


1 Introduction
Known as the cornerstones of contemporary nanoscience and nanotechnology, materials at the nanoscale, or commonly called nanomaterials in short, have been in the spotlight of modern physical sciences over decades[Reference Rahman1–Reference Borisenko and Ossicini3]. This is mainly due to their unique physical and chemical properties compared to their bulk counterparts. As these properties hold great promise in a wide spectrum of technological applications, it is highly desirable and imperative to gain in-depth understanding of these properties at the fundamental level. The properties of nanomaterials are usually tuned by adjusting the size, shape, and composition/architecture (in terms of composite nanomaterials)[Reference Cao and Wang2]. Among others, the optical properties, including linear and nonlinear absorption and light scattering, are of particular importance owing to their high relevance to the overall functionality of nanomaterials[Reference Zhang4].
The static spectroscopic techniques such as linear UV–Vis absorption, steady-state photoluminescence (PL), Raman, and some x-ray-based ones are indispensable for characterizing the optical properties of nanomaterials, as they can help to obtain basic information on the structure, size, shape, and surface/interface of nanomaterials[Reference Zhang4, Reference Kumar5]. Nevertheless, in order to look into and better understand the structure–function interplay as well as the pertinent working mechanisms in nanomaterials, one has to step further to glean dynamic information about, e.g., the lifetime of charge carriers or excitons and their associated relaxation pathways[Reference Othonos6–Reference Wu, Zhu and Lian10]. These key, mechanistic insights into the dynamic evolution of nanomaterials can in turn offer valuable guidance to the rational design, synthesis, and implementation of nanomaterials[Reference Bai, Jiang, Zhang and Xiong9] for many photochemical and photophysical applications, such as photocatalysis, photovoltaics, photoelectrochemistry and organic light-emitting diodes.
Considering that the charge carrier or exciton dynamics in nanomaterials evolves on a very short timescale, typically ranging from tens of femtoseconds to a few nanoseconds[Reference Wheeler and Zhang8], one needs to resort to ultrafast optical means[Reference Hannaford11–Reference Berera, Grondelle and Kennis14] to ‘visualize’ the dynamic processes of interest. In this respect, time-resolved femtosecond pump–probe spectroscopy has become one of the most extensively adopted techniques since the advent of ultrafast laser systems that produce pulses with femtosecond duration in the early 1990s. Indeed, the importance as well as the robustness of time-resolved femtosecond pump–probe spectroscopy in unraveling the ultrafast dynamics in nanomaterials have been widely recognized, especially over the past decade[Reference Othonos6–Reference Wu, Zhu and Lian10].
In this mini-review, we first deliver a brief description of the basic principle of ultrafast transient absorption (TA) spectroscopy within the time-resolved femtosecond pump–probe configuration[Reference Berera, Grondelle and Kennis14] (Section 2) and then present an overview of the recent advances achieved in our laboratory[15] regarding the investigations of ultrafast dynamics and related mechanisms in several, representative nanomaterials by means of ultrafast TA spectroscopy (Sections 3–5). In Sections 3 and 4, we address the ultrafast dynamics involved in the as-synthesized graphene oxide (GO) and a typical metal–organic framework (MOF) structure, respectively. In Section 5, we address ultrafast dynamics involved in three, representative, more complex nanosystems, i.e., MOF@TiO2 (a core–shell nanostructure), Pd–TiO2 (Pd nanocrystals deposited on TiO2 supports) and K4Nb6O17–TFA (K4Nb6O17 nanosheet with trifluoroacetic acid, TFA, as molecular co-catalysts). In each case study, we attempt to convey instructive, consistent information about the involved microscopic processes, pathways, dynamics and interactions in the investigated nanomaterial complex systems.
2 Ultrafast TA spectroscopy
In a typical ultrafast TA measurement, a fraction of the ground-state sample (nanomaterials or molecules) is promoted to an electronically excited state by a femtosecond pump (or excitation) pulse. Subsequently, a much weaker probe pulse is sent through the sample, with a time delay  ${\it\tau}$ relative to the pump pulse, so as to follow the ensuing response of the photoexcited sample. It is worth digressing here for a while that the probe pulse can be either from a center-wavelength-fixed femtosecond laser source (like the pump pulse) or from a broadband white-light continuum (WLC)[Reference Alfano16, Reference Brodeur and Chin17]; the configuration using a WLC probe, as adopted in our laboratory[15], possesses much higher efficiency in data acquisition[Reference Ge, Zhang, Jiang, Geng, Jiang, Fan, Guo, Hu, Chen, Chen, Wang and Luo18] and hence represents the most popular pump–probe configuration in ultrafast spectroscopic characterizations nowadays. Thus, a differential TA spectrum (
${\it\tau}$ relative to the pump pulse, so as to follow the ensuing response of the photoexcited sample. It is worth digressing here for a while that the probe pulse can be either from a center-wavelength-fixed femtosecond laser source (like the pump pulse) or from a broadband white-light continuum (WLC)[Reference Alfano16, Reference Brodeur and Chin17]; the configuration using a WLC probe, as adopted in our laboratory[15], possesses much higher efficiency in data acquisition[Reference Ge, Zhang, Jiang, Geng, Jiang, Fan, Guo, Hu, Chen, Chen, Wang and Luo18] and hence represents the most popular pump–probe configuration in ultrafast spectroscopic characterizations nowadays. Thus, a differential TA spectrum (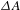 ${\it\Delta}A$) can be deduced by subtracting the absorption spectrum of the ground-state sample from the absorption spectrum of the excited sample. The
${\it\Delta}A$) can be deduced by subtracting the absorption spectrum of the ground-state sample from the absorption spectrum of the excited sample. The 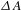 ${\it\Delta}A$ profile as a function of probe delay
${\it\Delta}A$ profile as a function of probe delay  ${\it\tau}$ and probe wavelength
${\it\tau}$ and probe wavelength  ${\it\lambda}$ can be registered by changing the
${\it\lambda}$ can be registered by changing the  ${\it\tau}$ between the pump and the probe. Normally, the obtained
${\it\tau}$ between the pump and the probe. Normally, the obtained 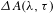 ${\it\Delta}A({\it\lambda},{\it\tau})$ is an ‘overlapping’ spectrum that consists of contributions from three, major processes, i.e., ground-state bleach (GSB), stimulated emission (SE), and excited-state absorption (ESA)[Reference Berera, Grondelle and Kennis14], as schematically illustrated in Figure 1.
${\it\Delta}A({\it\lambda},{\it\tau})$ is an ‘overlapping’ spectrum that consists of contributions from three, major processes, i.e., ground-state bleach (GSB), stimulated emission (SE), and excited-state absorption (ESA)[Reference Berera, Grondelle and Kennis14], as schematically illustrated in Figure 1.

Figure 1. Schematic illustration of the three, major contributions (i.e., GSB, SE, and ESA) to a typical 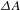 ${\it\Delta}A$ spectrum (at a certain time delay
${\it\Delta}A$ spectrum (at a certain time delay  ${\it\tau}$ between the pump and the probe), as marked in the left panel[Reference Berera, Grondelle and Kennis14], where the profile labeled ‘Sum’ (thickened solid line) represents the realistic ‘overlapping’
${\it\tau}$ between the pump and the probe), as marked in the left panel[Reference Berera, Grondelle and Kennis14], where the profile labeled ‘Sum’ (thickened solid line) represents the realistic ‘overlapping’ 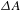 ${\it\Delta}A$ spectrum visualized by the spectrometer. The right panel depicts the corresponding, three processes monitored in a time-resolved pump–probe configuration (with a WLC probe, typically).
${\it\Delta}A$ spectrum visualized by the spectrometer. The right panel depicts the corresponding, three processes monitored in a time-resolved pump–probe configuration (with a WLC probe, typically).
According to the Pauli exclusion principle, the filling of quantized electronically excited states will lead to the bleaching of the corresponding optical transitions from the ground state to the excited state. In terms of a typical, photoresponsive semiconducting nanomaterial, the GSB signal basically reflects the population of electrons (holes) around the conduction (valence) band edge. Alternatively, the photoexcited electrons may return to the ground state through stimulated radiation induced by the probe light, yielding the SE signal. On the other hand, the photoexcited electrons in the excited state can also absorb the probe-light photons and get excited to certain higher levels of the excited state, generating the ESA signal. Generally, the ESA tracks the evolution of energized electrons in the excited state, while the SE accounts for the radiative recombination of energized electrons with holes. Apparently, in the 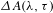 ${\it\Delta}A({\it\lambda},{\it\tau})$ spectrum the GSB and SE are negative while the ESA is positive in value. By analyzing the three types of
${\it\Delta}A({\it\lambda},{\it\tau})$ spectrum the GSB and SE are negative while the ESA is positive in value. By analyzing the three types of 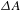 ${\it\Delta}A$ signals, one can follow in real time the charge carrier dynamics involved in nanomaterial systems[Reference Bai, Jiang, Zhang and Xiong9].
