


1. Introduction
Measures of relationship specify the probabilities that relatives share alleles identical by descent (ibd), with the actual or realized identity at individual loci binomially distributed due to Mendelian segregation. At individual loci, the actual identity by descent is binomially distributed, but because of the linkage, there are covariances in this quantity among loci; therefore, there is still variation in the proportion of alleles-shared ibd and hence in the actual or realized relationship, even assuming infinitely many genomic sites. In previous papers, formulae for this variance have been obtained (Stam, Reference Stam1980; Hill, Reference Hill1993a, Reference Hillb; Guo, Reference Guo1995; Visscher, Reference Visscher2009) and have recently been generalized to cover all relationships (Hill & Weir, Reference Hill and Weir2011, subsequently HW11). In the previous analyses, ancestors were assumed not to be inbred; although formulae for variation in the actual inbreeding have been obtained by adapting those for variation in relationship (HW11).
The magnitude of the variation in actual relationship is important in several contexts, discussed further by HW11. These include the need to allow for relationship in genomic data cleaning and in association studies (Laurie et al., Reference Laurie, Doheny, Mirel, Pugh, Bierut, Bhangale, Boehm, Caporaso, Edenberg, Gabriel, Harris, Hu, Jacobs, Kraft, Landi, Lumley, Manolio, McHugh, Painter, Paschall, Rice, Rice, Zheng and Weir2010) and the ability to assess the pedigree relationship using genome sharing rather than just genotypes at individual loci, thereby incorporating the correlation structure induced by linkage. In quantitative genetic applications, the accuracy of prediction of breeding values in genomic selection programmes (Meuwissen et al., Reference Meuwissen, Hayes and Goddard2001) and of estimation of quantitative genetic parameters from variation within families (Visscher et al., Reference Visscher, Medland, Ferreira, Morley, Zhu, Cornes, Montgomery and Martin2006) depend on the variation in actual relationship.
Partially inbred individuals are found in all populations, arising from matings of close relatives such as full-sibs, more distant ones such as second cousins, and innumerable complex situations. Data from dense SNP markers and sequencing enable shared identity of genomic regions of individuals to be established (Weir et al., Reference Weir, Anderson and Hepler2006). For example, inbred individuals are found in some of the GENEVA consortium data being used in human genome-wide association studies (Cornelis et al., Reference Cornelis, Agrawal, Cole, Hansel, Barnes, Beaty, Bennett, Bierut, Boerwinkle, Doheny, Feenstra, Feingold, Fornage, Haiman, Harris, Hayes, Heit, Hu, Kang, Laurie, Ling, Teri, Manolio, Marazita, Mathias, Mirel, Paschall, Pasquale, Pugh, Rice, Udren, van Dam, Wang, Wiggs, Williams and Yu2010), from which variation in actual relationship has been demonstrated (Laurie et al., Reference Laurie, Doheny, Mirel, Pugh, Bierut, Bhangale, Boehm, Caporaso, Edenberg, Gabriel, Harris, Hu, Jacobs, Kraft, Landi, Lumley, Manolio, McHugh, Painter, Paschall, Rice, Rice, Zheng and Weir2010; HW11). Among pairs of individuals with the same pedigrees, there can be considerable variation in the estimates of the proportions of loci at which they share zero, one or two pairs of alleles ibd. In addition to the non-zero levels of inbreeding found in natural populations, deliberate inbreeding is undertaken in some breeding programmes. We now extend the results on variation in identity states obtained for non-inbred ancestors to those where the common ancestors of relatives are inbred.
The notation and methodology used here are based heavily on that used previously (HW11). Basically, the probability that descendants each carry identical alleles at a pair of linked loci is computed dependent on the relationship among the parents. The excess of this probability over that assuming loci are unlinked provides an estimate of the covariance that single sites carry identical alleles, and integrating this covariance over all pairs of sites provides the variance of actual identity. The analysis is extended here to include the probability that the parent or parents share alleles at pairs of linked loci as a consequence of their relatedness and the inbreeding of their common ancestors.
2. Measures of identity by descent
The inbreeding coefficient FX, the probability of ibd alleles at a locus, of an individual X in a pedigree is known to follow from the path-counting equation ∑(1/2)tθA;A where t is the number of individuals in a pedigree loop linking the individual's parents to their common ancestor A, and θA;A is the coancestry of A with itself: the probability that two alleles transmitted by that individual are ibd. This coancestry is given by θA;A = (1+FA)/2, where FA is the inbreeding coefficient of A. The count t includes the two parents but excludes the common ancestor, the factor 1/2 is for the passage of an allele through each individual in the pedigree loop, and the sum is over all distinct loops to A and over all common ancestors A.
For two loci, with recombination rate c between them, the path-counting equation for the probability of X receiving alleles ibd at each locus, through transmission of the ibd segments including both loci, is [(1−c)/2]tθA;A* (c) where θA;A* (c) is the two-locus coancestry for A with itself. This has value
 $$\theta _{A\semi A} \lpar c\rpar \tab \equals F_{A} \plus \beta \lsqb 1 \minus 2F_{A} \plus F_{A} \lpar c\rpar \rsqb \comma $$
$$\theta _{A\semi A} \lpar c\rpar \tab \equals F_{A} \plus \beta \lsqb 1 \minus 2F_{A} \plus F_{A} \lpar c\rpar \rsqb \comma $$where β = [(1−c)2+c 2]/2, as shown in Table 1 (Weir & Cockerham, Reference Weir and Cockerham1969). Here, FA*(c) is the two-locus inbreeding coefficient, or the probability that A has ibd alleles at both loci. Note that when the loci are completely linked, c = 0 (β = 1/2), F*A(0)=FA and θA;A* (0) = θA;A. When the loci segregate independently, c = 1/2 (β = 1/4), F*A(1/2)=F2A and θA;A* (1/2) = θA;A2.
