


1. Introduction
Marfan syndrome (MFS) (OMIM#154700) is an autosomal dominant connective tissue disorder with a prevalence of 0·075 to 0·86 per 5000 individuals (von Kodolitsch et al., Reference von Kodolitsch, De Backer, Schuler, Bannas, Behzadi, Bernhardt, Hillebrand, Fuisting, Sheikhzadeh, Rybczynski, Kölbel, Püschel, Blankenberg and Robinson2015). MFS is a multisystemic disorder involving the ocular, skeletal and cardiovascular systems. Myopia and lens dislocation are the most common ocular features, while skeletal involvement is characterized by long bone overgrowth, pectus deformity and arachnodactyly (Fig. 1(a) and (b)). However, the most life-threatening complications in MFS are related to the cardiovascular system (Fig. 1(c)). These include aortic root dilatation primarily at the level of the sinuses of Valsalva, aortic dissection and rupture, mitral valve prolapse, mitral regurgitation and arrhythmias (Van Laer et al., Reference Van Laer, Proost and Loeys2013; Cherkas et al., Reference Cherkas and Zhuraev2016). In 1991, the fibrillin-1 gene (FBN1), which encodes a 350 kDa glycoprotein, was identified as the gene responsible for MFS (Dietz et al., Reference Dietz, Cutting, Pyeritz, Maslen, Sakai, Corson, Puffenberger, Hamosh, Nanthakumar and Curristin1991). Fibrillin-1, as a part of the extracellular matrix (ECM), provides elasticity and structural support to tissues and plays an important role in TGF-β signalling. Thus, mutations in FBN1 lead to a loss of ECM integrity and to a dysregulation of the downstream TGF-β signalling pathway (Neptune et al., Reference Neptune, Frischmeyer, Arking, Myers, Bunton, Gayraud, Ramirez, Sakai and Dietz2003; Ramachandra et al., Reference Ramachandra, Mehta, Guo, Wong, Tan and Shim2015).
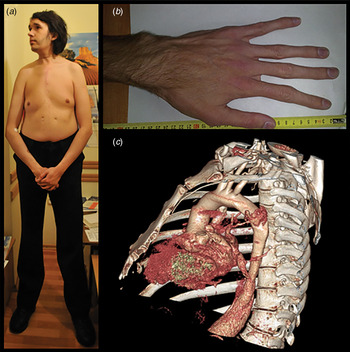
Fig. 1. Clinical features of MFS. Typical MFS patients with (a) disproportionate long bone overgrowth, (b) arachnodactyly and (c) thoracic aortic aneurysm.
In 80 to 100% of MFS patients, a FBN1 mutation can be identified (Loeys et al., Reference Loeys, De Backer, Van Acker, Wettinck, Pals, Nuytinck, Coucke and De Paepe2004; Faivre et al., Reference Faivre, Collod-Beroud, Ades, Arbustini, Child, Callewaert, Loeys, Binquet, Gautier, Mayer, Arslan-Kirchner, Grasso, Beroud, Hamroun, Bonithon-Kopp, Plauchu, Robinson, De Backer, Coucke, Francke, Bouchot, Wolf, Stheneur, Hanna, Detaint, De Paepe, Boileau and Jondeau2011; Radonic et al., Reference Radonic, de Witte, Groenink, de Bruin-Bon, Timmermans, Scholte, van den Berg, Baars, van Tintelen, Kempers, Zwinderman and Mulder2011; Sheikhzadeh et al., Reference Sheikhzadeh, Kade, Keyser, Stuhrmann, Arslan-Kirchner, Rybczynski, Bernhardt, Habermann, Hillebrand, Mir, Robinson, Berger, Detter, Blankenberg, Schmidtke and von Kodolitsch2011; Aalberts et al., Reference Aalberts, Thio, Schuurman, van Langen, van der Pol, van Tintelen and van den Berg2012; Yang et al., Reference Yang, Han, Jang, Moon, Sung, Chung, Lee, Ki and Kim2012; Proost et al., Reference Proost, Vandeweyer, Meester, Salemink, Kempers, Ingram, Peeters, Saenen, Vrints, Lacro, Roden, Wuyts, Dietz, Mortier, Loeys and Van Laer2015; von Kodolitsch et al., Reference von Kodolitsch, De Backer, Schuler, Bannas, Behzadi, Bernhardt, Hillebrand, Fuisting, Sheikhzadeh, Rybczynski, Kölbel, Püschel, Blankenberg and Robinson2015). Despite the presence of identical mutations, a large inter- and intra-familial phenotypic variability is observed, suggesting that modifiers may be involved (Van Laer et al., Reference Van Laer, Proost and Loeys2013). Genetic testing is important, as on the one hand, the identification of a pathogenic FBN1 mutation can be very helpful to establish an adequate treatment and management scheme for the proband and affected family members. On the other hand, unaffected family members can be reassured and be released from further clinical follow-up. Here, we present the first genetic testing in a Ukrainian cohort of 40 MFS probands and 10 family members.
