


1. Introduction
DNA sequencing has been one of the most important techniques in medical research and genetic diagnostics since chain termination sequencing was first described 38 years ago (Sanger et al., Reference Sanger, Nicklen and Coulson1977). Subsequently, PCR was introduced by Mullis et al. (Reference Mullis, Faloona, Scharf, Saiki, Horn and Erlich1986), broadening its applications. Automated Sanger sequencers based on capillary electrophoresis were an integral part of The Human Genome Project completed in 2003. The first human genome sequenced was the result of over 10 year's effort at an estimated cost of $2.7 billion (Lander et al., Reference Lander, Linton, Birren, Nusbaum, Zody, Baldwin, Devon, Dewar, Doyle, FitzHugh, Funke, Gage, Harris, Heaford, Howland, Kann, Lehoczky, LeVine, McEwan, McKernan, Meldrim, Mesirov, Miranda, Morris, Naylor, Raymond, Rosetti, Santos, Sheridan, Sougnez, Stange-Thomann, Stojanovic, Subramanian, Wyman, Rogers, Sulston, Ainscough, Beck, Bentley, Burton, Clee, Carter, Coulson, Deadman, Deloukas, Dunham, Dunham, Durbin, French, Grafham, Gregory, Hubbard, Humphray, Hunt, Jones, Lloyd, McMurray, Matthews, Mercer, Milne, Mullikin, Mungall, Plumb, Ross, Shownkeen, Sims, Waterston, Wilson, Hillier, McPherson, Marra, Mardis, Fulton, Chinwalla, Pepin, Gish, Chissoe, Wendl, Delehaunty, Miner, Delehaunty, Kramer, Cook, Fulton, Johnson, Minx, Clifton, Hawkins, Branscomb., Predki, Richardson, Wenning, Slezak, Doggett, Cheng, Olsen, Lucas, Elkin, Uberbacher, Frazier, Gibbs, Muzny, Scherer, Bouck, Sodergren, Worley, Rives, Gorrell, Metzker, Naylor, Kucherlapati, Nelson, Weinstock, Sakaki, Fujiyama, Hattori, Yada, Toyoda, Itoh, Kawagoe, Watanabe, Totoki, Taylor, Weissenbach, Heilig, Saurin, Artiguenave, Brottier, Bruls, Pelletier, Robert, Wincker, Smith, Doucette-Stamm, Rubenfield, Weinstock, Lee, Dubois, Rosenthal, Platzer, Nyakatura, Taudien, Rump, Yang, Yu, Wang, Huang, Gu, Hood, Rowen, Madan, Qin, Davis, Federspiel, Abola, Proctor, Myers, Schmutz, Dickson, Grimwood, Cox, Olson, Kaul, Raymond, Shimizu, Kawasaki, Minoshima, Evans, Athanasiou, Schultz, Roe, Chen, Pan, Ramser, Lehrach, Reinhardt, McCombie, de la Bastide, Dedhia, Blöcker, Hornischer, Nordsiek, Agarwala, Aravind, Bailey, Bateman, Batzoglou, Birney, Bork, Brown, Burge, Cerutti, Chen, Church, Clamp, Copley, Doerks, Eddy, Eichler, Furey, Galagan, Gilbert, Harmon, Hayashizaki, Haussler, Hermjakob, Hokamp, Jang, Johnson, Jones, Kasif, Kaspryzk, Kennedy, Kent, Kitts, Koonin, Korf, Kulp, Lancet, Lowe, McLysaght, Mikkelsen, Moran, Mulder, Pollara, Ponting, Schuler, Schultz, Slater, Smit, Stupka, Szustakowski, Thierry-Mieg, Thierry-Mieg, Wagner, Wallis, Wheeler, Williams, Wolf, Wolfe, Yang, Yeh, Collins, Guyer, Peterson, Felsenfeld, Wetterstrand, Patrinos, Morgan, de Jong, Catanese, Osoegawa, Shizuya, Choi and Chen2001; McPherson et al., Reference McPherson, Marra, Hillier, Waterston, Chinwalla, Wallis, Sekhon, Wylie, Mardis, Wilson, Fulton, Kucaba, Wagner-McPherson, Barbazuk, Gregory, Humphray, French, Evans, Bethel, Whittaker, Holden, McCann, Dunham, Soderlund, Scott, Bentley, Schuler, Chen, Jang, Green, Idol, Maduro, Montgomery, Lee, Miller, Emerling, Kucherlapati, Gibbs, Scherer, Gorrell, Sodergren, Clerc-Blankenburg, Tabor, Naylor, Garcia, de