${\it\Delta}A$ signals, one can follow in real time the charge carrier dynamics involved in nanomaterial systems[Reference Bai, Jiang, Zhang and Xiong9].

Figure 2. (a) Schematic optical layout of a typical ultrafast TA setup that features a tunable pump (delivered by a femtosecond optical parametric amplifier) and a broadband WLC probe (generated by focusing a small portion of the Ti:sapphire regenerative amplifier output in a transparent nonlinear crystal). (b) The CDP ExciPro femtosecond pump–probe system that operates in our ultrafast spectroscopy laboratory at USTC[15].
Figure 2(a) depicts a typical, optical layout of ultrafast TA setup that features a tunable femtosecond pump and a broadband WLC probe, as it exists in our laboratory[15]. The ultrafast TA measurements are carried out on a modified pump–probe system (CDP ExciPro; Figure 2(b)) in combination with an ultrafast amplified laser system (Coherent). The strong pump pulses (adjustable pulse energy 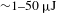 ${\sim}1{-}50~{\rm\mu}\text{J}$ at the sample cell) are delivered by an optical parametric amplifier (TOPAS-800-fs), which is excited by a Ti:sapphire regenerative amplifier (Legend Elite-1K-HE; center wavelength 800 nm, pulse duration 25 fs, pulse energy 3 mJ) seeded with a mode-locked Ti:sapphire laser (Micra 5) and pumped with a 1-kHz Nd:YLF laser (Evolution 30). The weak probe pulses are provided by a stable WLC that is generated by focusing the 800-nm beam (split from the regenerative amplifier by a portion of 10%) in a rotating CaF2 crystal or a Ti:sapphire crystal. The linear chirp of the WLC spectrum is routinely compensated with the aid of the cross-phase modulation signals recorded on a liquid hexane sample. The polarization of the pump and probe beams can be adjusted to meet different experimental requirements. The instrument response function is
${\sim}1{-}50~{\rm\mu}\text{J}$ at the sample cell) are delivered by an optical parametric amplifier (TOPAS-800-fs), which is excited by a Ti:sapphire regenerative amplifier (Legend Elite-1K-HE; center wavelength 800 nm, pulse duration 25 fs, pulse energy 3 mJ) seeded with a mode-locked Ti:sapphire laser (Micra 5) and pumped with a 1-kHz Nd:YLF laser (Evolution 30). The weak probe pulses are provided by a stable WLC that is generated by focusing the 800-nm beam (split from the regenerative amplifier by a portion of 10%) in a rotating CaF2 crystal or a Ti:sapphire crystal. The linear chirp of the WLC spectrum is routinely compensated with the aid of the cross-phase modulation signals recorded on a liquid hexane sample. The polarization of the pump and probe beams can be adjusted to meet different experimental requirements. The instrument response function is 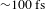 ${\sim}100~\text{fs}$, determined by cross-correlating the pump and probe pulses at the sample cell. Precise spatial overlap of the pump and probe beams (diameters
${\sim}100~\text{fs}$, determined by cross-correlating the pump and probe pulses at the sample cell. Precise spatial overlap of the pump and probe beams (diameters 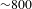 ${\sim}800$ and
${\sim}800$ and 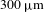 $300~{\rm\mu}\text{m}$, respectively) at the center of the 1.2-mm-thick sample cell (quartz) is attained by optimizing the
$300~{\rm\mu}\text{m}$, respectively) at the center of the 1.2-mm-thick sample cell (quartz) is attained by optimizing the 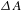 ${\it\Delta}A$ signals with the assistance of a laser beam analyzer (BG-USB-SP620, Ophir–Spiricon). The delay times between the pump and probe pulses are varied by a motorized optical delay line (minimum step
${\it\Delta}A$ signals with the assistance of a laser beam analyzer (BG-USB-SP620, Ophir–Spiricon). The delay times between the pump and probe pulses are varied by a motorized optical delay line (minimum step  ${\sim}1.6~\text{fs}$; maximum delay
${\sim}1.6~\text{fs}$; maximum delay 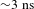 ${\sim}3~\text{ns}$). The delay time zero is determined in situ by cross-correlating the pump and probe pulses at the sample cell and also routinely cross-checked with some chemical samples such as the DCM and LDS698 dyes. A chopper operating at 500 Hz is used to modulate the pump pulses such that the
${\sim}3~\text{ns}$). The delay time zero is determined in situ by cross-correlating the pump and probe pulses at the sample cell and also routinely cross-checked with some chemical samples such as the DCM and LDS698 dyes. A chopper operating at 500 Hz is used to modulate the pump pulses such that the 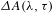 ${\it\Delta}A({\it\lambda},{\it\tau})$ spectra with and without the pump pulses can be recorded alternately. The WLC probe beam is first split into two tiny portions to synchronize the chopper and monitor the stability of the probe pulses using two separate photodiode detectors, and then separated into two parts (intensity ratio
${\it\Delta}A({\it\lambda},{\it\tau})$ spectra with and without the pump pulses can be recorded alternately. The WLC probe beam is first split into two tiny portions to synchronize the chopper and monitor the stability of the probe pulses using two separate photodiode detectors, and then separated into two parts (intensity ratio 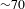 ${\sim}70$:30), with the 70% part focused on the sample cell and overlapped with the pump beam yielding a transmitted probe signal, while the 30% part focused onto another place of the sample cell to serve as a reference signal for achieving a best signal-to-noise ratio. The sample cell is mounted on a rotating stage so as to ensure that the photoexcited volume of the sample is kept fresh during the course of the measurements. The
${\sim}70$:30), with the 70% part focused on the sample cell and overlapped with the pump beam yielding a transmitted probe signal, while the 30% part focused onto another place of the sample cell to serve as a reference signal for achieving a best signal-to-noise ratio. The sample cell is mounted on a rotating stage so as to ensure that the photoexcited volume of the sample is kept fresh during the course of the measurements. The 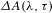 ${\it\Delta}A({\it\lambda},{\it\tau})$ spectral profiles are visualized by a 1024-pixel imaging spectrometer (CDP2022i) and further processed by the ExiPro 2.6 software equipped with the CDP pump–probe system.
${\it\Delta}A({\it\lambda},{\it\tau})$ spectral profiles are visualized by a 1024-pixel imaging spectrometer (CDP2022i) and further processed by the ExiPro 2.6 software equipped with the CDP pump–probe system.

Figure 3. (a) 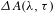 ${\it\Delta}A({\it\lambda},{\it\tau})$ spectra recorded on the highly oxidized, as-synthesized GO (
${\it\Delta}A({\it\lambda},{\it\tau})$ spectra recorded on the highly oxidized, as-synthesized GO ( $\text{C}/\text{O}\approx 2.7/1$) samples using a 570-nm pump and a WLC probe (490–530 nm for blue-shifted probing and 630–770 nm for red-shifted probing). (b) Schematic illustration of the band structure of the graphene–ORD model as well as the VB-hole blockade effect responsible for the type-I graphene-like dynamics observed in the blue-shifted probing regime. (c) A typical type-I kinetic trace extracted from the line cutting at 520 nm in (a). (d) A representative kinetic trace observed in pristine graphene[Reference Dawlaty, Shivaraman, Chandrasekhar, Rana and Spencer23] for comparison with (c). All the figures are adapted from Ref. [Reference Zhang, Zheng, Geng, Jiang, Ge, Fan, Duan, Chen, Wang and Luo20].
$\text{C}/\text{O}\approx 2.7/1$) samples using a 570-nm pump and a WLC probe (490–530 nm for blue-shifted probing and 630–770 nm for red-shifted probing). (b) Schematic illustration of the band structure of the graphene–ORD model as well as the VB-hole blockade effect responsible for the type-I graphene-like dynamics observed in the blue-shifted probing regime. (c) A typical type-I kinetic trace extracted from the line cutting at 520 nm in (a). (d) A representative kinetic trace observed in pristine graphene[Reference Dawlaty, Shivaraman, Chandrasekhar, Rana and Spencer23] for comparison with (c). All the figures are adapted from Ref. [Reference Zhang, Zheng, Geng, Jiang, Ge, Fan, Duan, Chen, Wang and Luo20].
