

Introduction
The experiences of parenting can cause stress related to work, life events, environment, marital relationships, daily hassles, and child rearing responsibilities [Reference Abidin1]. For this study, we define parenting stress as the stress that parents feel in their parenting role. Mothers of African Ancestry (AA) have higher levels of parenting stress than mothers of European Ancestry in the United States [Reference Nam, Wikoff and Sherraden2, Reference Cardoso, Padilla and Sampson3], which may contribute to adverse health outcomes in mothers and their children. Parenting stress among AA populations has been correlated with risk factors associated with health disparities observed in those of AA, such as hypertension. Parenting stress may contribute to overall maternal stress levels, influencing health outcomes of the mother and child similar to other types of psychological stress. In a previous study, we observed higher diastolic blood pressure in AA caregivers that report higher levels of parenting stress [Reference Taylor4]. A study by Hughes et al. reported that AA families experiencing higher levels of parenting stress are more likely to engage in uninvolved feeding styles with their children, which are associated with obesity in children [Reference Hughes5]. Although research evaluating the relationship of parenting stress to physiological outcomes in AA mothers and their children is limited, we hypothesized that parenting stress could be associated with epigenetic differences in stress response pathways, similar to those observed in other forms of psychological stress, and contribute to heath disparities observed in AA mothers and their children.
Epigenetic modifications to the genome do not alter the nucleotide sequence in DNA but are capable of altering gene expression and contributing to the pathogenesis of complex disease states [Reference Clark, Adamian and Taylor6]. Differences in DNA methylation, an epigenetic mark observed as a methyl group affixed to a cytosine residue in a cytosine-guanine dinucleotide, have been found in genes involved in stress response and inflammatory pathways in AA adults who experienced low socioeconomic status in childhood [Reference Needham7]. Stress related to finances has also been associated with increased DNA methylation at sites associated with accelerated epigenetic aging in AA women [Reference Simons8]. One recent study evaluated DNA methylation in adults relative to their perception of parenting and maternal stress during 3 developmental periods from their upbringing [Reference Naumova9]. The authors did not find any significant correlation between maternal stress and DNA methylation in adult children, although they did identify statistically significant associations between offspring’s perception of parenting from middle childhood to adulthood and the DNA methylation in offspring’s adult genomes. The authors also reported stronger associations with indicators of the child’s perceived parenting than with maternal reports of maternal parenting stress, suggesting that changes in perceived parental rejection have a stronger association with the offspring’s adult epigenome than measures of maternal parenting stress.
There has been extensive research investigating DNA methylation in children exposed to maternal stress in utero [Reference Monk, Spicer and Champagne10], although there is a gap in the literature evaluating the relationships among parenting stress and the epigenome of the mother or child. Schechter et al. evaluated DNA methylation at the nuclear receptor subfamily 3 group C member 1 (NR3C1) gene, a gene that encodes glucocorticoid receptor, related to post-traumatic stress disorder (PTSD) and parenting stress in mothers of young children (12–42 months old) [Reference Schechter11]. The authors suggest PTSD and parenting stress were negatively correlated with NR3C1 methylation, which could be associated with altered hypothalamic-pituitary-adrenal axis responsiveness. However, the authors noted that the study was not sufficiently powered to differentiate if the loss of DNA methylation at NR3C1 was associated with PTSD or higher levels of parenting stress independent of each other. Combined, the studies completed by Schechter and Naumova’s research groups suggest there may be differential DNA methylation in mothers and children associated specifically with parenting stress of the mother. Therefore, the purpose of this study was to examine the relationship among maternal parenting stress and epigenome-wide DNA methylation in AA mothers and their children.