2. Materials and methods
Probands in this study were consecutive patients derived from the two largest cardiovascular centres of Ukraine, accessible to all Ukranians. The probands were referred for evaluation of aortic root aneurysm or aortic root surgery. As such, this group is most probably biased towards more extreme cardiovascular phenotypes, but can be considered as representative for the Ukranian population (Zhuraev et al., Reference Zhuraev and Zerbino2014). All probands and their family members underwent a thorough clinical examination, including a slitlamp exam and physical exam. Based on the clinical findings, the systemic score was calculated according to www.marfan.org/dx/score. Twenty-eight patients met the diagnostic criteria for MFS, based on the original and revised Ghent nosology (www.marfan.org/dx/rules) (De Paepe et al., Reference De Paepe, Devereux, Dietz, Hennekam and Pyeritz1996; Loeys et al., Reference Loeys, Dietz, Braverman, Callewaert, De Backer, Devereux, Hilhorst-Hofstee, Jondeau, Faivre, Milewicz, Pyeritz, Sponseller, Wordsworth and De Paepe2010), while 12 were suspected of a MFS-related syndrome. None of the individuals refused inclusion in the study, but three probands were excluded because their DNA was of insufficient quality. The local ethical committee approved the clinical and genetic program for MFS.
Probands were screened with a next generation sequencing (NGS) panel, comprising 14 genes involved in thoracic aortic aneurysms (TAA) (Proost et al., Reference Proost, Vandeweyer, Meester, Salemink, Kempers, Ingram, Peeters, Saenen, Vrints, Lacro, Roden, Wuyts, Dietz, Mortier, Loeys and Van Laer2015). Enrichment of the regions of interest was performed with a custom Haloplex target enrichment kit according to the supplier's protocol (Agilent Technologies, Santa Clara, CA), followed by NGS on MiSeq (Illumina, San Diego, CA) using 150 bp paired-end sequencing reads. Next, data analysis was performed with a tailored pipeline and our in-house developed VariantDB was used to annotate and interpret the variants (Vandeweyer et al., Reference Vandeweyer, Van Laer, Loeys, Van den Bulcke and Kooy2014; Proost et al., Reference Proost, Vandeweyer, Meester, Salemink, Kempers, Ingram, Peeters, Saenen, Vrints, Lacro, Roden, Wuyts, Dietz, Mortier, Loeys and Van Laer2015). Decisions on the pathogenicity of variants were based on their presence in specific mutation databases, including Human Gene Mutation Database (HGMD) (www.hgmd.cf.ac.uk) and Universal Mutation Database (UMD) FBN1 (www.umd.be/FBN1/), which also contain the relevant links to the literature and/or on the functional importance of specific residues and their conservation across the TGF-β binding (TB) and the (calcium-binding) epidermal growth factor (EGF)-like domains as demonstrated in Supplementary Tables S1(a)–(c) (e.g., the conserved cysteine residues in EGF-like domains and the first four amino acids of the EGF-like domain, the so-called DIDE motif).
The variants found with NGS were confirmed by Sanger sequencing using the BigDye® Terminator Cycle Sequencing kit (Applied Biosystems, Life Technologies, Carlsbad, CA), followed by capillary electrophoresis on an ABI3130XL (Applied Biosystems). Multiplex ligation-dependent probe amplification (MLPA) was performed on samples that remained negative after TAA NGS panel testing.