Table 1. Two-locus coancestry† θA;A*(c) of individual A with itself as a function of the one- and two-locus inbreeding coefficients FA and FA*(c) of A. Individual A has genotype mimj/pipj at loci i, j.

† The probability that two gametes from A carry identical by descent (ibd) alleles at both loci.
The inbreeding coefficient of an individual is also the coancestry of its parents, so if X has parents V1, V2 (e.g. Figs 1 and 2) then FX = θV1;V2. Although these two quantities are equal, they have different reference points: the coancestries θ, θ* are for alleles on gametes transmitted by individuals, whereas the inbreeding coefficients F, F* are for alleles on gametes received by an individual, i.e. on gametes within an individual. There is need for this last perspective for more than one individual: ψY1;Y2 or ψY1;Y2* (c) are the probabilities of ibd for alleles at one or two loci on gametes received by individuals Y1 and Y2. Clearly, FX = ψX;X. The same path-counting equations hold for ψY1;Y2 as for θY1;Y2, but the count t then excludes Y1 and Y2.

Fig. 1. Pedigree for HS offspring Y1,Y2 of individual X, the offspring of HS parents.

Fig. 2. Pedigree for HS offspring Y1,Y2 of individual X, the offspring of FS parents.
(i) Inbred individual examples
Consider an inbred individual X, the offspring of a mating of half-sibs V1 and V2 who have common parent U2 (Fig. 1). The probability for alleles at any locus of X being ibd is the inbreeding coefficient FX = 1/8, and the variance in actual inbreeding among independent loci is FX(1−FX) = 7/64. For a recombination fraction between these sites of c, the two-locus inbreeding coefficient of X is given by F*X(c) = (1−c)2β/4 (Table 2), which reduces to 1/8 when c = 0 and to 1/64 when c = 1/2, i.e. FX and F2X, respectively. This argument is fairly easy to see because the probability of ibd for an individual at both sites is the same as the probability that two random haplotypes, sampled one from each parent, are ibd. Therefore, in this case where parents V1 and V2 are half-sibs, it is the probability that a pair of half-cousins, one with parent V1 and one with parent V2, share identical alleles at the two loci (HW11, Table 2). Weir & Cockerham (Reference Weir and Cockerham1969) presented a general algorithm for finding the probability of identity for alleles a, a′ and b, b′ at loci A and B, as shown in Appendix A.
Table 2. Correspondence between relationship and identity coefficients for common ancestor X at linked loci as a function of recombination rate c, β = [(1−c)2+c 2]/2 and of b = (1−c)/2.

Alternatively, consider an inbred individual X, the offspring of a mating of full-sibs V1 and V2 who have parents U1 and U2 (Fig. 2). The one- and two-locus inbreeding coefficients are FX = 1/4 and F*X(c) = (1−c)2β/2+c 2/8 (Table 2, Appendix A). The two-locus value reduces to 1/4 when c = 0 and to 1/16 when c = 1/2, i.e. FX and F2X, respectively. These results also follow as the probabilities of identity for alleles carried by two first cousins, one with parent V1 and one with parent V2 (HW11, Table 2).
3. Descendants of half-sibs
For unilineal relatives V1, V2 (e.g. Fig. 1), the inbreeding coefficient FX of their offspring X is the probability k 1 they share and transmit a pair of alleles ibd, and the path-counting equation is for identity resulting from that pair of alleles descending from common ancestor U2. The actual state of identity can be indicated by the variable ǩ 1 that takes the value 1 for identical alleles and 0 for non-identity. Taking expectations over all loci Ɛ(ǩ 1i)=k 1 and Var(ǩ 1i)=k 1(1−k 1). At two loci, i,j, the actual inbreeding coefficient is ![]() X*(c)=ǩ 1iǩ 1j and this has expectation FX*(c) = Ɛ(ǩ 1iǩ 1j)=F2X + Cov(ǩ 1i, ǩ 1j). The variance in the actual inbreeding of X averaged over the genome involves the sum of the variances at individual sites and the covariances at pairs of sites. With a large number of sites, it is the contribution of the covariances that dominates.
X*(c)=ǩ 1iǩ 1j and this has expectation FX*(c) = Ɛ(ǩ 1iǩ 1j)=F2X + Cov(ǩ 1i, ǩ 1j). The variance in the actual inbreeding of X averaged over the genome involves the sum of the variances at individual sites and the covariances at pairs of sites. With a large number of sites, it is the contribution of the covariances that dominates.