Materials and Methods
Data for this study were collected from our ongoing study: The Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure (InterGEN) [Reference Crusto12, Reference Taylor13]. This sample contains 74 mother-child dyads (n=148) involved in our current prospective study examining the intergenerational impact of genetic and psychological factors on blood pressure. Extensive details on recruitment and statistical methods have been described elsewhere [Reference Crusto12, Reference Taylor13]. In brief, after approval from the institutional review board was obtained, mothers were recruited from the community who had a child between the ages of 3 through 5 years of age and would be followed for 2 years to evaluate factors that may contribute to variations in blood pressure in mothers and children. Baseline demographic data and the Parenting Stress Index—Short Form (PSI-SF) [Reference Abidin14] was obtained via audio self-assisted interviewing software. Saliva samples for analyses with the Illumina Infinium Methylation EPIC (850 K) BeadChip [Reference Pidsley15] were collected from the mother and child at the time of the interview. After 2 years of data collection, 81 mother-child dyads have completed baseline interviews and provided saliva for DNAm analysis. Six mothers did not complete the PSI during the interview and 1 child could not provide saliva at the time of the interview for DNAm analysis; these dyads were excluded from these analyses (n=148 complete cases). Four mothers had 1 missing value from a PSI subscale. The mean for the subscale of the missed item for each individual was imputed and used for analyses per PSI protocol manual [Reference Abidin14]. Quality control and preprocessing analytic pipeline of 850 K DNAm has been described elsewhere [Reference Crusto12]. All individual samples passed the quality control procedures. In the epigenome-wide association study (EWAS), a total of 847,155 autosomal CpG sites were included. After merging with phenotypic data, 74 African American mothers and 74 children were retained so that only complete cases were analyzed. Epigenome-wide association analyses were conducted among mothers and children separately. Among children, the effects of maternal stress on children’s DNA methylation were examined. Linear regression models were applied with DNAm β-values as dependent variables, and total PSI-SF score as independent variables. Age and maternal smoking status were adjusted for potential confounding effects. Sex was also adjusted in the analyses among children. A reference-free EWAS method was used to account for potential heterogeneity in cell proportions from saliva samples [Reference Houseman, Molitor and Marsit16]. Genomic control approach [Reference Devlin and Roeder17] adjustment was applied to address potential unmeasured confounding and inflation of type I error. Genomic control is a statistical method to measure the global deviation of a distribution of test statistics from a null distribution [Reference Devlin and Roeder17]. This method is not limited to a specific type of trait or study design, and does not require a case-control design. The method of genomic control has been widely applied to genome-wide association studies and EWAS of both quantitative and qualitative traits to measure and correct for the global inflation of low p-values to reduce false positive finding [Reference Pearson and Manolio18, Reference Shenker19]. False discovery rate was applied to account for multiple testing of epigenome-wide CpG sites. All analyses were conducted in the R statistical computing environment [20] and a selection of packages from Bioconductor.
Results
The sample population is comprised of self-identified African American/Black women and one of their biological children who are between the ages of 3 through 5 years old. Demographic data are listed in Table 1 and in online Supplementary Table S1. The mean age of our maternal participants was 32.4 years old and 24% indicated that they currently smoke cigarettes. The mean age of our children was 3.7 years old, and 35% of children were male.
Table 1 Descriptive statistics of study sample (n=74 dyads)
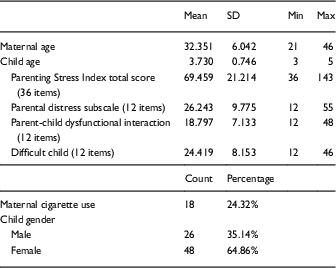
Parenting Stress Index items were scored on a 5-point Likert scale, with larger scores corresponding to increased parenting stress.