Jong, Catanese, Nowak, Osoegawa, Qin, Rowen, Madan, Dors, Hood, Trask, Friedman, Massa, Cheung, Kirsch, Reid, Yonescu, Weissenbach, Bruls, Heilig, Branscomb, Olsen, Doggett, Cheng, Hawkins, Myers, Shang, Ramirez, Schmutz, Velasquez, Dixon, Stone, Cox, Haussler, Kent, Furey, Rogic, Kennedy, Jones, Rosenthal, Wen, Schilhabel, Gloeckner, Nyakatura, Siebert, Schlegelberger, Korenberg, Chen, Fujiyama, Hattori, Toyoda, Yada, Park, Sakaki, Shimizu, Asakawa, Kawasaki, Sasaki, Shintani, Shimizu, Shibuya, Kudoh, Minoshima, Ramser, Seranski, Hoff, Poustka, Reinhardt and Lehrach2001; Sachidanandam et al., Reference Sachidanandam, Weissman, Schmidt, Kakol, Stein, Marth, Sherry, Mullikin, Mortimore, Willey, Hunt, Cole, Coggill, Rice, Ning, Rogers, Bentley, Kwok, Mardis, Yeh, Schultz, Cook, Davenport, Dante, Fulton, Hillier, Waterston, McPherson, Gilman, Schaffner, Van Etten, Reich, Higgins, Daly, Blumenstiel, Baldwin, Stange-Thomann, Zody, Linton, Lander and Altshuler2001). Automated Sanger sequencing, also called ‘first-generation DNA sequencing’, is the most widely used technology worldwide today for variant detection through sequencing. Although currently the gold standard for DNA sequencing, there are some limitations in using this method for high-throughput applications, including read length, runtime and per base cost (Rizzo & Buck, Reference Rizzo and Buck2012). Fortunately, a high-throughput, powerful technology known as next-generation sequencing (NGS) (or massively parallel sequencing (MPS)) has been developed over the past 10 years. This revolutionary sequencing technique is capable of sequencing millions of small fragments covering the whole genome or large regions of interest, such as the entire coding portion of the genome (i.e. exome), at a reasonable cost and reduced runtime compared to Sanger sequencing (Yan et al., Reference Yan, Tekin, Blanton and Liu2013 a ; Rabbani et al., Reference Rabbani, Tekin and Mahdieh2014).
Elucidation of the genetic basis of human diseases is vital for understanding the underlying pathology, making an early diagnosis, developing prevention and/or better treatment regimens, and improving genetic counselling (Gilissen et al., Reference Gilissen, Hoischen, Brunner and Veltman2012). Although traditional laboratory approaches, including karyotyping and copy number variation (CNV) analysis, and computational methods, including linkage and association, have led to great insights into human diseases over the past few decades, there is still a substantial gap between disease phenotype and the underlying genetic involvements (Bamshad et al., Reference Bamshad, Ng, Bigham, Tabor, Emond, Nickerson and Shendure2011; Gilissen et al., Reference Gilissen, Hoischen, Brunner and Veltman2011). Limiting elements of traditional gene-discovery strategies, such as the necessity of large families, and the existence of reduced penetrance, variable expressivity and locus heterogeneity have always been a problem for geneticists (Bamshad et al., Reference Bamshad, Ng, Bigham, Tabor, Emond, Nickerson and Shendure2011). The development of NGS, however, has made the identification of causative genes easier, even for small families and diseases with extensive locus heterogeneity (Duman & Tekin, Reference Duman and Tekin2012). Furthermore, NGS can also be used to analyse genomic functions through transcriptome, methylome and chromatin structure studies.