The relatedness of unilineal relatives also depends only on the measure k 1. If ǩ 1i indicates actual ibd status at locus i for the half-sibs Y1,Y2 with common parent X (Fig. 1)
 $${\cal{E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \rpar \equals \theta _{X\semi X} \equals {1 \over 2}\lpar 1 \plus F_{X} \rpar \comma $$
$${\cal{E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \rpar \equals \theta _{X\semi X} \equals {1 \over 2}\lpar 1 \plus F_{X} \rpar \comma $$ $${\cal{E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \theta _{X\semi X}{ \ast } \lpar c\rpar \equals F_{X} \plus \beta \lsqb 1 \minus 2F_{X} \plus F_{X}{\ast} \lpar c\rpar \rsqb \comma $$
$${\cal{E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \theta _{X\semi X}{ \ast } \lpar c\rpar \equals F_{X} \plus \beta \lsqb 1 \minus 2F_{X} \plus F_{X}{\ast} \lpar c\rpar \rsqb \comma $$ $${\rm {Cov}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \comma \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \theta _{X\semi X}{ \ast } \lpar c\rpar \minus \theta _{X\semi X}^{\setnum{2}} \equals \theta _{X\semi X}{\ast \lpar c\rpar \minus \theta _{X\semi X} \lpar 1\sol 2\rpar .$$
$${\rm {Cov}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \comma \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \theta _{X\semi X}{ \ast } \lpar c\rpar \minus \theta _{X\semi X}^{\setnum{2}} \equals \theta _{X\semi X}{\ast \lpar c\rpar \minus \theta _{X\semi X} \lpar 1\sol 2\rpar .$$To predict the sharing of ibd pairs of alleles by individuals who are descendants of Y1 and Y2 but are otherwise unrelated, note that the probability of a gametic pair of alleles is transmitted from parent to offspring is (1−c)/2 and to t-th generation descendants is [(1−c)/2]t. For example, t = 1 is for half-uncle nephew (e.g. Y1 and the offspring of Y2) and t = 2 is for half-cousins (e.g offspring of Y1 and Y2) or half-great uncle-great nephew (e.g. Y1 with a grandson of Y2). For descendants Z1, Z2 of Y1, Y2 such that there are t individuals (excluding Z1, Z2, X) in the loop from Z1 to X to Z2, Ɛ(ǩ 1i, ǩ 1j) = ψZ1;Z2* (c) and
 $${{\cal E}}\lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \tab \equals \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{t} \theta _{X\semi X}{ \ast } \lpar c\rpar .$$
$${{\cal E}}\lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \tab \equals \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{t} \theta _{X\semi X}{ \ast } \lpar c\rpar .$$To facilitate calculations over multiple generations, and to integrate over the chromosomes, we adopt methods used previously (HW11). Details are given in Appendix B. Letting b = (1−c)/2, we can write the right-hand side of eqn (2) as ∑nαnbn, and recognizing that setting c = 1/2, b = 1/4 (independent loci) gives the product of expected values Ɛ(ǩ 1i), Ɛ(ǩ 1j):
 $${\rm {Cov}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \comma \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \mathop\sum\limits_{n} \,\alpha _{n} \left[ {b^{n} \minus \mathop {\left( {{1 \over 4}} \right)}\nolimits^{n} } \right].$$
$${\rm {Cov}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \comma \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \mathop\sum\limits_{n} \,\alpha _{n} \left[ {b^{n} \minus \mathop {\left( {{1 \over 4}} \right)}\nolimits^{n} } \right].$$The range of values of n, and the values of αn, depend on the pedigree of the common ancestor X and we give common examples of θX;X* (c) in Table 2 (essentially for t = 1).
Assuming Haldane's mapping function, for a chromosome of length l Morgans, and computing the variance of actual relationship as the mean covariance over all pairs of loci,
 $${\rm{Var}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}} \comma l\rpar \equals \sum\limits_{n} \,\alpha _{n} \phi _{n} \lpar l\rpar \comma $$
$${\rm{Var}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}} \comma l\rpar \equals \sum\limits_{n} \,\alpha _{n} \phi _{n} \lpar l\rpar \comma $$where (Appendix B)
 $$\phi _{n} \lpar l\rpar \tab \equals \left\{ {\openup6\matrix{ \displaystyle{{1 \over {2l^{\setnum{2}} }}\mathop \displaystyle{\left( {{1 \over 4}} \right)}\nolimits^{n} \sum\limits_{r \equals \setnum{1}}^{n} \,\left( {n}\atop{r} \right)\displaystyle{{2rl \minus 1 \plus e^{ \minus \setnum{2}rl} } \over {r^{\setnum{2}} }}\comma } \tab {n\ges 1\comma } \cr {0\comma } \tab {n\les 0.} \cr} } \right.$$
$$\phi _{n} \lpar l\rpar \tab \equals \left\{ {\openup6\matrix{ \displaystyle{{1 \over {2l^{\setnum{2}} }}\mathop \displaystyle{\left( {{1 \over 4}} \right)}\nolimits^{n} \sum\limits_{r \equals \setnum{1}}^{n} \,\left( {n}\atop{r} \right)\displaystyle{{2rl \minus 1 \plus e^{ \minus \setnum{2}rl} } \over {r^{\setnum{2}} }}\comma } \tab {n\ges 1\comma } \cr {0\comma } \tab {n\les 0.} \cr} } \right.$$For the genome as a whole, letting li be the map length of chromosome i and ∑ili=L, the variance is ∑ili 2Var(ǩ 1, li)/L 2.