3 Ultrafast dynamics in GO
GO is an attractive alternative for large-scale production of graphene, but its general structure is still under debate due to its complicated non-stoichiometric nature[Reference Loh, Bao, Eda and Chhowalla19]. In a recent work by Zhang et al. [Reference Zhang, Zheng, Geng, Jiang, Ge, Fan, Duan, Chen, Wang and Luo20], a set of well-designed femtosecond pump–probe experiments on as-synthesized GO was performed to extrapolate in situ the realistic domain structural information of GO. The contour map in Figure 3(a) shows a typical set of 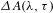 ${\it\Delta}A({\it\lambda},{\it\tau})$ spectra recorded on the highly oxidized, as-synthesized GO (
${\it\Delta}A({\it\lambda},{\it\tau})$ spectra recorded on the highly oxidized, as-synthesized GO (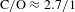 $\text{C}/\text{O}\approx 2.7/1$) samples using a 570-nm pump and a WLC probe. With respect to the pump at 570 nm, the WLC probe ‘scan’ was elaborately designed into two regimes, i.e., blue-shifted probing (490–530 nm) and red-shifted probing (630–770 nm), which distinguishes this work from the previous pump–probe studies on GO[Reference Kumar, Anija, Kamaraju, Vasu, Subrahmanyam, Sood and Rao21, Reference Kaniyankandy, Achary, Rawalekar and Ghosh22].
$\text{C}/\text{O}\approx 2.7/1$) samples using a 570-nm pump and a WLC probe. With respect to the pump at 570 nm, the WLC probe ‘scan’ was elaborately designed into two regimes, i.e., blue-shifted probing (490–530 nm) and red-shifted probing (630–770 nm), which distinguishes this work from the previous pump–probe studies on GO[Reference Kumar, Anija, Kamaraju, Vasu, Subrahmanyam, Sood and Rao21, Reference Kaniyankandy, Achary, Rawalekar and Ghosh22].
In the context of the newly proposed graphene–ORD model (ORD denotes the large-area, oxygen-rich domain in GO) whose corresponding band structure is schematically presented in Figure 3(b), it is expected that the localized impurity states at around the Fermi level (light-blue bar in Figure 3(b)) would not affect the carrier dynamics if a blue-shifted probing scheme is used. This is understandable because the pump-generated valence-band (VB) holes (including the low-energy tail of their Fermi–Dirac distribution inside VB) will set a blockade to effectively impede electron transfer from the localized impurity states to where the probe monitors (outside the blockade), as depicted in Figure 3(b). Such a blockade effect is automatically mandated by the momentum conservation as a result of the symmetrical, linear band structure of graphene despite a small band-gap opening in the graphene-like domain. In light of this expectation, the focus was placed on the probe scan in the blue-shifted regime.
As expected, a set of complete graphene-type transients (type-I) over a wide blue-shifted region of 490–530 nm (Figure 3(a), lower panel) was observed. As normally observed in pristine graphene, this type of 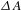 ${\it\Delta}A$ transients manifests itself as probe bleach, which is well understood as a consequence of the so-called phase-state filling or Pauli blocking effect. Figure 3(c) exhibits a typical kinetic trace probed at 520 nm (corresponding to the lower cut line in Figure 3(a)). The bi-exponential fit yields two recovery time constants –
${\it\Delta}A$ transients manifests itself as probe bleach, which is well understood as a consequence of the so-called phase-state filling or Pauli blocking effect. Figure 3(c) exhibits a typical kinetic trace probed at 520 nm (corresponding to the lower cut line in Figure 3(a)). The bi-exponential fit yields two recovery time constants – 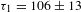 ${\it\tau}_{1}=106\pm 13$ fs and
${\it\tau}_{1}=106\pm 13$ fs and 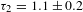 ${\it\tau}_{2}=1.1\pm 0.2$ ps – on the same timescales as their counterparts for graphene as well as reduced GO (rGO). As a comparison, a representative kinetic trace observed in pristine graphene[Reference Dawlaty, Shivaraman, Chandrasekhar, Rana and Spencer23] is also given (Figure 3(d)). Notably, the observed type-I kinetics turns out to be independent of the pump wavelength (500–650 nm), which nicely conforms to the fact that the aforementioned blockade effect (Figure 3(b)) should not depend on where the pump is applied as long as a blue-shifted probing scheme is used.
${\it\tau}_{2}=1.1\pm 0.2$ ps – on the same timescales as their counterparts for graphene as well as reduced GO (rGO). As a comparison, a representative kinetic trace observed in pristine graphene[Reference Dawlaty, Shivaraman, Chandrasekhar, Rana and Spencer23] is also given (Figure 3(d)). Notably, the observed type-I kinetics turns out to be independent of the pump wavelength (500–650 nm), which nicely conforms to the fact that the aforementioned blockade effect (Figure 3(b)) should not depend on where the pump is applied as long as a blue-shifted probing scheme is used.
This striking finding indicates that within the highly oxidized, as-synthesized GO does exist isolated graphene-like domain that essentially preserves the intrinsic electronic properties of graphene, thereby providing direct in situ evidence for the validity of the proposed graphene–ORD domain structural model of GO. Furthermore, rich dynamics (types II and III) were also mapped out with a red-shifted probing in 630–770 nm (Figure 3(a), upper panel), which involve a kinetics crossover behavior and pronounced electron–phonon interactions (detailed discussions omitted here)[Reference Zhang, Zheng, Geng, Jiang, Ge, Fan, Duan, Chen, Wang and Luo20].
To summarize, this ultrafast dynamics study on GO[Reference Zhang, Zheng, Geng, Jiang, Ge, Fan, Duan, Chen, Wang and Luo20] allows for reaching an important conclusion that the highly oxidized, as-synthesized GO consists of two types of domain, i.e., carbon-rich graphene-like domain and oxygen-rich domain, thereby creating a new understanding of the realistic domain structure and properties of as-synthesized GO as well as offering useful guidance for future applications based on chemically modified or functionalized graphene materials.
4 Ultrafast dynamics in MOF
MOFs have recently been extensively studied and demonstrated to promise important applications in many fields, especially in CO2 capture, thanks to their high porosity and well tunable interactions with CO2 molecules[Reference Sumida, Rogow, Mason, McDonald, Bloch, Herm, Bae and Long24]. It is also realized that the metal clusters in MOFs can behave as inorganic semiconductor quantum entities and the organic linkers can serve as antenna to activate these metal clusters upon photoexcitation, thus making ‘MOF-based photocatalysis’ possible. Given the versatile and tailorable characters of MOF structures, the utilization of solar energy by MOFs, especially in the Visible region, could be feasible by judiciously selecting metal ions and organic linkers. However, the current modulation of light adsorption over MOFs is limited to a narrow range by tethering the ligand with one or two additional amine groups[Reference Fu, Sun, Chen, Huang, Ding, Fu and Li25]. To efficiently harvest Visible light, it is critical to develop MOFs with absorption covering the Visible region as wide as possible.
In a very recent work by Xu et al. [Reference Xu, Hu, Wang, Li, Zhang, Luo, Yu and Jiang26], a type of broadband photoactive (200–800 nm), porphyrin-involved, semiconducting MOF, i.e., PCN-222 (also called MOF-545; Figure 4(a)) transformed from the ligand molecule of H2TCPP (H2TCPP denotes tetrakis(4-carboxyphenyl)porphyrin), was demonstrated to be capable of selectively capturing and further photoreducing CO2 into 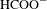 $\text{HCOO}^{-}$ (in the presence of triethanolamine, TEOA, as an sacrificial agent) with high efficiency under Visible-light irradiation. The greatly enhanced photoreduction performance of PCN-222 compared to its ligand H2TCPP (Figure 4(b)) is expected to originate from its high efficiency in electron–hole separation. Apart from the electron spin resonance (ESR) and PL characterizations, ultrafast TA spectroscopy was employed to verify this prediction.
$\text{HCOO}^{-}$ (in the presence of triethanolamine, TEOA, as an sacrificial agent) with high efficiency under Visible-light irradiation. The greatly enhanced photoreduction performance of PCN-222 compared to its ligand H2TCPP (Figure 4(b)) is expected to originate from its high efficiency in electron–hole separation. Apart from the electron spin resonance (ESR) and PL characterizations, ultrafast TA spectroscopy was employed to verify this prediction.