Hearing loss (HL) is the most common sensory deficit, affecting millions of people worldwide, with an incidence of 1/1000 newborns in the US. At least half of congenital deafness is due to genetic factors, the vast majority of which is monogenic. Autosomal recessive, autosomal dominant and X-linked inheritance are observed in 77, 22 and 1% of genetic cases, respectively (Morton, Reference Morton1991). Approximately 70% of congenital deafness is nonsyndromic (nonsyndromic hearing loss; NSHL), while the remaining 30% is comprised of many different syndromes in which deafness is a feature (Shearer & Smith, Reference Shearer and Smith2012; Yan et al., Reference Yan, Tekin, Blanton and Liu2013 a ). Current research estimates that 1% of all human genes have a function in hearing (Teek et al., Reference Teek, Kruustuk, Zordania, Joost, Kahre, Tonisson, Nelis, Zilina, Tranebjaerg, Reimand and Ounap2013). To date, mutations in over 80 genes, with more than 1000 mutations, have been found to cause NSHL making hereditary hearing loss one of the most genetically heterogeneous traits (http://hereditaryhearingloss.org). Except for those recurrently reported in a few genes, such as GJB2 (MIM 121011) and SLC26A4 (MIM 605646), most deafness mutations are extremely rare and are only seen in either a single or a very few families (Diaz-Horta et al., Reference Diaz-Horta, Duman, Foster, Sirmaci, Gonzalez, Mahdieh, Fotouhi, Bonyadi, Cengiz, Menendez, Ulloa, Edwards, Züchner, Blanton and Tekin2012).
Here, we review recent developments on the usage of NGS for hereditary HL, with an emphasis on whole-exome sequencing (WES). The extreme genetic heterogeneity of NSHL makes it the ideal disorder to use to demonstrate the full potential of NGS.
2. NGS platforms and techniques
While there are five commercially available NGS platforms in the marketplace: Roche/454 FLX, the Illumina HiSeq Series, the Applied Biosystems (ABI) SOLiD Analyzer, the Polonator G.007 and the Helicos HeliScope, the first three platforms currently dominate the area (Yan et al., Reference Yan, Tekin, Blanton and Liu2013 a ). Although these platforms each have their own strengths and weaknesses originating from the technologies used, they all include three main stages: template preparation, sequencing/imaging and data analysis. Template preparation is the first step and determines the part of the genome to be sequenced, i.e. whole-genome sequencing (WGS), WES or targeted next-generation sequencing (Yan et al., Reference Yan, Tekin, Blanton and Liu2013 a ). The first platform introduced, Roche/454 GS FLX, uses pyrosequencing. When DNA polymerase incorporates a nucleotide into the growing DNA strand and ATP hydrolysis occurs, the pyrophosphate release is recorded (Shearer et al., Reference Shearer, Hildebrand, Sloan and Smith2011). The Illumina platform, based on cyclic reversible termination (CRT) technology, is the most widely used NGS platform. In CRT, fluorescent labelled chain-terminating nucleotides are incorporated into the growing strand, caught and imaged by the sequencer, and then the fluorescent terminator is cleaved off the nucleotide (Ju et al., Reference Ju, Kim, Bi, Meng, Bai, Li, Li, Marma, Shi, Wu, Edwards, Romu and Turro2006; Shearer et al., Reference Shearer, Hildebrand, Sloan and Smith2011). The SOLiD platform uses sequence-by-ligation (SBL) technology. A fluorescently labelled probe hybridizes to the DNA template to be sequenced. The probe then is joined to the growing strand by DNA ligase, and then imaged by the sequencer (Shearer et al., Reference Shearer, Hildebrand, Sloan and Smith2011).