Significant variation in maternal DNA methylation at 95 CpG sites were associated with levels of parenting stress after genomic control (false discovery rate adjusted p<0.05) (Fig. 1). Notably, the relationship between parenting stress and DNA methylation was inversely proportional at most sites (83 out of 95 sites, 87%) and had decreased DNA methylation with higher levels of parenting stress (online Supplementary Table S2). We did not identify any epigenome-wide significant differences in DNA methylation in children related to the level of maternal parenting stress after genomic control. We compared the top 20 epigenetic associations with maternal parenting stress levels among children with the 95 significant CpG sites discovered among mothers. Summarized in Table 2, 4 CpG sites were found to meet epigenome-wide significant among mothers and were also marginally significant among children (p<5×10−7). We identified a change in DNA methylation associated with poly (ADP-ribose) polymerase-1 (PARP-1), which plays a key role in stress signaling. One of the CpG sites maps to 2 genes, kelch like family member 35 (KLHL35) and glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5). KLHL35, is involved in the ubiquitination processes, although its specific role in the process has not yet been characterized. GDPD5, contributes to glycerol metabolism and plays a role in neurogenesis and differentiation of neurons. The 2 remaining sites, cg14574088 and cg15575851, map to uncharacterized sites on the human genome. In addition, the observed DNA methylation patterns in the children are consistent with DNAm patterns observed in their mothers (Fig. 2).
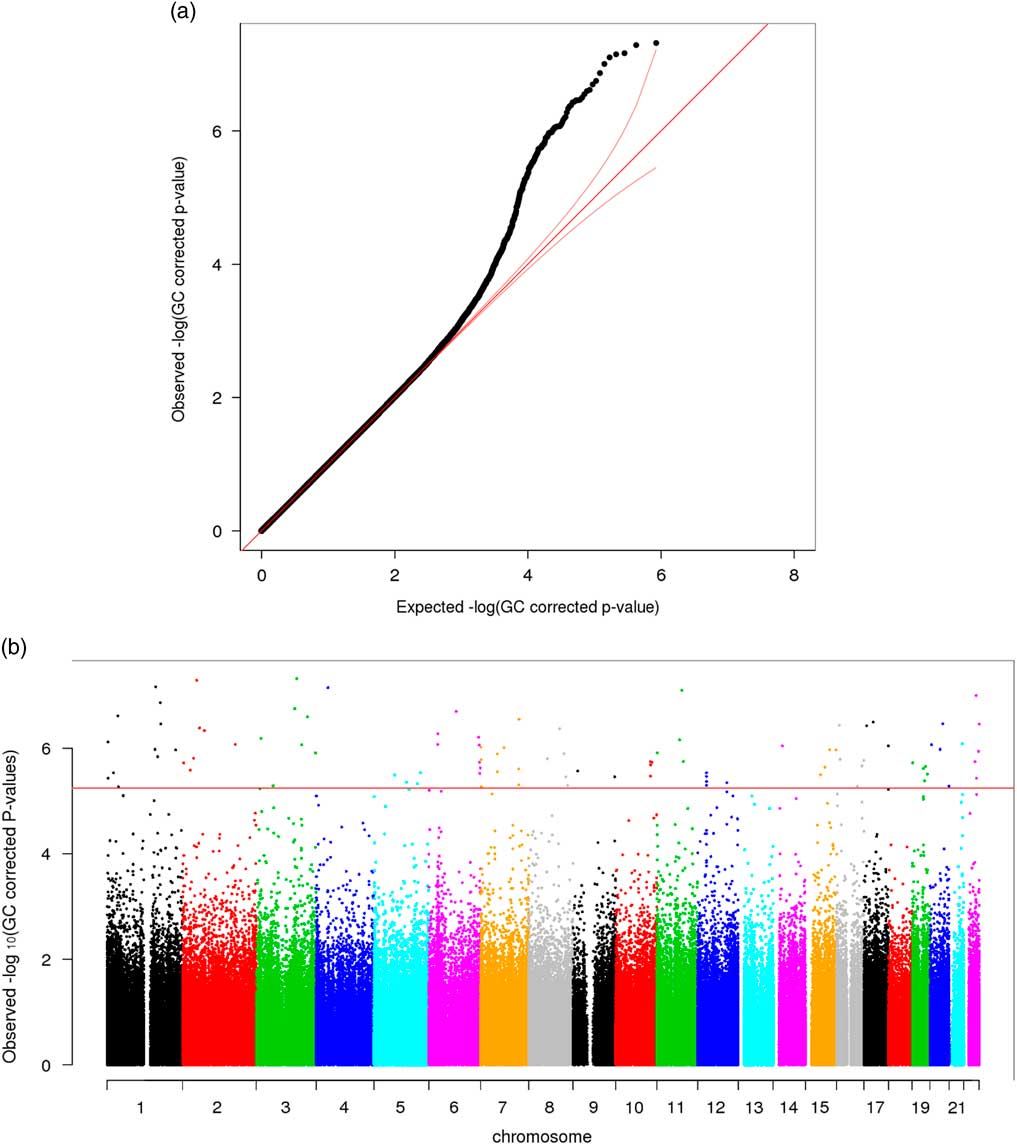
Fig. 1 (a) QQ plot—epigenome-wide association analysis with total parenting stress index score among 74 African Ancestry (AA) mothers [genomic control (GC) approach was applied on raw p-values]. (b) Manhattan plot—epigenome-wide association analysis with total parenting stress index score among 74 AA mothers (GC approach was applied on raw p-values; red line: false discovery rate adjusted p-value=0.05).
