61.48%) and F. hepatica (62.18%), but lower than that of Es. miyagawai (65.3%). ATG and TAG are the most frequently used initiation and termination codons, respectively, which is the case in many other trematodes, including F. hepatica, Es. miyagawai and Ec. japonicus (Le et al., Reference Le, Blair and McManus2001, Reference Le, Nguyen and Nguyen2016).
The AT/GC-skews in each mt genome gene or region of the four Echinostomata trematodes are listed in supplementary table S5. AT-skew values of the four trematodes are generally negative, and the majority of GC-skew values are positive. However, the AT-skew value for the T. cymbius NCR (−0.500) is lower than the other three (Ec. japonicus, F. hepatica and Es. miyagawai), which range from −0.386 to −0.214. GC-skew values in the four species have a large range of variation, with the lowest (0.238) in Ec. japonicus and the highest (0.676) in T. cymbius. Although some genes in T. cymbius and Ec. japonicus possess different skew values, the overall pattern of nucleotide skew in these two species is quite close.
Phylogenetic analyses
Phylogenetic analyses of the nucleotide sequences of the 12 concatenated coding regions from 39 trematode mt genomes were performed using two approaches (BI and ML).
The phylogenetic results of the two methods are somewhat different (See figs 1 and 2). The BI phylogenetic tree splits into two large clades. One clade contains seven members of Schistosomatidae, and the other clade contains 32 members of another 15 families (fig. 1). Interestingly, although the families Dipostomidae and Clinotomidae are both traditionally placed in the suborder Diplostomata, these species did not group with Schistosomatidae, which is also traditionally placed in the order Diplostomida. Rather, these species cluster basal within the order Plagiorchiida, paraphyletic to Diplostomida. This result is consistent with a previous study using mt sequences and ultra-conserved genomic elements to analyse the validity of the Diplostomoidea and Diplostomida (Locke et al., Reference Locke, Van Dam and Caffara2018). However, our result is inconsistent with another earlier