If X is the result of a parent-offspring (PO) mating or a full-sib (FS) mating, for example, FX = 1/4; but we show in Table 2 that the θX;X* (c) values are different unless c = 0 or c = 1/2. This leads to different variances of the actual identities for half-sib progeny of X and pairs of their descendants:
 $$\eqalign{\tab{\rm {Var}}\lpar \mathop {\v{k} }\nolimits_{\setnum{1}} \comma l\rpar\cr \tab \equals \left\{\openup3pt {\matrix{\! {{\rm{PO}}\colon } \tab\!\!\! {16\phi _{t \plus \setnum{5}} \lpar l\rpar \minus 16\phi _{t \plus \setnum{4}} \lpar l\rpar}\hfill\cr\tab\ensp{ \plus8\phi _{t \plus \setnum{3}} \lpar l\rpar \minus {3 \over 4}\phi _{t \plus \setnum{1}} \lpar l\rpar \plus {1 \over 2}\phi _{t} \lpar l\rpar \comma }\hfill \cr{{\rm {FS}}\colon } \tab\!\!\! {32\phi _{t \plus \setnum{6}} \lpar l\rpar \minus 32\phi _{t \plus \setnum{5}} \lpar l\rpar \plus 18\phi _{t \plus \setnum{4}} \lpar l\rpar }\hfill\cr\tab\ensp { \minus 7\phi _{t \plus \setnum{3}} \lpar l\rpar \plus {{17} \over 4}\phi _{t \plus \setnum{2}} \lpar l\rpar \minus {3 \over 2}\phi _{t \plus \setnum{1}} \lpar l\rpar \plus {9 \over {16}}\phi _{t} \lpar l\rpar } \right.$$
$$\eqalign{\tab{\rm {Var}}\lpar \mathop {\v{k} }\nolimits_{\setnum{1}} \comma l\rpar\cr \tab \equals \left\{\openup3pt {\matrix{\! {{\rm{PO}}\colon } \tab\!\!\! {16\phi _{t \plus \setnum{5}} \lpar l\rpar \minus 16\phi _{t \plus \setnum{4}} \lpar l\rpar}\hfill\cr\tab\ensp{ \plus8\phi _{t \plus \setnum{3}} \lpar l\rpar \minus {3 \over 4}\phi _{t \plus \setnum{1}} \lpar l\rpar \plus {1 \over 2}\phi _{t} \lpar l\rpar \comma }\hfill \cr{{\rm {FS}}\colon } \tab\!\!\! {32\phi _{t \plus \setnum{6}} \lpar l\rpar \minus 32\phi _{t \plus \setnum{5}} \lpar l\rpar \plus 18\phi _{t \plus \setnum{4}} \lpar l\rpar }\hfill\cr\tab\ensp { \minus 7\phi _{t \plus \setnum{3}} \lpar l\rpar \plus {{17} \over 4}\phi _{t \plus \setnum{2}} \lpar l\rpar \minus {3 \over 2}\phi _{t \plus \setnum{1}} \lpar l\rpar \plus {9 \over {16}}\phi _{t} \lpar l\rpar } \right.$$ The above results give the variance of ǩ 1. As Y1 and Y2 and their descendants cannot share both genes at a locus (i.e. k 2 = 0), the variation in actual relationship 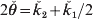 $2{\skew3\v\theta } \equals \v{k} _{\setnum{2}} \plus \v{k} _{\setnum{1}}\sol 2}$ is given by Var(ǩ 1, l)/4 and in actual co-ancestry
$2{\skew3\v\theta } \equals \v{k} _{\setnum{2}} \plus \v{k} _{\setnum{1}}\sol 2}$ is given by Var(ǩ 1, l)/4 and in actual co-ancestry  ${\skew3\v\theta } \equals {\v k}_{\setnum{2}} \sol 2 \plus \v{k} _{\setnum{1}} \sol 4$ by Var(ǩ 1, l)/16.
${\skew3\v\theta } \equals {\v k}_{\setnum{2}} \sol 2 \plus \v{k} _{\setnum{1}} \sol 4$ by Var(ǩ 1, l)/16.
4. Descendants of full-sibs
We now consider the case of matings between female X1 and male X2, unrelated to each other but with inbreeding coefficients F X1 and F X2, respectively, and evaluate the variance in actual relationship among their full-sib progeny Y1 and Y2 and descendants of these such as first cousins.
Full-sibs can share 0, 1 or 2 alleles at each locus. As haplotypes are transmitted independently by the two parents, the variance in relationship among full-sibs is simply the sum of the components from paternal and maternal half-sibs with relevant inbreeding coefficients.
The actual state for Y1 and Y2 sharing pairs of alleles at each of two loci, i and j, is ǩ 2iǩ 2j=ǩ 1im ǩ 1ip ǩ 1jm ǩ 1jp where m and p denote maternally and paternally derived alleles. Hence, from the definition of the two-locus coancestry,
 $${\cal{E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{2}i} \mathop {\v{k} }\nolimits_{\setnum{2}j} \rpar \equals {\cal{E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i}^{m} \mathop {\v{k} }\nolimits_{\setnum{1}j}^{m} \rpar {\cal{E}}\lpar \mathop {\v{k} }\nolimits_{\setnum{1}i}^{p} \mathop {\v{k} }\nolimits_{\setnum{1}j}^{p} \rpar \equals \theta _{X\setnum{1}\semi X\setnum{1}}{ \ast } \lpar c\rpar \theta _{X\setnum{2}\semi X\setnum{2}}{ \ast } \lpar c\rpar \comma$$
$${\cal{E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{2}i} \mathop {\v{k} }\nolimits_{\setnum{2}j} \rpar \equals {\cal{E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i}^{m} \mathop {\v{k} }\nolimits_{\setnum{1}j}^{m} \rpar {\cal{E}}\lpar \mathop {\v{k} }\nolimits_{\setnum{1}i}^{p} \mathop {\v{k} }\nolimits_{\setnum{1}j}^{p} \rpar \equals \theta _{X\setnum{1}\semi X\setnum{1}}{ \ast } \lpar c\rpar \theta _{X\setnum{2}\semi X\setnum{2}}{ \ast } \lpar c\rpar \comma$$which reduces to θX1;X1θX2;X2 = (1+F X1)(1+F X2)/4 if c = 0, β = 1/2 and to the square of that if c = 1/2, β = 1/4. Evaluation depends on the pedigrees of X1 and X2, but is straightforward by expansion in terms of coefficients b as above and in Table 2.