Figure 4. (a) Schematic of the three-dimensional network of PCN-222 that features large channels running through the  $c$-axis. (b) The amount of
$c$-axis. (b) The amount of  $\text{HCOO}^{-}$ produced as a function of the time of Visible-light irradiation over (i) PCN-222, (ii) H2TCPP, (iii) no PCN-222, (iv) no TEOA, and (v) no CO2. A Xe lamp was filtered to produce light in 420–800 nm. (c)
$\text{HCOO}^{-}$ produced as a function of the time of Visible-light irradiation over (i) PCN-222, (ii) H2TCPP, (iii) no PCN-222, (iv) no TEOA, and (v) no CO2. A Xe lamp was filtered to produce light in 420–800 nm. (c) 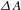 ${\it\Delta}A$ spectra of PCN-222 recorded at different probe delays (pump at 500 nm). (d) A representative
${\it\Delta}A$ spectra of PCN-222 recorded at different probe delays (pump at 500 nm). (d) A representative 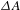 ${\it\Delta}A$ kinetic trace of PCN-222 probed at 430 nm. (e) Schematic illustration of the photoexcited dynamics involved in H2TCPP (left) and PCN-222 (right). All the figures are adapted from Ref. [Reference Xu, Hu, Wang, Li, Zhang, Luo, Yu and Jiang26].
${\it\Delta}A$ kinetic trace of PCN-222 probed at 430 nm. (e) Schematic illustration of the photoexcited dynamics involved in H2TCPP (left) and PCN-222 (right). All the figures are adapted from Ref. [Reference Xu, Hu, Wang, Li, Zhang, Luo, Yu and Jiang26].
In the TA measurements, the pump was set at 500 nm (i.e., 2.48 eV), a wavelength that is suitable for effectively photoinducing an interband transition in the semiconducting PCN-222 (bandgap  ${\sim}1.75$ eV). Figure 4(c) displays the
${\sim}1.75$ eV). Figure 4(c) displays the 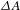 ${\it\Delta}A$ spectra recorded at different probe delays (0.2–3000 ps), showing negative GSB signals given that the WLC probe (400–490 nm) is blue-shifted relative to the pump (500 nm). As the WLC probe yields essentially the same
${\it\Delta}A$ spectra recorded at different probe delays (0.2–3000 ps), showing negative GSB signals given that the WLC probe (400–490 nm) is blue-shifted relative to the pump (500 nm). As the WLC probe yields essentially the same  ${\it\Delta}A$ kinetics in this spectral region, only a typical one taken at 430 nm is shown in Figure 4(d). Within the probe-delay limit of the pump–probe spectrometer (
${\it\Delta}A$ kinetics in this spectral region, only a typical one taken at 430 nm is shown in Figure 4(d). Within the probe-delay limit of the pump–probe spectrometer (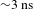 ${\sim}3~\text{ns}$), the
${\sim}3~\text{ns}$), the  ${\it\Delta}A$ recovery converges to an asymptote (dashed line in Figure 4(d)) with a non-zero value of roughly
${\it\Delta}A$ recovery converges to an asymptote (dashed line in Figure 4(d)) with a non-zero value of roughly 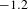 $-1.2$ mOD (OD denotes optical density). The multi-exponential fitting to this asymptotic recovery gives rise to two characteristic time constants:
$-1.2$ mOD (OD denotes optical density). The multi-exponential fitting to this asymptotic recovery gives rise to two characteristic time constants: 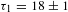 ${\it\tau}_{1}=18\pm 1$ ps (47%) and
${\it\tau}_{1}=18\pm 1$ ps (47%) and 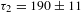 ${\it\tau}_{2}=190\pm 11$ ps (53%). Notably, the nearly perfect parallelism between the asymptote and the
${\it\tau}_{2}=190\pm 11$ ps (53%). Notably, the nearly perfect parallelism between the asymptote and the 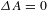 ${\it\Delta}A=0$ line (dashed-dotted line in Figure 4(d)) suggests that the eventual recovery to
${\it\Delta}A=0$ line (dashed-dotted line in Figure 4(d)) suggests that the eventual recovery to 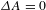 ${\it\Delta}A=0$ (a process that should take place) features an extremely long lifetime (
${\it\Delta}A=0$ (a process that should take place) features an extremely long lifetime (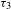 ${\it\tau}_{3}$), probably a few tens to hundreds of nanoseconds or even microsecond long. The two components in the picosecond domain (
${\it\tau}_{3}$), probably a few tens to hundreds of nanoseconds or even microsecond long. The two components in the picosecond domain (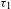 ${\it\tau}_{1}$ and
${\it\tau}_{1}$ and 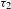 ${\it\tau}_{2}$) may reflect the electron dynamics associated with the electron trap states that are energetically located within the bandgap of PCN-222. Given that the lifetimes of such trap states (reflecting the electron-detrapping rates) are typically in the nanosecond domain, the time-resolved PL characterizations were further performed (excitation at 515 nm; emission at 712 nm). The PL lifetime of H2TCPP is determined to be
${\it\tau}_{2}$) may reflect the electron dynamics associated with the electron trap states that are energetically located within the bandgap of PCN-222. Given that the lifetimes of such trap states (reflecting the electron-detrapping rates) are typically in the nanosecond domain, the time-resolved PL characterizations were further performed (excitation at 515 nm; emission at 712 nm). The PL lifetime of H2TCPP is determined to be 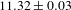 $11.32\pm 0.03$ ns. Similar to the TA case, two lifetimes with sizable components were also observed for the PL of PCN-222:
$11.32\pm 0.03$ ns. Similar to the TA case, two lifetimes with sizable components were also observed for the PL of PCN-222:  ${\it\tau}_{1}=0.38\pm 0.02$ ns (46%) and
${\it\tau}_{1}=0.38\pm 0.02$ ns (46%) and 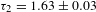 ${\it\tau}_{2}=1.63\pm 0.03$ ns (54%), or
${\it\tau}_{2}=1.63\pm 0.03$ ns (54%), or 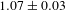 $1.07\pm 0.03$ ns on average (not shown here)[Reference Xu, Hu, Wang, Li, Zhang, Luo, Yu and Jiang26].
$1.07\pm 0.03$ ns on average (not shown here)[Reference Xu, Hu, Wang, Li, Zhang, Luo, Yu and Jiang26].
On the basis of the combined results of TA and PL, the pertinent mechanisms underlying the photoexcited dynamics involved in the two systems are illustrated in Figure 4(e). For the molecular system of H2TCPP ligand (left panel), the picture is rather straightforward: the 500-nm laser pulse launches an electronic transition from the ground state S0 to a certain excited state Sn, followed by rapid intramolecular deactivation to the lowest-lying excited state S1, as dictated by Kasha’s rule. As a result, the PL from S1 to S0 with a lifetime of 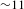 ${\sim}11$ ns was detected. On the other hand, in the semiconducting PCN-222 system (right panel), the existence of two distinct lifetimes for the PL emissions monitored at 712 nm (i.e., 1.74 eV, slightly below the bandgap energy of 1.75 eV) suggests that the radiative electron–hole recombination could originate from two trap states (acting as PL emission centers)[Reference Wheeler and Zhang8] that are located within the bandgap and in the very close proximity of the CB bottom (depicted as a whole in the right panel of Figure 4(e)). These two, near band-edge, trap states receive and accumulate the photogenerated electrons transferred from the CB bottom in a bi-exponential relaxation manner, reflecting their different trap depths. In terms of the bi-exponential component proportions, the two electron-trapping processes (47% for
${\sim}11$ ns was detected. On the other hand, in the semiconducting PCN-222 system (right panel), the existence of two distinct lifetimes for the PL emissions monitored at 712 nm (i.e., 1.74 eV, slightly below the bandgap energy of 1.75 eV) suggests that the radiative electron–hole recombination could originate from two trap states (acting as PL emission centers)[Reference Wheeler and Zhang8] that are located within the bandgap and in the very close proximity of the CB bottom (depicted as a whole in the right panel of Figure 4(e)). These two, near band-edge, trap states receive and accumulate the photogenerated electrons transferred from the CB bottom in a bi-exponential relaxation manner, reflecting their different trap depths. In terms of the bi-exponential component proportions, the two electron-trapping processes (47% for 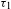 ${\it\tau}_{1}$ and 53% for
${\it\tau}_{1}$ and 53% for 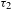 ${\it\tau}_{2}$) seem to nicely coincide with the PL emissions (46% for
${\it\tau}_{2}$) seem to nicely coincide with the PL emissions (46% for 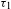 ${\it\tau}_{1}$ and 54% for
${\it\tau}_{1}$ and 54% for 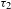 ${\it\tau}_{2}$), providing collateral evidence for the validity of the above assignments. As compared to the S1 state of H2TCPP, the two trap states of PCN-222 are relatively short-lived (
${\it\tau}_{2}$), providing collateral evidence for the validity of the above assignments. As compared to the S1 state of H2TCPP, the two trap states of PCN-222 are relatively short-lived (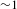 ${\sim}1$ ns on average for PCN-222 versus
${\sim}1$ ns on average for PCN-222 versus 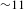 ${\sim}11$ ns for H2TCPP), implying relatively shallow depths. Nevertheless, the eventual recovery (to
${\sim}11$ ns for H2TCPP), implying relatively shallow depths. Nevertheless, the eventual recovery (to 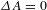 ${\it\Delta}A=0$) featuring an extremely long lifetime
${\it\Delta}A=0$) featuring an extremely long lifetime 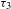 ${\it\tau}_{3}$ most likely correlates to a third trap state that possesses a rather deep depth. The detrapping of electrons from this deep trap state turns out to be so slow that its corresponding radiative electron–hole recombination is dramatically suppressed, which reasonably explains the absence of the long-lifetime component in the time-resolved PL kinetics (not shown here)[Reference Xu, Hu, Wang, Li, Zhang, Luo, Yu and Jiang26]. Thus it is safe to conclude that, as a direct result of the transformation from the H2TCPP ligand to its MOF architecture (i.e., PCN-222), the emergence of such a unique, deep trap state enables effective accumulation of long-lived, photogenerated electrons ready for the photocatalytic reduction of CO2 molecules captured by the porous PCN-222.