3. Why WES?
Nowadays, sequencing the whole genome is becoming available for wider usage; however, it was not practical until recently because of cost and the amount of data produced. To discover the genes causing Mendelian diseases, especially those having genetic heterogeneity, WES represents a relatively easy-to-use alternative method in the research area (Yan et al., Reference Yan, Tekin, Blanton and Liu2013 a ). While the coding parts of the human genome, i.e. exons of (most) genes or the exome, constitute only about 1% of the entire human genome, 85% of mutations known to cause Mendelian diseases are located in the coding region or in canonical splice sites (Choi et al., Reference Choi, Scholl, Ji, Liu, Tikhonova, Zumbo, Nayir, Bakkaloğlu, Ozen, Sanjad, Nelson-Williams, Farhi, Mane and Lifton2009). The targeted genomic enrichment required for WES has some challenges, such as incomplete knowledge about all truly protein-coding exons, variable efficiencies of capture probes used and targeting sequence issues (e.g. microRNAs, promoters, pseudogenes, repetitive elements and ultra-conserved elements) (Bamshad et al., Reference Bamshad, Ng, Bigham, Tabor, Emond, Nickerson and Shendure2011). Despite these limitations, exome sequencing has clearly proven to be a powerful tool for discovering the underlying genetic etiology of known or suspected Mendelian disorders. Moreover, by increasing read length and coverage, these problems seem relatively likely to be solved.
4. Data analysis
On average, WES identifies approximately 22 000 single nucleotide variants (SNVs) in a sample. More than 95% of these variants are already known polymorphisms. Strategies for identifying causal SNVs vary depending on a number of factors such as the putative mode of inheritance of the trait, the pedigree structure and the presence of locus heterogeneity in the trait (Bamshad et al., Reference Bamshad, Ng, Bigham, Tabor, Emond, Nickerson and Shendure2011).
Analysis of the WES data begins with filtering out the variants against a set of polymorphisms that are frequent ( > 0·5%) in public (e.g. dbSNP and 1000 Genomes Project) or internal databases (e.g. GEM.App) (Gonzalez et al., Reference Gonzalez, Lebrigio, Van Booven, Ulloa, Powell, Speziani, Tekin, Schüle and Züchner2013). This step narrows the number of variants to a manageable fraction (2% on average) of the SNVs identified in an individual. Variations in conserved regions, their functional class (frameshifts, nonsense, splice site) and missense are prioritized in descendant order. Candidate variants are also prioritized according to predicted effects on the protein function (e.g. SIFT (Kumar et al., Reference Kumar, Henikoff and Ng2009) and PolyPhen2 (Adzhubei et al., Reference Adzhubei, Schmidt, Peshkin, Ramensky, Gerasimova, Bork, Kondrashov and Sunyaev2010)) and conservation scores (e.g. GERP (Davydov et al., Reference Davydov, Goode, Sirota, Cooper, Sidow and Batzoglou2010) and PHAST (Hubisz et al., Reference Hubisz, Pollard and Siepel2011)) (Bamshad et al., Reference Bamshad, Ng, Bigham, Tabor, Emond, Nickerson and Shendure2011).
Analysis of the segregation of candidate variants in the family is also an essential filter, even if mapping data are not available. Sequencing the two most distantly related individuals with the phenotype of interest can substantially reduce the genomic search space for candidate causal alleles. Multiplex families and/or inbred families are more informative than simplex families for inherited causes (Bamshad et al., Reference Bamshad, Ng, Bigham, Tabor, Emond, Nickerson and Shendure2011) (Fig. 1).

Fig. 1. Typical workflow for gene discovery using whole-exome sequencing. WES, whole-exome sequencing.
5. Gene discovery in NSHL: the impact of WES
Mutations in POU3F4 (MIM 300039), which was mapped to the X-chromosome in 1988 (Wallis et al., Reference Wallis, Ballo, Wallis, Beighton and Goldblatt1988), were identified in 1995 (Wallis et al., Reference Wallis, Ballo, Wallis, Beighton and Goldblatt1988; de Kok et al., Reference de Kok, van der Maarel, Bitner-Glindzicz, Huber, Monaco, Malcolm, Pembrey, Ropers and Cremers1995). It was the first gene discovered as a cause of NSHL. In 1992, the first autosomal dominant locus was mapped to chromosome 5q31 (Leon et al., Reference Leon, Raventos, Lynch, Morrow and King1992). Five years later, a mutation was identified in DIAPH1 at this locus (MIM 602121) (Leon et al., Reference Leon, Raventos, Lynch, Morrow and King1992; Lynch et al., Reference Lynch, Lee, Morrow, Welcsh, Leon and King1997). The first locus for autosomal recessive NSHL was mapped to 13q12 (Guilford et al., Reference Guilford, Ben Arab, Blanchard, Levilliers, Weissenbach, Belkahia and Petit1994), which subsequently led to the identification of mutations in GJB2 (MIM 121011) (Kelsell et al., Reference Kelsell, Dunlop, Stevens, Lench, Liang, Parry, Mueller and Leigh1997). This gene is considered to be responsible for up to 50% of autosomal recessive NSHL in childhood in many parts of the world (Guilford et al., Reference Guilford, Ben Arab, Blanchard, Levilliers, Weissenbach, Belkahia and Petit1994; Popov et al., Reference Popov, Stancheva, Kachakova, Rangachev, Konov, Varbanova, Mitev, Kaneva and Popova2014). Prior to 2010, despite intense efforts by leading laboratories over nearly two decades, only 60 deafness genes had been discovered. Since 2010, by employing NGS technologies, 21 genes have been added to the NSHL gene list (http://hereditaryhearingloss.org) (Table 1 and Fig. 2).