The sharing of single copies among descendants of the full-sibs can be evaluated extending the methods for descendants of half-sibs. Suppose that parents X1, X2 have full-sib offspring Y1, Y2 and Y2 has offspring Z2. Then Y1 and Z2 are uncle and nephew and they can have only one ibd allele at each locus. Either X1 or X2 can transmit an entire haplotype to both Y1 and Y2 and the latter haplotype can be transmitted to Z2. This probability of the event is [θX1;X1*(c) + θX2;X2*(c)](1−c)/2 and it results in Y1 and Z2 sharing the haplotype. Alternatively, X1 can transmit ibd alleles at one locus and X2 can transmit ibd alleles at the other locus so Y1,Y2 share two pairs of ibd alleles: if Y2 then transmits these ibd alleles to Z2 then uncle and nephew again share ibd alleles at both loci. The probability of this event is cθX1;X1θX2;X2 so
 $$\eqalign{{\cal E} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals{\displaystyle{1 \over 2}}\lpar 1 \!\minus\! c\rpar \lsqb \theta _{X\setnum{1}\semi X\setnum{1}}{\ast } \lpar c\rpar \plus \theta _{X\setnum{2}\semi X\setnum{2}}{ \ast } \lpar c\rpar \rsqb \plus c\theta _{X\setnum{1}\semi X\setnum{1}} \theta _{X\setnum{2}\semi X\setnum{2}} \comma $$
$$\eqalign{{\cal E} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals{\displaystyle{1 \over 2}}\lpar 1 \!\minus\! c\rpar \lsqb \theta _{X\setnum{1}\semi X\setnum{1}}{\ast } \lpar c\rpar \plus \theta _{X\setnum{2}\semi X\setnum{2}}{ \ast } \lpar c\rpar \rsqb \plus c\theta _{X\setnum{1}\semi X\setnum{1}} \theta _{X\setnum{2}\semi X\setnum{2}} \comma $$which reduces to (θX1;X1 + θX2;X2)/2 = (2+F X1+F X2)/4 if c = 0, and to the square of that if c = 1/2. For great uncle-great nephew and more distant uncle–nephew relationships, the probabilities are obtained as products of terms in eqn (5) by powers of (1−c)/2.
Similarly, for cousins Z1,Z2, the offspring of Y1,Y2 and the grand-offspring of X1,X2
 $$\eqalign{ {{\cal E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \tab {1 \over 2}\mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}}\times \lsqb \theta _{X\setnum{1}\semi X\setnum{1}}{ \ast } \lpar c\rpar \plus \theta _{X\setnum{2}\semi X\setnum{2}}{ \ast } \lpar c\rpar \rsqb \cr \tab \plus {1 \over 2}c^{\setnum{2}} \theta _{X\setnum{1}\semi X\setnum{1}} \theta _{X\setnum{2}\semi X\setnum{2}} . \cr} $$
$$\eqalign{ {{\cal E}} \lpar \mathop {\v{k} }\nolimits_{\setnum{1}i} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \tab {1 \over 2}\mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}}\times \lsqb \theta _{X\setnum{1}\semi X\setnum{1}}{ \ast } \lpar c\rpar \plus \theta _{X\setnum{2}\semi X\setnum{2}}{ \ast } \lpar c\rpar \rsqb \cr \tab \plus {1 \over 2}c^{\setnum{2}} \theta _{X\setnum{1}\semi X\setnum{1}} \theta _{X\setnum{2}\semi X\setnum{2}} . \cr} $$Values for later descendants are obtained by scaling eqn (6), for example, by (1−c)/2 for cousins once removed and by [(1−c)/2]2 for second cousins or cousins twice removed. All expressions for Ɛ(ǩ 1iǩ 1j) can be written as polynomials in b and evaluated accordingly.
5. Mapping functions, map length and physical genome length
In the analysis undertaken in this paper and in previous analyses of variation in genome sharing (HW11 and references cited therein) and indeed in other studies on other statistics such as distribution of lengths of shared regions (Stam, Reference Stam1980; Donnelly, 1983), the Haldane mapping function (Haldane, Reference Haldane1919), c = (1−e −2l)/2, has been used. Not least, this is mathematically tractable, and explicit integration of the formulae-relating recombination fraction to map length is feasible, as in eqn (3). Haldane's function does not allow for interference, however, and various others have been constructed to incorporate interference. The importance of this assumption in variances of genome sharing, whether or not parents are inbred, has not been checked.
In mammalian studies, the Kosambi mapping function (Kosambi, Reference Kosambi1944), c = (1−e −4l)/[2(1+e −4l)] is most widely used, including in the published human linkage map (Matise et al., Reference Matise, Chen, Chen, De la Vega, Hansen, He, Hyland, Kennedy, Kong, Murray, Ziegle, Stewart and Buyske2007). For both functions c → l as l → 0 and c → 0·5 as l → ∞ but, for intermediate values of l, c is relatively larger for the Kosambi function: for example, for l = 0·5, where the absolute difference is near its maximum, for Haldane c = 0·316 and for Kosambi c = 0·381. To assess the dependence of the variation in genome sharing on the mapping function, numerical integration was used to evaluate Appendix eqn (B2), replacing the term (1+e −2(x−y))/4 for b = (1−c)/2 by [2− (1−e −4(x−y))/(1+e −4(x−y))]/4. Numerical integration using bivariate Simpson's rule was used, and precision was checked by concurrent numerical integration of the Haldane function.
The variance of actual relationship is smaller with the Kosambi than the Haldane mapping function (Appendix C), as would be expected because the recombination fraction is, for given map length, larger with the former. The disparity increases the longer the chromosome, but it already differs little between l of 2 and 3M. Although the degree of relationship and type of relationship, for example, lineal or collateral, have some effect, it is rather small. Hence, as an approximate conclusion, the SD of relationship for l of 0·5, 1, 2 and 3M is about 4, 7, 10 and 11% smaller, respectively, with the Kosambi function incorporating interference (Appendix C). Although these are clear differences, the qualitative impact is rather small, and likely to be a little under 10% for the human genome as a whole.
Observations of genomic identity between chromosomes at the molecular level are initially likely to be in terms of the physical length, measured in Megabases not map lengths. Most or all calculations in this and other work on prediction of lengths of genome sharing are at the level of map distance. The conversion from one to the other then depends on the correspondence of the physical and linkage maps. This varies among chromosomes and species, around the typical mammalian figure of 1 cm/Mb, depending inter alia on positions of centromeres and repetitive regions, and the ratio of Morgans to Mb depends on chromosome length and differs among chromosomes; for example, the chicken has a very high M/Mb ratio relative to mammals and indeed relative to the zebra finch, but for both species of birds, the recombination rates on the microchromosomes are relatively high (Stapley et al., Reference Stapley, Birkhead, Burke and Slate2008). For human chromosomes, although the linkage map is not far from linearly related to the physical map for the longer metacentric chromosomes, the relationship is somewhat sigmoidal; whereas for the shortest acrocentric chromosomes, no recombination are seen for over 25% of the centromeric end (Matise et al., Reference Matise, Chen, Chen, De la Vega, Hansen, He, Hyland, Kennedy, Kong, Murray, Ziegle, Stewart and Buyske2007 and http://compgen.rutgers.edu/RutgersMap/MapBrowser.aspx). Generalizations are therefore difficult, but it does imply a need to convert the initially observed lengths of shared regions into map distances before drawing inferences from analyses such as that presented here.