${\it\tau}_{3}$ most likely correlates to a third trap state that possesses a rather deep depth. The detrapping of electrons from this deep trap state turns out to be so slow that its corresponding radiative electron–hole recombination is dramatically suppressed, which reasonably explains the absence of the long-lifetime component in the time-resolved PL kinetics (not shown here)[Reference Xu, Hu, Wang, Li, Zhang, Luo, Yu and Jiang26]. Thus it is safe to conclude that, as a direct result of the transformation from the H2TCPP ligand to its MOF architecture (i.e., PCN-222), the emergence of such a unique, deep trap state enables effective accumulation of long-lived, photogenerated electrons ready for the photocatalytic reduction of CO2 molecules captured by the porous PCN-222.
To summarize, this ultrafast dynamics study on MOF[Reference Xu, Hu, Wang, Li, Zhang, Luo, Yu and Jiang26] discloses that the emergence of an extremely long-lived electron trap state in MOF is responsible for the substantial suppression of detrimental electron–hole recombination, thereby boosting its photocatalytic efficiency. This work sheds light on the electron-transfer mechanism involved in MOF, thereby providing instructive guidance for developing other similar MOF systems for high-efficiency photocatalytic applications.
5 Ultrafast dynamics in more complex nanosystems
In the above two sections, two relatively less complex, unary nanosystems have been addressed from the ultrafast dynamics perspective. We now turn to the other three, more complex, binary nanosystems, in which one can find that the ultrafast dynamics associated with interface states, Schottky junction versus plasmonic effects, and electron/hole defect states in these representative nanosystems play a central role in governing their photocatalytic performance.
5.1 MOF@TiO2
As mentioned in Section 4, the MOF materials have been identified as a class of excellent porous materials for gas capture and storage, owing to their capability of offering adsorptive sites for gas uptake[Reference Liu, Thallapally, McGrail, Brown and Liu27]. Nevertheless, the photocatalytic performance of MOFs is often limited when compared to the widely used inorganic semiconductors. To improve the photocatalytic efficiency for gaseous reactions using an MOF material, it would be ideal to functionally integrate the gas adsorption to MOF with the exciton generation by inorganic semiconductor. The key to this desirable integration lies in whether the photoexcited electrons can be effectively transferred from inorganic semiconductor to MOF.
In a recent work by Li et al. [Reference Li, Hu, Deng, Wang, Wang, Hu, Jiang, Jiang, Zhang, Xie and Xiong28], a proof-of-concept MOF@TiO2 core–shell structure was designed and fabricated, in which the MOF unit is Cu3(BTC)2 (BTC denotes benzene-1,3,5-tricarboxylate). TiO2 is known as an inorganic semiconductor with photocatalytic activity for converting CO2 to CH4 (with the aid of H2O), while Cu3(BTC)2 has been proven an excellent material for CO2 storage. Such a Cu3(BTC)2@TiO2 core–shell architecture (Figure 5(a)) is anticipated to possess unique, two-fold functions: (i) TiO2 is formed as the outer shells so as to be well photoexcited to generate excitons; (ii) the macroporous TiO2 shells may facilitate the capture of gas molecules in the MOF cores and provide sufficient surface area for photocatalysis. Indeed, this hybrid system was found to exhibit superior efficiency in CO2 conversion to its counterparts (Figure 5(b)). It is predicted that the enhanced photoreduction performance of Cu3(BTC)2@TiO2 (compared to bare TiO2) may originate from its high efficiency in electron–hole separation. To verify this prediction, apart from the photocurrent characterization, ultrafast TA spectroscopy was employed to interrogate Cu3(BTC)2@TiO2 in reference to bare TiO2 and bare Cu3(BTC)2.
Given that the bandgap of TiO2 is  ${\sim}3.2$ eV, the pump at 350 nm (i.e.,
${\sim}3.2$ eV, the pump at 350 nm (i.e., 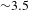 ${\sim}3.5$ eV) was used. The probing region was set in 400–750 nm (1.6–3.1 eV). As the
${\sim}3.5$ eV) was used. The probing region was set in 400–750 nm (1.6–3.1 eV). As the  ${\it\Delta}A$ kinetics turn out to be nearly independent of the probe wavelengths, a set of representative data taken at 450, 600 and 600 nm (center wavelength) for bare TiO2, bare Cu3(BTC)2 and Cu3(BTC)2@TiO2, respectively, is shown in Figure 5(c). The relevant mechanism is summarized in Figure 5(d). For bare TiO2, the SE signals build up within the instrument response function (
${\it\Delta}A$ kinetics turn out to be nearly independent of the probe wavelengths, a set of representative data taken at 450, 600 and 600 nm (center wavelength) for bare TiO2, bare Cu3(BTC)2 and Cu3(BTC)2@TiO2, respectively, is shown in Figure 5(c). The relevant mechanism is summarized in Figure 5(d). For bare TiO2, the SE signals build up within the instrument response function (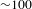 ${\sim}100$ fs) and then decay with a typical recovery time constant of
${\sim}100$ fs) and then decay with a typical recovery time constant of 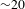 ${\sim}20$ ps. The former process reflects the instantaneous electron transfer from the CB to the surface states lying below the CB bottom (not depicted in Figure 5(d)), while the latter is responsible for the electron–hole recombination that would undesirably consume the photoexcitation energy. For bare Cu3(BTC)2, however, no
${\sim}20$ ps. The former process reflects the instantaneous electron transfer from the CB to the surface states lying below the CB bottom (not depicted in Figure 5(d)), while the latter is responsible for the electron–hole recombination that would undesirably consume the photoexcitation energy. For bare Cu3(BTC)2, however, no 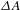 ${\it\Delta}A$ signals are discernable over the entire probing region albeit it can absorb the 350-nm light; this null-signal behavior could be ascribed to the absence of electronic states of Cu3(BTC)2 on resonance with the probe in 400–750 nm.
${\it\Delta}A$ signals are discernable over the entire probing region albeit it can absorb the 350-nm light; this null-signal behavior could be ascribed to the absence of electronic states of Cu3(BTC)2 on resonance with the probe in 400–750 nm.

Figure 5. (a) Schematic illustration of the synthesized Cu3(BTC)2@TiO2 core–shell structure. (b) Production yields of CH4 and H2 from CO2 using Cu3(BTC)2@TiO2 as photocatalysts under the UV irradiation for 4 h, in reference to bare TiO2 and bare Cu3(BTC)2. All the samples were carefully pretreated to remove any carbon contaminants. (c) Representative 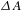 ${\it\Delta}A$ kinetic traces for bare TiO2, bare Cu3(BTC)2, and Cu3(BTC)2@TiO2, probed at 450, 600, and 600 nm, respectively (pump at 350 nm). (d) Schematic illustration of the mechanisms involving the interface states-related electron transfer and relaxation dynamics. All the figures are adapted from Ref. [Reference Li, Hu, Deng, Wang, Wang, Hu, Jiang, Jiang, Zhang, Xie and Xiong28].