study using nuclear small-subunit ribosomal and large-subunit ribosomal DNA sequences to analyse the phylogeny of Digeneans (Olson et al., Reference Olson, Cribb and Tkach2003). In the non-Diplostomata clade of our BI tree, trematodes of the suborder Pronocephalata form a clade basal to all the other suborders (just more recent than the aberrant Dipostomidae and Clinotomidae clade). In the Echinostomata branch, all of the Echinostomatidae species cluster together within the Echinostomatidae clade, except Es. hortense. Echinostoma hortense clusters with the Fasciola, Fascioloides and Fasciolopsis species used in our study. The result is consistent with a recent study by Li et al. (Reference Li, Qiu and Zeng2019), who used mtDNA sequences to analyse the phylogenetic relationships of Es. miyagawai with other species of Echinostomata (Li et al., Reference Li, Qiu and Zeng2019).
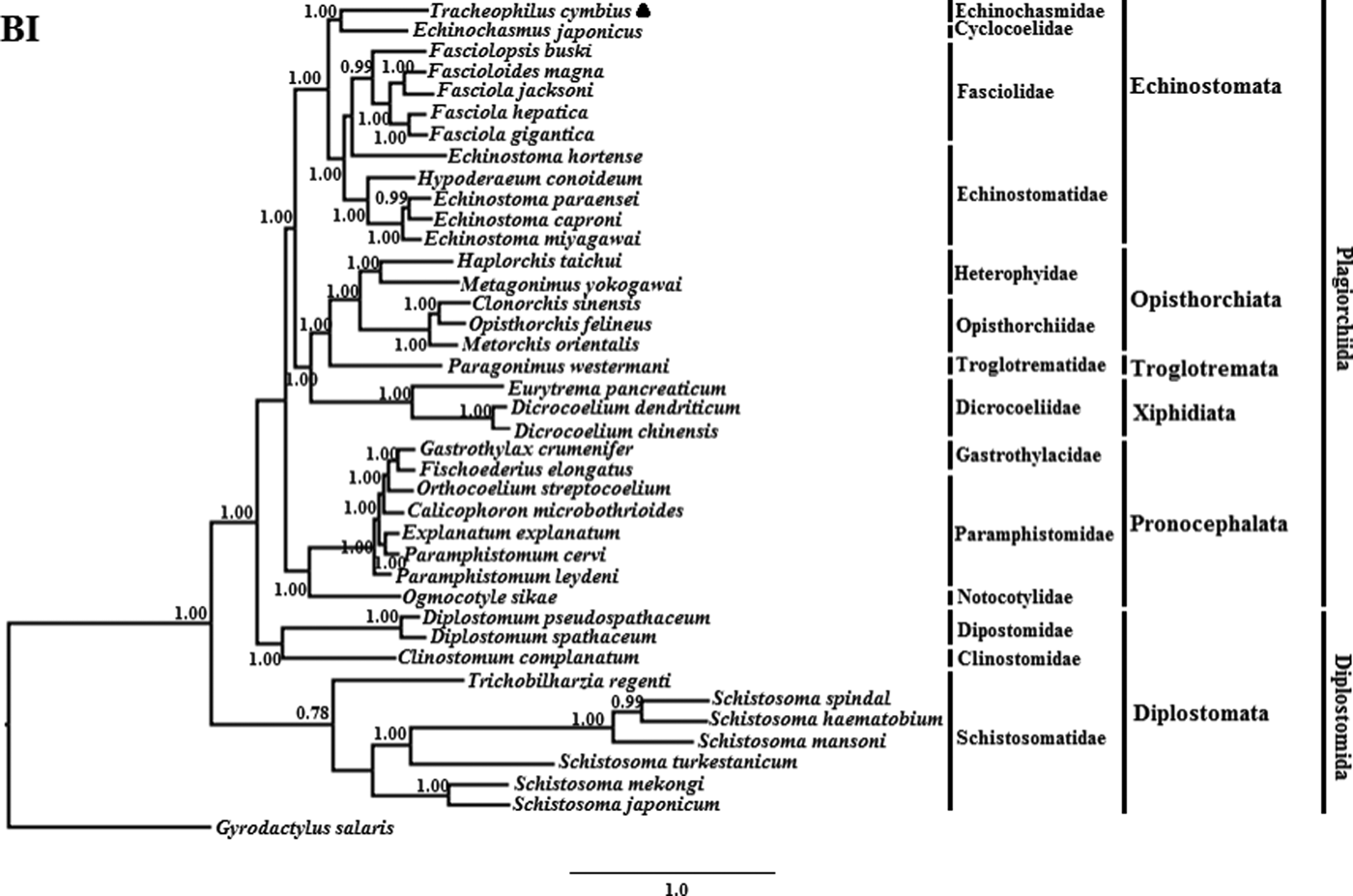
Fig. 1. Genetic relationships of Tracheophilus cymbius with other representative trematodes based on mitochondrial nucleotide data. Phylogenetic analysis based on the concatenated mitochondrial sequence data representing 12 protein-coding genes was conducted using Bayesian inference (BI), with Gyrodactylus salaris as the outgroup.
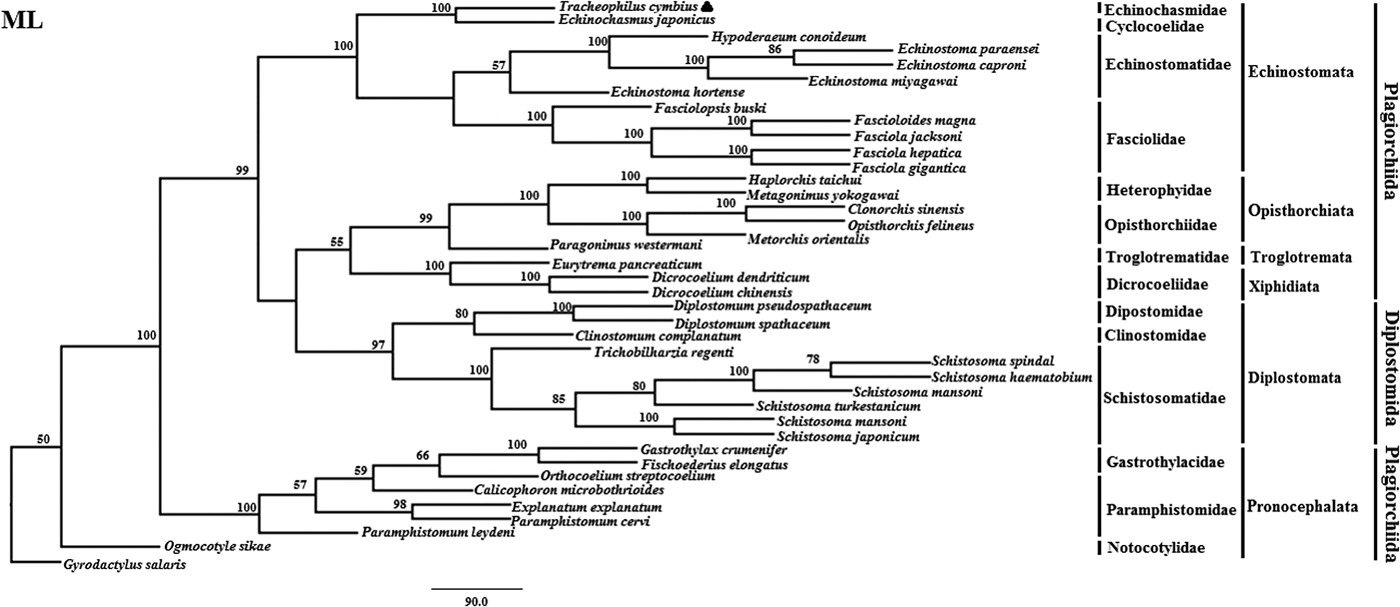
Fig. 2. Genetic relationships of Tracheophilus cymbius with other representative trematodes based on mitochondrial nucleotide data. Phylogenetic analysis based on the concatenated mitochondrial sequence data representing 12 protein-coding genes was conducted using maximum likelihood (ML), with Gyrodactylus salaris as the outgroup.
The ML phylogenetic (fig. 2) tree differs from the BI phylogenetic tree in a couple of key respects. Suborders Echinostomata, Opisthorchiata, Troglotremata, Xiphidiata and Pronocephalata form independent, monophyletic groups in both phylogenetic trees. Furthermore, the family Schistosomatidae forms a monophyletic clade in both phylogenetic trees, yet the suborder Diplostomata (Dipostomidae + Clinostomidae + Schistosomatidae) is an independent monophyletic clade in the ML tree and paraphyletic in the BI