6. Discussion
The methodology given here fills a small lacuna in the analysis of variation in actual relationship, but to our knowledge has not been analysed previously. The formulae may be complicated, but the algorithms are easy to use.
As an example, consider the case of variance, expressed as sd, in actual relationship of half-sibs when the common parent of these sibs has undergone inbreeding (Fig. 3 a) by one of several routes. The sd is not greatly reduced by the parental inbreeding, even in the case of selfing (F = 0·5), but the coefficient of variation CV (Fig. 3 b) is reduced substantially more, because the expected relationship increases with F. The values differ only very slightly according to the mode of inbreeding for given F for example, by an offspring-parent compared with a full-sib mating.

Fig. 3. (a) sd and (b) CV of actual relationship as a function of map length (l) of HS offspring of individuals whose parents were unrelated (F = 0), or obtained by uncle-niece or HS mating (F = 1/8), or obtained by FS or PO mating (F = 1/4), or by selfing (F = 1/2). Expected relationships for these values of F are 0·25, 0·281, 0·312 and 0·375, respectively.
Also consider comparisons between different levels of relationship according to the degree of inbreeding of the parent. For a single locus, or completely linked loci, from eqn (2) setting c = 0 and hence F(X)=F*X(c),Var(ǩ 1, 0) = (1/2)t +1(1+FX)[1 − (1/2)t +1(1+FX)]. Thus, for half-sib offspring (t = 0), the variance is highest when FX = 0; but for t > 0, it is highest when FX = 1. Examples shown in Fig. 4 comparing variances for half-sibs and half-cousins as a function of map length and degree of inbreeding of the parent indicate that, as map length increases, the ranking of variances remains the same, i.e. decreasing with FX for half-sibs and increasing with FX for half-cousins. The (likely) explanation is that all half-sib offspring inherit a haplotype from their parent, which are therefore increasingly similar the more inbred is the parent. In contrast, a grandoffspring has a 50% chance of inheriting no haplotype from the inbred parent, and the similarity of these is more than outweighed by the divergence between the inbred and non-inbred parent.

Fig. 4. sd of actual relationship as a function of map length (l) of HS offspring and half-cousin (HC) descendants of individuals whose parents were unrelated (none, F = 0), or got by HS mating (F = 1/8), or got by FS mating (F = 1/4), or by selfing (self, F = 1/2).
For offspring of full-sib matings, however, where for individual loci or no recombination Var(ǩ 1, 0)∝(2+F X1+F X2)[1 − (2+F X1+F X2)/4] (from eqns (5) or (6)), the variance of relationship falls as inbreeding of either parent rises. This is as would be expected from the preceding argument on half-sibs because the grandoffspring must inherit from one or other grandparent.
This work was supported in part by NIH grant (GM 075091) and the Leverhulme Trust. The authors thank Ian White for helpful comments.
Appendix A. Derivation of two-locus descent measures
Weir & Cockerham (Reference Weir and Cockerham1969) presented a general algorithm for finding the probability of identity by descent for alleles a, a′ and b, b′ at loci A and B, respectively. Depending on whether these four alleles are transmitted on two gametes (ab from one individual U1 and a′b′ from another individual U2), or three gametes (ab from one individual U1, a′ from a second individual U2 and b′ from a third individual U3), or four gametes (a, b, a′, b′ from different individuals U1, U2, U3, U4) the probabilities are written as θU1;U2*(c), γU1;U2,U3*(c) or δU1,U2;U3,U4*(c), respectively. Calculation of any of these probabilities proceeds by tracing alleles back to founding individuals in a pedigree, taking recombination into account when necessary.