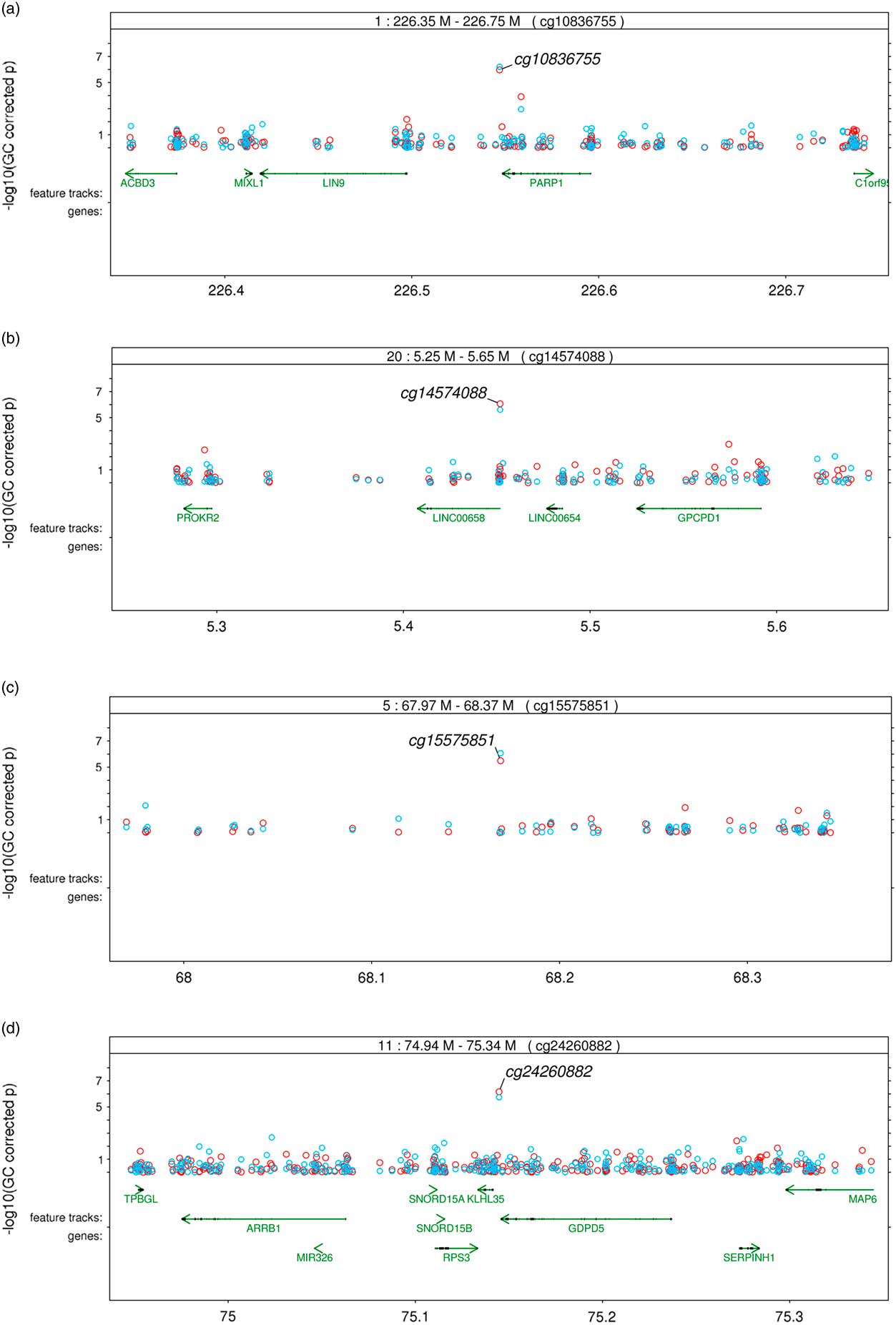
Fig. 2 Regional plots of common significant epigenetic associations with maternal parenting stress in African Ancestry mothers and children (red circles: epigenetic associations in mother; blue circles: epigenetic associations in children). (a) through (d) correspond to the CpG sites listed in Table 2; CpGs sites are listed at the top of each figure within the box annotating the location.
Table 2 Epigenome-wide associations with parenting stress identified among African Ancestry mothers and replicated among children
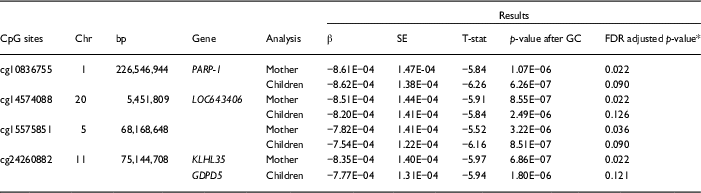
bp, base pair; GC, genomic control; FDR, false discovery rate.
* FDR adjustment applied after GC approach.
Discussion
Our study results suggest that levels of parenting stress are associated with differential DNA methylation in mothers, but not significantly in their young children. Of particular importance, we observed that higher levels of parenting stress was associated with hypomethylation in CpG sites located in the 3-UTR and gene body of PARP-1 (Fig. 2a ). In this study, we conducted an EWAS of parenting stress, in which we interrogated individual CpG sites to identify significant epigenetic associations based on test for single CpG site, rather than test for multiple associated CpG sites within a local genomic region (ie, differential methylation regions). We presented regional plots of 4 regions harboring significant epigenetic associations in both mothers and children. Only the PARP-1 region has additional epigenetic associations, not the other 3 regions. Multiple associations from the same region may increase the confidence of true epigenetic associations. However, single associated CpG site within a region does not indicate a false positive given the stringent significant threshold and replication between mothers and children. The density of the EPIC array, ~3.7 kb mean interval between CpG sites does not guarantee the coverage of co-methylated sites. In our experience, significant and replicated epigenetic associations can be either clustered or stand-alone. In the case of cigarette smoking, one of the most established epigenetic modifiers, AHRR region has multiple strongly smoking-associated CpG sites, and F2RL3 contains a single smoking-associated CpG site using 450 K chip [Reference Sun21]. To further explore the regional associations, we plotted all regions harboring significant association with parenting stress among mothers (online Supplementary Figure). Fifty out of 95 regions contain more than one suggestive associations (p<0.001).