tree. Both methods' trees confidentially group T. cymbius and Ec. japonicus as sister taxa with high support values (BI 1.0, ML 100%), basal to the other Echinosomata. Both phylogenies show T. cymbius to be most closely related to Echinochasmidae, with a common ancestor basal to a clade containing Echinostomatidae and Fasciolidae. This is inconsistent with a previous study using the nuclear 28S rRNA gene (Tkach et al., Reference Tkach, Kudlai and Kostadinova2016) in which the Cyclocoelidae are more closely related to Echinostomatidae and Fasciolidae than to Echinochasmidae. Inconsistency between nuclear and mt phylogenies is not uncommon though, perhaps here due to a lower phylogenetic signal/noise ratio in the mt genomes of digeneans than in nuclear genomes, aggravated by the effect of incomplete taxon sampling (Philippe et al., Reference Philippe, Brinkmann and Lavrov2011; Locke et al., Reference Locke, Van Dam and Caffara2018).
In conclusion, the T. cymbius complete mtDNA genome sequence has been determined and reported for the first time. This represents the first mt genome available from any member of the family Cyclocoelidae. These data will provide novel mtDNA markers for studying the molecular epidemiology and population genetics of Cyclocoelidae and other trematodes.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X19000932.
Acknowledgements
The authors would like to thank Mr. Zhong Fu Wang who helped in the collection of trematodes, and Mr. Steven M. Thompson, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of drafts of this manuscript.
Financial support
This work was supported by a grant from the National Natural Science Foundation of China (grant number 31972703) and the National Key Research and Development Program of China (grant number 2017YFD0501300).
Conflicts of interest
None.
Ethical standards
This study was performed strictly according to the recommendations of the Guide for the Care and Use of Laboratory Animals of the Ministry of Health, China, and the protocol was reviewed and approved by the Research Ethics Committee of Heilongjiang Bayi Agricultural University, China.