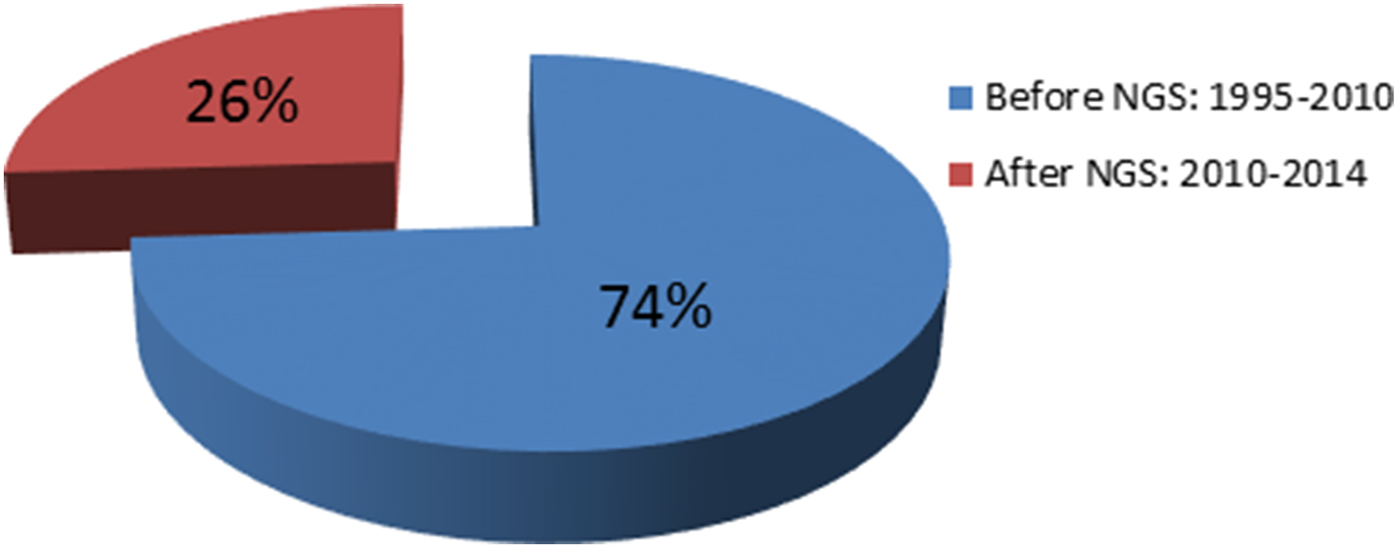
Fig. 2. The impact of next-generation sequencing on gene discovery for nonsyndromic deafness. NGS, next-generation sequencing.