${\it\Delta}A$ kinetic traces for bare TiO2, bare Cu3(BTC)2, and Cu3(BTC)2@TiO2, probed at 450, 600, and 600 nm, respectively (pump at 350 nm). (d) Schematic illustration of the mechanisms involving the interface states-related electron transfer and relaxation dynamics. All the figures are adapted from Ref. [Reference Li, Hu, Deng, Wang, Wang, Hu, Jiang, Jiang, Zhang, Xie and Xiong28].
Care must be taken in the distinctly different 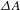 ${\it\Delta}A$ kinetics of Cu3(BTC)2@TiO2, which features positive ESA signals with an initial build-up (
${\it\Delta}A$ kinetics of Cu3(BTC)2@TiO2, which features positive ESA signals with an initial build-up (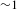 ${\sim}1$ ps) followed by a long-time recovery (
${\sim}1$ ps) followed by a long-time recovery (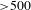 ${>}500$ ps), as shown in Figure 5(c). Thus it would be safe to infer that the formation of the interface states between TiO2 and Cu3(BTC)2 most likely leads to effective shuttering of the electron-transfer channels from the TiO2 CB to its surface states; if otherwise, the negative SE signals (associated with the surface states of TiO2) would by no means be overwhelmed by the positive ESA signals. Consequently, the photoexcited electrons in the TiO2 CB should transfer to (and then relax within) the adjacent interface states of Cu3(BTC)2@TiO2, and are subsequently probed via ESA to the higher-lying excited states of the Cu3(BTC)2@TiO2 hybrid (Figure 5(d)). As such, the observed
${>}500$ ps), as shown in Figure 5(c). Thus it would be safe to infer that the formation of the interface states between TiO2 and Cu3(BTC)2 most likely leads to effective shuttering of the electron-transfer channels from the TiO2 CB to its surface states; if otherwise, the negative SE signals (associated with the surface states of TiO2) would by no means be overwhelmed by the positive ESA signals. Consequently, the photoexcited electrons in the TiO2 CB should transfer to (and then relax within) the adjacent interface states of Cu3(BTC)2@TiO2, and are subsequently probed via ESA to the higher-lying excited states of the Cu3(BTC)2@TiO2 hybrid (Figure 5(d)). As such, the observed 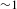 ${\sim}1$-ps build-up time (much longer than the instrument response function of
${\sim}1$-ps build-up time (much longer than the instrument response function of 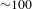 ${\sim}100$ fs) should account for this electron-transfer/relaxation process. On the other hand, the absence of SE signals suggests that the designed hybrid system is intrinsically robust in improving the efficiency of electron–hole separation. Note also that the subsequent recovery of the ESA signals appears rather slow (
${\sim}100$ fs) should account for this electron-transfer/relaxation process. On the other hand, the absence of SE signals suggests that the designed hybrid system is intrinsically robust in improving the efficiency of electron–hole separation. Note also that the subsequent recovery of the ESA signals appears rather slow (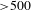 ${>}500$ ps), indicating that the photoexcited electrons localized in the interface states of Cu3(BTC)2@TiO2 could be long-lived, which is apparently beneficial for the activation of CO2 adsorbed on the MOF cores.
${>}500$ ps), indicating that the photoexcited electrons localized in the interface states of Cu3(BTC)2@TiO2 could be long-lived, which is apparently beneficial for the activation of CO2 adsorbed on the MOF cores.
To summarize, this ultrafast dynamics study on the proof-of-concept MOF@semiconductor system[Reference Li, Hu, Deng, Wang, Wang, Hu, Jiang, Jiang, Zhang, Xie and Xiong28] elucidates that the photogenerated electrons can be effectively transferred from the semiconductor to the MOF, thereby facilitating charge separation in the semiconductor and concomitantly supplying energized electrons to gas molecules adsorbed on MOF. This work enables a deeper understanding on the interface states-related dynamics and hence opens a new window to implementing MOF structures in the design of hybrid photocatalysts for gaseous reactions.
5.2 Pd–TiO2
The activation of molecular oxygen (O2) is a critical step in various oxidation reactions. The charge state of Pd surface has been proven a critical parameter for tuning the ability of Pd nanocrystals to activate O2 to generate a species that behaves like singlet O2 both chemically and physically[Reference Long, Mao, Ye, Yan, Huang, Wang, Fu, Wang, Wu, Xie and Xiong29]. Motivated by this finding, Long et al. [Reference Long, Mao, Gong, Zhou, Hu, Zhi, You, Bai, Jiang, Zhang, Wu and Xiong30] recently developed a metal–semiconductor hybrid system, Pd–TiO2, in which the Pd nanocrystals (enclosed by 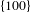 $\{100\}$ facets that are active for producing singlet-O2-analogous species) are deposited on the TiO2 supports, and further demonstrated that the generation of singlet-O2-analogous species is a vital step in the glucose oxidation catalyzed by Pd-based solid catalysts.
$\{100\}$ facets that are active for producing singlet-O2-analogous species) are deposited on the TiO2 supports, and further demonstrated that the generation of singlet-O2-analogous species is a vital step in the glucose oxidation catalyzed by Pd-based solid catalysts.
Interestingly, it was found that in catalyzing the oxidation of glucose a  ${\it\Lambda}$-shaped relationship accounts for the production yield of gluconic acid as a function of the power intensity of incident UV light (Figure 6(a)). As is known, driven by the Schottky junction formed between the metal Pd and the
${\it\Lambda}$-shaped relationship accounts for the production yield of gluconic acid as a function of the power intensity of incident UV light (Figure 6(a)). As is known, driven by the Schottky junction formed between the metal Pd and the  $n$-type semiconductor TiO2, the TiO2 supports can provide photogenerated electrons for metal catalysts under illumination. From the observed
$n$-type semiconductor TiO2, the TiO2 supports can provide photogenerated electrons for metal catalysts under illumination. From the observed 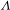 ${\it\Lambda}$-shaped relationship, one may hypothesize that the Pd components could also be activated by strong illumination to reversely inject plasmonic hot electrons into the TiO2 CB, lowering the electron density of Pd surface. To check the validity of this hypothesis, ultrafast TA spectroscopy was employed to examine the competition between the Schottky-junction-driven and plasmonic effects on charge transfer in this Pd–TiO2 hybrid system.
${\it\Lambda}$-shaped relationship, one may hypothesize that the Pd components could also be activated by strong illumination to reversely inject plasmonic hot electrons into the TiO2 CB, lowering the electron density of Pd surface. To check the validity of this hypothesis, ultrafast TA spectroscopy was employed to examine the competition between the Schottky-junction-driven and plasmonic effects on charge transfer in this Pd–TiO2 hybrid system.

Figure 6. (a) Production yield of gluconic acid (in 2 h) versus the UV-light incident power using Pd–TiO2 as catalysts (in reference to Pd nanocubes and bare TiO2), showing a 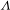 ${\it\Lambda}$-shaped relationship. (b) Representative
${\it\Lambda}$-shaped relationship. (b) Representative 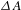 ${\it\Delta}A$ kinetic traces (left: 0–2 ns, right: 0–30 ps; probe at 520 nm) recorded with a 480-nm pump. (c) Schematic illustration of the mechanisms involving the electron transfer and relaxation dynamics under the 480-nm photoexcitation. (d) Representative
${\it\Delta}A$ kinetic traces (left: 0–2 ns, right: 0–30 ps; probe at 520 nm) recorded with a 480-nm pump. (c) Schematic illustration of the mechanisms involving the electron transfer and relaxation dynamics under the 480-nm photoexcitation. (d) Representative 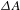 ${\it\Delta}A$ kinetic traces (left: 0–2 ns, right: 0–30 ps; probe at 520 nm) recorded with a 350-nm pump. (e) Schematic illustration of the mechanisms involving the electron transfer and relaxation dynamics under the 350-nm photoexcitation. All the figures are adapted from Ref. [Reference Long, Mao, Gong, Zhou, Hu, Zhi, You, Bai, Jiang, Zhang, Wu and Xiong30].
${\it\Delta}A$ kinetic traces (left: 0–2 ns, right: 0–30 ps; probe at 520 nm) recorded with a 350-nm pump. (e) Schematic illustration of the mechanisms involving the electron transfer and relaxation dynamics under the 350-nm photoexcitation. All the figures are adapted from Ref. [Reference Long, Mao, Gong, Zhou, Hu, Zhi, You, Bai, Jiang, Zhang, Wu and Xiong30].