For individual X in Fig. 1, the offspring of half-sib parents, the two pairs of alleles ab, a′b′ are from individuals V1, V2 and may all have descended from U2 so
 $$\eqalign{ F_{X}{ \ast } \lpar c\rpar \equals \tab \theta _{V\setnum{1}\semi V\setnum{2}}{ \ast } \lpar c\rpar \cr \equals \tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \theta _{U\setnum{1}\semi U\setnum{2}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{1}\semi U\setnum{3}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{2}\semi U\setnum{2}}{ \ast } \lpar c\rpar\cr\tab \plus \theta _{U\setnum{2}\semi U\setnum{3}}{ \ast } \lpar c\rpar \rsqb \plus \left( {{{1 \minus c} \over 2}} \right)\left( {{c \over 2}} \right)\lsqb \gamma _{U\setnum{1}\semi U\setnum{2}\comma U\setnum{3}}{ \ast } \lpar c\rpar\cr \tab \plus \gamma _{U\setnum{1}\semi U\setnum{3}\comma U\setnum{2}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{2}\comma U\setnum{3}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{3}\comma U\setnum{2}}{ \ast } \lpar c\rpar \cr \tab \plus \gamma _{U\setnum{2}\semi U\setnum{1}\comma U\setnum{2}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{3}\semi U\setnum{1}\comma U\setnum{2}}{ \ast } \lpar c\rpar \cr \tab \plus \gamma _{U\setnum{3}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \rsqb \plus \mathop {\left( {{c \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{2}\comma U\setnum{3}}{ \ast } \lpar c\rpar \cr \tab\plus \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{3}\comma U\setnum{2}}{ \ast } \lpar c\rpar \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{2}\comma U\setnum{3}}{ \ast } \lpar c\rpar \cr \tab \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{3}\comma U\setnum{2}}{ \ast } \lpar c\rpar \rsqb . \cr} $$
$$\eqalign{ F_{X}{ \ast } \lpar c\rpar \equals \tab \theta _{V\setnum{1}\semi V\setnum{2}}{ \ast } \lpar c\rpar \cr \equals \tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \theta _{U\setnum{1}\semi U\setnum{2}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{1}\semi U\setnum{3}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{2}\semi U\setnum{2}}{ \ast } \lpar c\rpar\cr\tab \plus \theta _{U\setnum{2}\semi U\setnum{3}}{ \ast } \lpar c\rpar \rsqb \plus \left( {{{1 \minus c} \over 2}} \right)\left( {{c \over 2}} \right)\lsqb \gamma _{U\setnum{1}\semi U\setnum{2}\comma U\setnum{3}}{ \ast } \lpar c\rpar\cr \tab \plus \gamma _{U\setnum{1}\semi U\setnum{3}\comma U\setnum{2}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{2}\comma U\setnum{3}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{3}\comma U\setnum{2}}{ \ast } \lpar c\rpar \cr \tab \plus \gamma _{U\setnum{2}\semi U\setnum{1}\comma U\setnum{2}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{3}\semi U\setnum{1}\comma U\setnum{2}}{ \ast } \lpar c\rpar \cr \tab \plus \gamma _{U\setnum{3}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \rsqb \plus \mathop {\left( {{c \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{2}\comma U\setnum{3}}{ \ast } \lpar c\rpar \cr \tab\plus \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{3}\comma U\setnum{2}}{ \ast } \lpar c\rpar \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{2}\comma U\setnum{3}}{ \ast } \lpar c\rpar \cr \tab \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{3}\comma U\setnum{2}}{ \ast } \lpar c\rpar \rsqb . \cr} $$Ignoring the terms that are zero (those with alleles at the same site coming from different ancestors), and using eqn (1) with FU 2=FU 2*(c) = 0
 $$\eqalign{ F_{X}{ \ast } \lpar c\rpar \equals \tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \theta _{U\setnum{2}\semi U\setnum{2}}{ \ast } \lpar c\rpar \cr \tab \equals {1 \over 4}\beta \lpar 1 \minus c\rpar ^{\setnum{2}} \comma \cr} $$
$$\eqalign{ F_{X}{ \ast } \lpar c\rpar \equals \tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \theta _{U\setnum{2}\semi U\setnum{2}}{ \ast } \lpar c\rpar \cr \tab \equals {1 \over 4}\beta \lpar 1 \minus c\rpar ^{\setnum{2}} \comma \cr} $$where β = [(1−c)2+c 2]/2. Setting c = 0 gives the one-locus result FX = 1/8, and setting c = 1/2 gives the square of that.
For individual X in Fig. 2, the offspring of full-sib parents, the two pairs of alleles ab, a′b′ are from individuals V1, V2 and then from U1 and U2 so
 $$\eqalign{ F_{X}{ \ast } \lpar c\rpar \tab \equals \theta _{V\setnum{1}\semi V\setnum{2}}{ \ast } \lpar c\rpar \cr \equals \tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \theta _{U\setnum{1}\semi U\setnum{1}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{1}\semi U\setnum{2}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{2}\semi U\setnum{1}}{ \ast } \lpar c\rpar \cr\tab\plus \theta _{U\setnum{2}\semi U\setnum{2}}{ \ast } \lpar c\rpar \rsqb \plus 2\left( {{{1 \minus c} \over 2}} \right)\left( {{c \over 2}} \right)\lsqb \gamma _{U\setnum{1}\semi U\setnum{1}\comma U\setnum{2}}{ \ast } \lpar c\rpar \cr \tab \plus \gamma _{U\setnum{1}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{1}\comma U\setnum{2}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \rsqb \cr \tab \plus \mathop {\left( {{c \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{1}\comma U\setnum{2}}{ \ast} \lpar c\rpar \plus \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{2}\comma U\setnum{1}}{ \ast} \lpar c\rpar \cr \tab \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{1}\comma U\setnum{2}}{ \ast} \lpar c\rpar \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \rsqb . \cr} $$
$$\eqalign{ F_{X}{ \ast } \lpar c\rpar \tab \equals \theta _{V\setnum{1}\semi V\setnum{2}}{ \ast } \lpar c\rpar \cr \equals \tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \theta _{U\setnum{1}\semi U\setnum{1}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{1}\semi U\setnum{2}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{2}\semi U\setnum{1}}{ \ast } \lpar c\rpar \cr\tab\plus \theta _{U\setnum{2}\semi U\setnum{2}}{ \ast } \lpar c\rpar \rsqb \plus 2\left( {{{1 \minus c} \over 2}} \right)\left( {{c \over 2}} \right)\lsqb \gamma _{U\setnum{1}\semi U\setnum{1}\comma U\setnum{2}}{ \ast } \lpar c\rpar \cr \tab \plus \gamma _{U\setnum{1}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{1}\comma U\setnum{2}}{ \ast } \lpar c\rpar \plus \gamma _{U\setnum{2}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \rsqb \cr \tab \plus \mathop {\left( {{c \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{1}\comma U\setnum{2}}{ \ast} \lpar c\rpar \plus \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{2}\comma U\setnum{1}}{ \ast} \lpar c\rpar \cr \tab \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{1}\comma U\setnum{2}}{ \ast} \lpar c\rpar \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{2}\comma U\setnum{1}}{ \ast } \lpar c\rpar \rsqb . \cr} $$Ignoring the terms that are zero, and using eq (1) with FU 1=FU 1*(c)=FU 2=FU 2*(c) = 0.