Table 1. Nonsyndromic deafness genes discovered via whole-exome sequencing
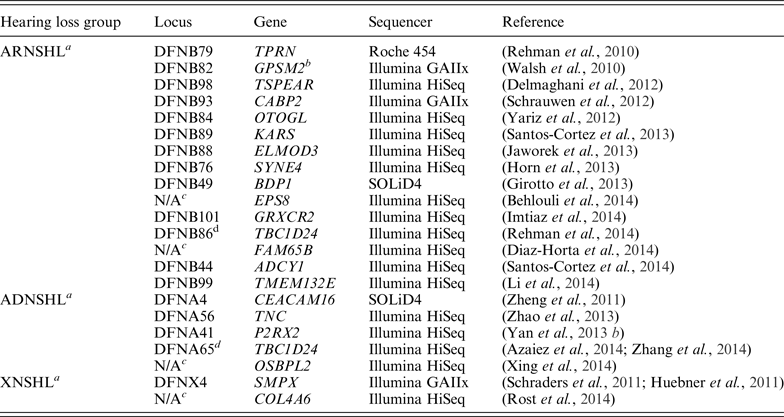
a ADNSHL, autosomal dominant nonsyndromic hearing loss; ARNSHL, autosomal recessive nonsyndromic hearing loss; XNSHL, X-linked nonsyndromic hearing loss.
b GPSM2 mutations have been subsequently shown to cause Chudley-McCullough syndrome (Doherty et al., Reference Doherty, Chudley, Coghlan, Ishak, Innes, Lemire, Rogers, Mhanni, Phelps, Jones, Zhan, Fejes, Shahin, Kanaan, Akay, Tekin, Triggs-Raine. and Zelinski2012).
c N/A, not available.
d Mutations in the TBC1D24 gene are responsible for ARNSHL or ADNSHL.
In 2010, the first example of NGS used to identify a Mendelian disease gene was the application of exome sequencing to reveal a mutation in DHODH (MIM 126064) as the cause of Miller syndrome (MIM 263750) (Ng et al., Reference Ng, Buckingham, Lee, Bigham, Tabor, Dent, Huff, Shannon, Jabs, Nickerson, Shendure and Bamshad2010). The first gene associated with NSHL identified through NGS was TPRN (MIM 613354), which causes NSHL at the DFNB79 locus. This locus was mapped to 9q34·3 in a Pakistani family with autosomal recessive NSHL (ARNSHL). In that study, homozygosity mapping was combined with exome sequencing to identify the causative mutation (Rehman et al., Reference Rehman, Morell, Belyantseva, Khan, Boger, Shahzad, Ahmed, Riazuddin, Khan, Riazuddin and Friedman2010). This groundbreaking report was closely followed by another study that used WES as a means to identify a nonsense variant in a novel NSHL gene, GPSM2 (MIM 609245), in a consanguineous Palestinian family (Walsh et al., Reference Walsh, Shahin, Elkan-Miller, Lee, Thornton, Roeb, Abu Rayyan, Loulus, Avraham, King and Kanaan2010). Using WES, mutations in GPSM2 were later shown to cause Chudley-McCullough syndrome (MIM 604213), characterized by brain anomalies associated with deafness (Doherty et al., Reference Doherty, Chudley, Coghlan, Ishak, Innes, Lemire, Rogers, Mhanni, Phelps, Jones, Zhan, Fejes, Shahin, Kanaan, Akay, Tekin, Triggs-Raine. and Zelinski2012). These initial reports were important examples of using WES in ARNSHL. Following these studies, in 2011, Zheng et al. (Reference Zheng, Miller, Yang, Hildebrand, Shearer, DeLuca, Scheetz, Drummond, Scherer, Legan, Goodyear, Richardson, Cheatham, Smith and Dallos2011) were the first to use NGS to identify a mutation for autosomal dominant NSHL (ADNSHL), CEACAM16 (MIM 614591).
Analysis of the data obtained through WES is a complicated process, and filters must be applied to simplify the process. Identifying the responsible locus using homozygosity (a.k.a. autozygosity) mapping has been a very efficient way of narrowing the analytic field. With the decreasing cost of WES and understanding of the technology continually increasing, NGS without previously obtained mapping information is positioned to play a pivotal role in novel gene discovery. Behlouli et al. (Reference Behlouli, Bonnet, Abdi, Bouaita, Lelli, Hardelin, Schietroma, Rous, Louha, Cheknane, Lebdi, Boudjelida, Makrelouf, Zenati and Petit2014) identified a homozygous stop codon mutation in EPS8 (MIM 600206), encoding an actin-binding protein of cochlear hair cell stereocilia, as the cause of ARNSHL in two affected siblings from a consanguineous family. Similarly, Diaz-Horta et al. (Reference Diaz-Horta, Subasioglu-Uzak, Grati, DeSmidt, Foster, Cao, Bademci, Tokgoz-Yilmaz, Duman, Cengiz, Abad, Mittal, Blanton, Liu, Farooq, Walz, Lu and Tekin2014) reported another novel ARNSHL gene, FAM65B (MIM 611410), which encodes a membrane-associated protein of hair cell stereocilia. A homozygous splice site mutation was identified in a large consanguineous family. In both these latter cases, WES was employed without prior studies identifying the loci.