Figure 7. (a) Schematic illustration of molecular co-catalysis strategy for accelerating hole transfer. Homogeneous molecular co-catalysts use reversible redox couple and highly active free radical reactions to promote hole transfer, unlike the conventional, supported heterogeneous co-catalysts that are constrained by finite contact areas between co-catalysts and reactants. (b) Photocatalytic activity on the K4Nb6O17 nanosheet catalysts with different molar ratio of TFA/K4Nb6O17. Reaction conditions: 50 mg catalyst, 200 mL 20 vol% methanol aqueous solution, and a specific amount of TFA, under irradiation of a 300-W Xe lamp. Representative 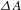 ${\it\Delta}A$ kinetics probed at 500 nm (pump at 300 nm) for K4Nb6O17 in (c) the absence and (d) the presence of TFA. (e) Steady-state and (f) time-resolved PL spectra (excitation at 315 nm; emission at 430 nm) for both K4Nb6O17 and K4Nb6O17–TFA. (g) Schematic illustration of the charge separation mechanisms involving the electron/hole defect states-related dynamics. All the figures are adapted from Ref. [Reference Bi, Li, Zhang, Jin, Zhang, Zhang, Luo, Wu and Xie32].
${\it\Delta}A$ kinetics probed at 500 nm (pump at 300 nm) for K4Nb6O17 in (c) the absence and (d) the presence of TFA. (e) Steady-state and (f) time-resolved PL spectra (excitation at 315 nm; emission at 430 nm) for both K4Nb6O17 and K4Nb6O17–TFA. (g) Schematic illustration of the charge separation mechanisms involving the electron/hole defect states-related dynamics. All the figures are adapted from Ref. [Reference Bi, Li, Zhang, Jin, Zhang, Zhang, Luo, Wu and Xie32].
In the TA measurements, two pumping scenarios with different center wavelengths at 480 and 350 nm were used to excite the samples of bare TiO2, bare Pd and Pd–TiO2. Under photoexcitation at 480 nm with only Pd excited (Figure 6(b)), bare TiO2 produces no 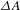 ${\it\Delta}A$ signal due to its
${\it\Delta}A$ signal due to its 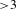 ${>}3$-eV bandgap, while bare Pd exhibits the pronounced plasmon damping effect in its
${>}3$-eV bandgap, while bare Pd exhibits the pronounced plasmon damping effect in its 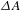 ${\it\Delta}A$ kinetic trace. However, Pd–TiO2 exhibits a rather different dynamic behavior with recovery much slower than bare Pd. This can be understood as a result of hot-electron injection from Pd to TiO2, followed by electron relaxation within the TiO2 CB to yield the SE signals (Figure 6(c)). To further verify this effect, a 350-nm pump was then used to simultaneously excite Pd and TiO2 (Figure 6(d)). For bare TiO2, there appears apparent competition between SE and ESA, with the initial build-up and the subsequent decay features of ESA superimposed on the predominant SE trace, reflecting the excitation of TiO2. Similar plasmon damping effect to the case of pumping at 480 nm was observed for bare Pd, indicating the activation of Pd at 350 nm. For the hybrid Pd–TiO2 system of interest, the SE process induced by the injection of hot electrons from Pd to TiO2 turns out to be so strong as to completely suppress the ESA processes on both Pd and TiO2 (Figure 6(e)). On the basis of the above results and consistent analyses, the aforementioned hypothesis can be validated.
${\it\Delta}A$ kinetic trace. However, Pd–TiO2 exhibits a rather different dynamic behavior with recovery much slower than bare Pd. This can be understood as a result of hot-electron injection from Pd to TiO2, followed by electron relaxation within the TiO2 CB to yield the SE signals (Figure 6(c)). To further verify this effect, a 350-nm pump was then used to simultaneously excite Pd and TiO2 (Figure 6(d)). For bare TiO2, there appears apparent competition between SE and ESA, with the initial build-up and the subsequent decay features of ESA superimposed on the predominant SE trace, reflecting the excitation of TiO2. Similar plasmon damping effect to the case of pumping at 480 nm was observed for bare Pd, indicating the activation of Pd at 350 nm. For the hybrid Pd–TiO2 system of interest, the SE process induced by the injection of hot electrons from Pd to TiO2 turns out to be so strong as to completely suppress the ESA processes on both Pd and TiO2 (Figure 6(e)). On the basis of the above results and consistent analyses, the aforementioned hypothesis can be validated.
To summarize, this ultrafast dynamics study on the hybrid Pd–TiO2 system[Reference Long, Mao, Gong, Zhou, Hu, Zhi, You, Bai, Jiang, Zhang, Wu and Xiong30] verifies that the injection of plasmonic hot electrons can take the opposite direction to the charge migration driven by Schottky junction. This work highlights the importance of rationally tailoring the charge state of metal surface as well as carefully modulating the function of plasmonic nanocrystals in O2 activation for promoting the organic oxidation reactions under proper light illumination.
5.3 K4Nb6O17–TFA
In artificial photocatalysis, sluggish kinetics of hole transfer and the resulting high charge recombination rate are well known as the Achilles’ heel of photocatalytic conversion efficiency[Reference Simon, Bouchonville, Berr, Vaneski, Adrović, Volbers, Wyrwich, Döblinger, Susha, Rogach, Jäckel, Stolarczyk and Feldmann31]. In a very recent work by Bi et al. [Reference Bi, Li, Zhang, Jin, Zhang, Zhang, Luo, Wu and Xie32], to address this challenge a new strategy using water-soluble molecules as co-catalysts to accelerate hole transfer for improved photocatalytic H2 evolution activity was proposed and implemented. TFA, by virtue of its reversible redox couple 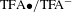 $\text{TFA}\bullet /\text{TFA}^{-}$, was used to serve as a homogeneous co-catalyst that not only maximizes the contact areas between co-catalysts and reactants but also greatly promotes hole transfer, as schematically illustrated in Figure 7(a). As a proof of concept, the K4Nb6O17 nanosheet catalyst was demonstrated to achieve drastically increased photocatalytic H2 production rate in the presence of TFA, up to 32 times with respect to the blank case (i.e., in the absence of TFA), as shown in Figure 7(b). Apart from pertinent theoretical analysis and other characterizations such as ESR, UV–Vis and PL, ultrafast TA spectroscopy was employed to evaluate how the addition of TFA influences the charge separation involved in this K4Nb6O17-based photocatalysis.
$\text{TFA}\bullet /\text{TFA}^{-}$, was used to serve as a homogeneous co-catalyst that not only maximizes the contact areas between co-catalysts and reactants but also greatly promotes hole transfer, as schematically illustrated in Figure 7(a). As a proof of concept, the K4Nb6O17 nanosheet catalyst was demonstrated to achieve drastically increased photocatalytic H2 production rate in the presence of TFA, up to 32 times with respect to the blank case (i.e., in the absence of TFA), as shown in Figure 7(b). Apart from pertinent theoretical analysis and other characterizations such as ESR, UV–Vis and PL, ultrafast TA spectroscopy was employed to evaluate how the addition of TFA influences the charge separation involved in this K4Nb6O17-based photocatalysis.