 $$\eqalign{ F_{X}{ \ast } \lpar c\rpar \equals\tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \theta _{U\setnum{1}\semi U\setnum{1}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{2}\semi U\setnum{2}}{ \ast} \lpar c\rpar \rsqb \cr \tab \plus \mathop {\left( {{c \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{1}\comma U\setnum{2}}{ \ast} \lpar c\rpar \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{2}\comma U\setnum{1}}{ \ast} \lpar c\rpar \rsqb \cr \equals \tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \lpar 1 \minus c\rpar ^{\setnum{2}} \plus \lpar c\rpar ^{\setnum{2}} \rsqb \plus \mathop {\left( {{c \over 2}} \right)}\nolimits^{\setnum{2}} 2\mathop {\left( {{1 \over 2}} \right)}\nolimits^{\setnum{2}} \cr \equals \tab{1 \over 2}\beta \lpar 1 \minus c\rpar ^{\setnum{2}} \plus {1 \over 8}c^{\setnum{2}} . \cr} $$
$$\eqalign{ F_{X}{ \ast } \lpar c\rpar \equals\tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \theta _{U\setnum{1}\semi U\setnum{1}}{ \ast } \lpar c\rpar \plus \theta _{U\setnum{2}\semi U\setnum{2}}{ \ast} \lpar c\rpar \rsqb \cr \tab \plus \mathop {\left( {{c \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \delta _{U\setnum{1}\comma U\setnum{2}\semi U\setnum{1}\comma U\setnum{2}}{ \ast} \lpar c\rpar \plus \delta _{U\setnum{2}\comma U\setnum{1}\semi U\setnum{2}\comma U\setnum{1}}{ \ast} \lpar c\rpar \rsqb \cr \equals \tab \mathop {\left( {{{1 \minus c} \over 2}} \right)}\nolimits^{\setnum{2}} \lsqb \lpar 1 \minus c\rpar ^{\setnum{2}} \plus \lpar c\rpar ^{\setnum{2}} \rsqb \plus \mathop {\left( {{c \over 2}} \right)}\nolimits^{\setnum{2}} 2\mathop {\left( {{1 \over 2}} \right)}\nolimits^{\setnum{2}} \cr \equals \tab{1 \over 2}\beta \lpar 1 \minus c\rpar ^{\setnum{2}} \plus {1 \over 8}c^{\setnum{2}} . \cr} $$Setting c = 0 gives the one-locus result FX = 1/4, and setting c = 1/2 gives the square of that.
Appendix B. Evaluation of covariances (based on HW11)
Let b = (1−c)/2, the probability a pair of loci are jointly transmitted between generations, and express powers of c as polynomials in b:
 $$c^{n} \equals \mathop\sum\limits_{i \equals \setnum{0}}^{n} \,\left( {\matrix{ n \cr i \cr} } \right)\lpar \minus 2b\rpar ^{i} .$$
$$c^{n} \equals \mathop\sum\limits_{i \equals \setnum{0}}^{n} \,\left( {\matrix{ n \cr i \cr} } \right)\lpar \minus 2b\rpar ^{i} .$$Writing θX;X*(c) = Ɛ(ǩ i 1ǩ 1j) as a polynomial (examples in Table 2)
 $${\cal E} \lpar \mathop {\v{k} }\nolimits_{i\setnum{1}} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \mathop\sum\limits_{n \equals \setnum{0}}^{N} \,\alpha _{n} b^{n} \comma $$
$${\cal E} \lpar \mathop {\v{k} }\nolimits_{i\setnum{1}} \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \equals \mathop\sum\limits_{n \equals \setnum{0}}^{N} \,\alpha _{n} b^{n} \comma $$and noting that the covariance is zero for unlinked loci (b = 1/4),
 $${\rm {Cov}}\lpar \mathop {\v{k} }\nolimits_{i\setnum{1}} \comma \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \tab \equals \mathop\sum\limits_{n \equals \setnum{0}}^{N} \,\alpha _{n} \left[ {b^{n} \minus \mathop {\left( {{1 \over 4}} \right)}\nolimits^{n} } \right].$$
$${\rm {Cov}}\lpar \mathop {\v{k} }\nolimits_{i\setnum{1}} \comma \mathop {\v{k} }\nolimits_{\setnum{1}j} \rpar \tab \equals \mathop\sum\limits_{n \equals \setnum{0}}^{N} \,\alpha _{n} \left[ {b^{n} \minus \mathop {\left( {{1 \over 4}} \right)}\nolimits^{n} } \right].$$Assuming Haldane's mapping function, b = (1−c)/2 = (1+e −2d)/4 where d is the map distance between the loci, so bn−(1/4)n = (1/4)n[(1+e− 2d)n−1]. Integrating over all pairs of loci, we define
 $$\eqalign{\phi _{n} \lpar l\rpar \tab \equals {2 \over {l^{\setnum{2}} }}\mathop {\left( {{1 \over 4}} \right)}\nolimits^{n} \int_{x \equals \setnum{0}}^{l} \,\int_{y \equals \setnum{0}}^{x} \,\lsqb \lpar 1 \plus e^{ \minus \setnum{2}\lpar x \minus y\rpar } \rpar ^{n} \minus 1\rsqb \, dy\,dx.$$
$$\eqalign{\phi _{n} \lpar l\rpar \tab \equals {2 \over {l^{\setnum{2}} }}\mathop {\left( {{1 \over 4}} \right)}\nolimits^{n} \int_{x \equals \setnum{0}}^{l} \,\int_{y \equals \setnum{0}}^{x} \,\lsqb \lpar 1 \plus e^{ \minus \setnum{2}\lpar x \minus y\rpar } \rpar ^{n} \minus 1\rsqb \, dy\,dx.$$Integration of eqn (B2) gives eqn (3) in the text.
Appendix C Effect of mapping function
sd of actual relationship computed using Kosambi mapping function divided by sd of actual relationship computed using the Haldane mapping function for different map lengths and pedigree relationships, including cases where the common ancestor is inbred.