Altered DNA methylation associated with PARP-1 warrants further follow up in human population as the gene is known to play a key role in cellular function and response to stress. Alterations in the PARP-1 gene have been associated with genome instability, shorter lifespans, and spontaneous tumors in animal models [Reference Luo and Kraus22]. Gene activity is often induced by DNA strand breaks and other conditions that signal the cell may come under stress, such as inflammation or changes in metabolic conditions. Up-regulation of PARP-1 can result in death of the cells by decreasing the amount of ATP and β-nicotinamide adenine dinucleotide (NAD+) available to carry out cellular processes [Reference Ha, Hester and Snyder23]. Furthermore, PARP-1 functions in the regulation of DNA methylation modifications and is necessary for reprogramming somatic cells [Reference Doege24]. PARP-1 is widely expressed when DNA damage or other threats to the cell are present and deficiencies in PARP-1 have been associated with increased levels of DNA methylation [Reference Doege24]. Since high levels of PARP-1 are associated with cell death, and extreme deficiencies of PARP-1 leads to reduced transcription of genes that help repair DNA damage in response to cellular threats, the production of PARP-1 must be maintained in a specific range for optimal functioning. Alteration in DNA methylation at the promoter site of PARP-1 has resulted in altered gene expression in laboratory settings [Reference Gao25, Reference Gong26]. Further studies are needed to determine if differences in DNA methylation at PARP-1 are associated with altered protein production or other measureable health effects in clinical populations. Since altered expression levels of PARP-1 are associated modification of DNA methylation and gene expression at other sites in the genome, differential methylation at this site in clinical populations may be precursor to other alterations that could have a lasting impact on future health outcomes.
Although the specific role of KLHL35 has not yet been identified in humans, others have observed differential methylation at CpG sites of the gene. Methylation at a cluster of CpGs near the second exon of KLHL35 was decreased in children that were born of older mothers [Reference Markunas27]. The researchers also determined that the differential methylation persisted into adulthood and was replicable in another smaller sample. Since the role of KLHL35 is not yet known, the biological significance of the observed changes in DNA methylation are unclear. Similar to Markunas’ [Reference Markunas27] results on maternal age at delivery, we identified a decrease in DNA methylation of KLHL35 with increased parenting stress. Conversely, hypermethylation of the KLHL35 sites has been associated with liver and renal cancers [Reference Morris28, Reference Shitani29]. It is unclear what health implications for hypomethylation associated with KLHL35 entail in our population or Markunas’ study. Although downstream protein production was not evaluated in our nor Markunas’ study, the observed DNA methylation mark may serve as a biomarker for parenting stress.
We chose to use saliva sampling in this study due to the ease of access and reduction of invasive procedures while working with young children. Our protocol produced high quality samples that passed QC protocols for the new EPIC array technology. One drawback to the use of saliva for DNA methylation analysis is the heterogeneity of DNA sources in saliva samples (eg, epithelial cells, leukocytes, and even foreign bacteria). However, a recent study comparing saliva and blood samples using the Illumina HumanMethylation450 BeadChip, found that DNA methylation levels in saliva may be more similar to brain tissue than blood, and the authors advocate for further exploration of the utility of saliva samples in psychiatric genetics [Reference Smith30]. Furthermore, our study detected differences in DNA methylation within the stress response pathway using saliva, a biofluid historically used to measure stress hormones such as cortisol. Future studies may choose to use saliva samples to concurrently measure DNA methylation and other biomarkers of stress with minimally invasive sample collection.
Because the study did not collect multiple samples for DNA methylation over time, it is unclear if the DNA methylation differences observed were a direct result of parenting stress or were preexisting differences that contribute to the mother’s response or perceived level of parenting stress. We also did not collect protein samples and are unable to evaluate if the differences in DNA methylation observed related to parenting stress are associated with differences in expression of PARP-1 or KLHL35. Since transcribed levels PARP-1 that are too high and too low have been associated with negative outcomes in laboratory models, it will be important to evaluate implications in clinical settings related to health outcomes. Further research on the function of KLHL35 is necessary to determine if alterations contribute to changes in health or merely mark differences between groups.
Acknowledgments
This work was supported by the National Institutes of Health, National Institute of Nursing Research R01NR013520.
Disclosures
The authors have no conflicts of interest to declare.
Supplementary materials
To view supplementary material for this article, please visit https://doi.org/10.1017/cts.2018.3







 Open access
Open access