In the TA measurements, K4Nb6O17 in the presence of TFA (denoted K4Nb6O17–TFA in short) was interrogated in reference to bare K4Nb6O17. The pump was chosen at 300 nm, which can effectively photoinduce an interband transition in the semiconductor K4Nb6O17 system. Given that the 390–610 nm WLC probe yields essentially the same 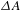 ${\it\Delta}A$ kinetics for each sample, a set of representative data taken at 500 nm is presented in Figures 7(c) and 7(d). Both samples exhibit ESA signals, whose relaxations are characterized by two time constants:
${\it\Delta}A$ kinetics for each sample, a set of representative data taken at 500 nm is presented in Figures 7(c) and 7(d). Both samples exhibit ESA signals, whose relaxations are characterized by two time constants: 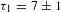 ${\it\tau}_{1}=7\pm 1$ ps and
${\it\tau}_{1}=7\pm 1$ ps and 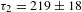 ${\it\tau}_{2}=219\pm 18$ ps for K4Nb6O17 while
${\it\tau}_{2}=219\pm 18$ ps for K4Nb6O17 while 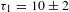 ${\it\tau}_{1}=10\pm 2$ ps and
${\it\tau}_{1}=10\pm 2$ ps and 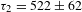 ${\it\tau}_{2}=522\pm 62$ ps for K4Nb6O17–TFA (all with a roughly 40:60 percentage for the two exponential components). The long-time decay component(s) cannot be identified due to the probe-delay limit of the pump–probe spectrometer. The observed two components in the picosecond domain may reflect the electron dynamics associated with the defect states that are energetically located within the bandgap of K4Nb6O17. The fact that such defect states could be long-lived (in the nanosecond domain) necessitates the complementary PL characterizations. The steady-state PL spectra (excitation at 315 nm; Figure 7(e)) indicates that the addition of TFA leads to substantial PL quenching, suggesting suppressed radiative electron–hole recombination in K4Nb6O17–TFA with respect to K4Nb6O17. Figure 7(f) compares the PL lifetimes recorded for the two samples at the emission wavelength of 430 nm (i.e., the PL intensity maximum in Figure 7(e)). Similar to the TA case, two PL lifetimes with sizable components are identified for both samples:
${\it\tau}_{2}=522\pm 62$ ps for K4Nb6O17–TFA (all with a roughly 40:60 percentage for the two exponential components). The long-time decay component(s) cannot be identified due to the probe-delay limit of the pump–probe spectrometer. The observed two components in the picosecond domain may reflect the electron dynamics associated with the defect states that are energetically located within the bandgap of K4Nb6O17. The fact that such defect states could be long-lived (in the nanosecond domain) necessitates the complementary PL characterizations. The steady-state PL spectra (excitation at 315 nm; Figure 7(e)) indicates that the addition of TFA leads to substantial PL quenching, suggesting suppressed radiative electron–hole recombination in K4Nb6O17–TFA with respect to K4Nb6O17. Figure 7(f) compares the PL lifetimes recorded for the two samples at the emission wavelength of 430 nm (i.e., the PL intensity maximum in Figure 7(e)). Similar to the TA case, two PL lifetimes with sizable components are identified for both samples: 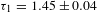 ${\it\tau}_{1}=1.45\pm 0.04$ ns (81%) and
${\it\tau}_{1}=1.45\pm 0.04$ ns (81%) and 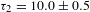 ${\it\tau}_{2}=10.0\pm 0.5$ ns (19%) for K4Nb6O17 while
${\it\tau}_{2}=10.0\pm 0.5$ ns (19%) for K4Nb6O17 while  ${\it\tau}_{1}=4.9\pm 0.1$ ns (87%) and
${\it\tau}_{1}=4.9\pm 0.1$ ns (87%) and 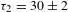 ${\it\tau}_{2}=30\pm 2$ ns (13%) for K4Nb6O17–TFA.
${\it\tau}_{2}=30\pm 2$ ns (13%) for K4Nb6O17–TFA.
On the basis of the combined results of TA and PL, a schematic mechanism depicting the involved photoexcited carrier dynamics is illustrated in Figure 7(g). The observation of PL emissions peaking at 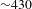 ${\sim}430$ nm (i.e., 2.88 eV, red-shifted with respect to the 3.56-eV bandgap) with two lifetimes suggests that the radiative electron–hole recombination responsible for PL could originate from two interfacial defect states (probably with different trap depths) near the CB bottom (labeled
${\sim}430$ nm (i.e., 2.88 eV, red-shifted with respect to the 3.56-eV bandgap) with two lifetimes suggests that the radiative electron–hole recombination responsible for PL could originate from two interfacial defect states (probably with different trap depths) near the CB bottom (labeled  $e$-DS as a whole in Figure 7(g)) that act as the PL emission centers[Reference Wheeler and Zhang8]. These defect states receive and accumulate the photogenerated electrons transferred from the CB bottom in a bi-exponential relaxation manner, as evidenced by the observed
$e$-DS as a whole in Figure 7(g)) that act as the PL emission centers[Reference Wheeler and Zhang8]. These defect states receive and accumulate the photogenerated electrons transferred from the CB bottom in a bi-exponential relaxation manner, as evidenced by the observed 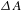 ${\it\Delta}A$ kinetics which are obviously commensurate with the time-resolved PL results. On the other hand, the photogenerated holes are transferred from the VB to the defect states near the VB top (labeled
${\it\Delta}A$ kinetics which are obviously commensurate with the time-resolved PL results. On the other hand, the photogenerated holes are transferred from the VB to the defect states near the VB top (labeled  $h$-DS in Figure 7(g)). The addition of TFA can be expected to accelerate the subsequent hole transfer from
$h$-DS in Figure 7(g)). The addition of TFA can be expected to accelerate the subsequent hole transfer from  $h$-DS to hole scavengers in the solution (including methanol as well as TFA itself). This is understandable because the situation of
$h$-DS to hole scavengers in the solution (including methanol as well as TFA itself). This is understandable because the situation of 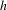 $h$-DS being effectively vacated via the TFA anion-accelerated hole transfer will eventually decelerate the processes of electron–hole recombination and electron transfer from CB to
$h$-DS being effectively vacated via the TFA anion-accelerated hole transfer will eventually decelerate the processes of electron–hole recombination and electron transfer from CB to  $e$-DS, as verified by the observed, significantly altered carrier dynamics. Notably, in the presence of TFA the average time describing the overall electron transfer from CB to
$e$-DS, as verified by the observed, significantly altered carrier dynamics. Notably, in the presence of TFA the average time describing the overall electron transfer from CB to  $e$-DS is increased by a factor of
$e$-DS is increased by a factor of 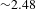 ${\sim}2.48$ (
${\sim}2.48$ (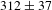 $312\pm 37$ ps for K4Nb6O17–TFA versus
$312\pm 37$ ps for K4Nb6O17–TFA versus  $126\pm 11$ ps for K4Nb6O17) and meanwhile that describing the overall radiative electron–hole recombination by a factor of
$126\pm 11$ ps for K4Nb6O17) and meanwhile that describing the overall radiative electron–hole recombination by a factor of  ${\sim}2.73$ (
${\sim}2.73$ (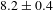 $8.2\pm 0.4$ ns for K4Nb6O17–TFA versus
$8.2\pm 0.4$ ns for K4Nb6O17–TFA versus 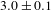 $3.0\pm 0.1$ ns for K4Nb6O17). On the basis of the self-consistent results of PL and TA observations, it would be safe to assert that TFA mainly influences the hole-transfer process and that the increased electron lifetimes in the system could offer more opportunities for photogenerated electrons to take effect in the photocatalytic H2 evolution reaction.
$3.0\pm 0.1$ ns for K4Nb6O17). On the basis of the self-consistent results of PL and TA observations, it would be safe to assert that TFA mainly influences the hole-transfer process and that the increased electron lifetimes in the system could offer more opportunities for photogenerated electrons to take effect in the photocatalytic H2 evolution reaction.
To summarize, this ultrafast dynamics study on the K4Nb6O17–TFA system[Reference Bi, Li, Zhang, Jin, Zhang, Zhang, Luo, Wu and Xie32] evidences that the proposed molecular co-catalyst strategy is rather effective in accelerating the sluggish kinetics of hole transfer, a well-known bottleneck in many realistic photocatalytic systems, thereby significantly improving the charge separation efficiency in the system. This work establishes a new, fertile ground for developing high-efficiency photocatalytic nanosystems that are free of noble metal co-catalysts.
6 Conclusion and outlook
Ultrafast spectroscopy has been widely recognized as a robust, routine tool for tracking in real time the ultrafast dynamics involved in various nanosystems, especially the semiconductor nanomaterials. The mechanistic information deduced from such ultrafast dynamics studies can usually offer key, fundamentally instructive guidance for the rational and targeted design of nanomaterials towards achieving significantly improved performance in, e.g., photocatalysis, photovoltaics and photoelectrochemistry. In this mini-review, we have presented an overview of the recent advances in our ultrafast spectroscopy laboratory on the study and understanding of ultrafast dynamics and related mechanisms involved in several representative nanomaterial complex systems by means of femtosecond TA spectroscopy. An attempt has been made to convey a consistent, physical picture of various important processes, pathways, dynamics and interactions involved in the investigated nanomaterial complex systems, most of which hold great promise in photocatalysis applications. Last but not least, this mini-review can certainly not cover all of the newly emerging aspects related to a vast number of photocatalytic nanosystems, e.g., the intriguing plasmonic photocatalysis[Reference Zhang, Chen, Liu and Tsai33, Reference Kang, Gong, Hu, Li, Qiu, Zhu, Ajayan and Fang34], in which a step forward has recently been made in our laboratory[Reference Huang, Zhang, Lv, Long, Zhang, Lin, Wei, Wang, Chen, Li, Zhang, Luo and Xiong35] that features an effective incorporation of ultrafast spectroscopy into the relevant mechanistic elucidation.
Acknowledgments
The authors gratefully acknowledge support from the National Natural Science Foundation of China (21573211 and 21421063), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB01020200) and the Fundamental Research Funds for the Central Universities of China (WK2340000063).








 Open access
Open access