6. Diagnostic usage of NGS in NSHL
In clinical diagnostics for HL, targeted genomic enrichment followed by NGS has been shown to be an efficient strategy. In this scenario, a group of genes or a specific region are sequenced. Brownstein et al. (Reference Brownstein, Friedman, Shahin, Oron-Karni, Kol, Abu Rayyan, Parzefall, Lev, Shalev, Frydman, Davidov, Shohat, Rahile, Lieberman, Levy-Lahad, Lee, Shomron, King, Walsh, Kanaan and Avraham2011), using this strategy, screened 246 genes known to be responsible for human or mouse deafness in 11 probands of Middle Eastern origin and identified causative mutations in six of them. Shearer et al. (2013) suggested that this strategy can provide comprehensive genetic testing on a large number of patients with presumed genetic deafness. In their study, they used a panel that included 89 known deafness genes and yielded a diagnostic rate of 31% in ADNSHL and 56% in ARNSHL (Shearer et al., Reference Shearer, Black-Ziegelbein, Hildebrand, Eppsteiner, Ravi, Joshi, Guiffre, Sloan, Happe, Howard, Novak, Deluca, Taylor, Scheetz, Braun, Casavant, Kimberling, Leproust and Smith2013). In another study to investigate the diagnostic utility of targeted genomic enrichment followed by NGS in China, a panel designed to target 80 common deafness genes was used in 12 multiplex families with NSHL and causative variants were identified in four families (Wu et al., Reference Wu, Lin, Lu, Chen, Yang, Hsu and Chen2013). Finally, Vozzi et al. (Reference Vozzi, Morgan, Vuckovic, D'Eustacchio, Abdulhadi, Rubinato, Badii, Gasparini and Girotto2014) found the causative gene variants in four out of 12 families from Italy and Qatar and confirmed the usefulness of a targeted sequencing approach using a panel including 96 known genes related to HL.
On the other hand, WES has been shown to be effective in identifying causative mutations. In our recent WES study of 20 families prescreened for mutations in GJB2, 60% (12/20) of the cases had a mutation in a known gene; we were able to identify the causative mutation in the remaining eight families (Diaz-Horta et al., Reference Diaz-Horta, Duman, Foster, Sirmaci, Gonzalez, Mahdieh, Fotouhi, Bonyadi, Cengiz, Menendez, Ulloa, Edwards, Züchner, Blanton and Tekin2012). While targeted NGS panels provide higher coverage for individual genes, a significant lowering of costs, ease-of-analysis and ease-of-counselling, WES eliminates the need of continued development and validation of custom panels. Importantly, it offers a direct access to novel gene discovery in people who do not have mutations in known deafness genes (Diaz-Horta et al., Reference Diaz-Horta, Duman, Foster, Sirmaci, Gonzalez, Mahdieh, Fotouhi, Bonyadi, Cengiz, Menendez, Ulloa, Edwards, Züchner, Blanton and Tekin2012).
7. Conclusion
NGS is now an accepted clinical and research tool for the study of genetic deafness. It is a powerful approach for identifying patient-specific etiologies in genetically heterogeneous disorders and/or undiagnosed Mendelian phenotypes and has the potential to dramatically change the delivery of patient care. Hereditary HL presents as one of the most suitable disorders for the application of this ‘cutting-edge’ technology. NGS should be strongly considered, especially in cases where etiological diagnosis is inconclusive following established single gene testing, or when NGS presents as a faster and less expensive option for accurate genetic diagnosis.
This study was supported by National Institutes of Health Grant R01DC009645 to M.T. T.A. was supported by The Scientific and Technological Research Council of Turkey (TUBITAK) (project no: 1059B191401904).
Declaration of Interest
None.