We performed statistical analysis for possible genotype/phenotype correlations in the FBN1 mutation positive and mutation negative groups using the Mann–Whitney U-test for continuous variables and the Fisher's exact test for categorical variables. The patients carrying a variant of unknown significance (VUS) in TGFBR1 and FLNA (patient 1 and 22, respectively) were excluded from this analysis. The patients carrying both a FBN1 mutation and one or more VUS were placed in the mutation positive group.
3. Results and discussion
Of the 40 probands, 27 had causal mutations in FBN1, one patient had a VUS in TGFBR1, and one patient had a VUS in FLNA. In addition to the FBN1 mutation, four patients had additional variants in either FBN1, SMAD3, FLNA or NOTCH1 (Table 1). At this moment, we cannot exclude that these VUS may modify the phenotype caused by the FBN1 mutation. Of the 27 mutations in FBN1, 12 were missense, 11 predicted a premature termination codon (nine nonsense and two frameshifts) and four affected splice sites. Ten of these FBN1 variants were novel. Except one (c.3845A > G; p.Asn1282Ser in patient 17), the FBN1 mutations were not present in the ExAC database (Table 1). No large deletions/duplications could be detected by MLPA.
Table 1. Mutation or VUS positive probands.

Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. Patient numbers are in accordance with the patient numbers in Table 2.
/ : not present in ExAC B: benign; NA: not available; P: pathogenic.
a Numbering of domains is based on Uniprot entry: P35555 (see Supplementary Table S1(a)–(c) for the relevant alignments).
b dbSNFP, integrated into the VariantDB annotation tool (Vandeweyer et al., Reference Vandeweyer, Van Laer, Loeys, Van den Bulcke and Kooy2014), was used to automatically generate the prediction scores of MutationTaster, Polyphen-2 and SIFT respectively, as described by Liu et al., Reference Liu, Jian and Boerwinkle2011.
c In the UMD FBN1 (www.umd.be/FBN1/) or in the HGMD (public part: www.hgmd.cf.ac.uk/ac).
d DIDE consensus sequence: Asp–Ile–Asp–Glu (see Supplementary Table S1(a)).
As our TAA NGS assay has a validated high sensitivity, and as MLPA excluded the presence of large deletions/insertions, we can largely rule out the possibility of false negatives in the coding regions. Only deep intronic mutations and mutations in the 5´- and 3´-untranslated regions will remain undetected with the applied methodology, but we expect that these account for only a minor fraction of all MFS patients. Thus, a possible explanation for the relatively low yield may be the fact that several mutation negative probands did not fulfill the diagnostic criteria for MFS. Indeed, of the 27 FBN1-positive probands, only 15% (four out of 27) had a systemic score below seven, while this was 67% (six out of nine) for the nine FBN1-negative probands. Furthermore, only one of the 15 FBN1 negative patients did present ectopia lentis. As such, more Marfan-like than true Marfan (fulfilling clinical diagnostic criteria) presentations were present in the FBN1-negative group. Moreover, Marfan-like patients that remained negative with the gene panel, may carry mutations in more recently identified TAA genes or yet to be identified TAA genes.
For six of the probands, DNA of family members was available and segregation analysis was performed. The p.Cys570Tyr mutation (patient 2) was found in three additional affected family members (Table 2). Although all family members presented with ectopia lentis, only two have undergone aortic surgery (ages 20 years and 33 years). Also the p.Ile2585Thr (patient 11), the p.Arg2394* (patient 23) and the p.Cys85Tyr (patient 29) mutations segregated with the MFS phenotype in four additional family members. The truncating mutation (p.Lys1983*) identified in patient 12 was also found in his mother. She had no aortic root dilatation, but presented with mitral valve prolapse and skeletal features including wrist sign, pectus carinatum and tall stature. As a direct result of our genetic testing, she is now in regular cardiovascular follow-up and has been started on losartan in order to delay future aortic surgery.
Table 2. Clinical data of 40 MFS probands and their family members.

Patient numbers are in accordance with the patient numbers in Table 1 and suffixes indicate family members.
/ : no surgery.
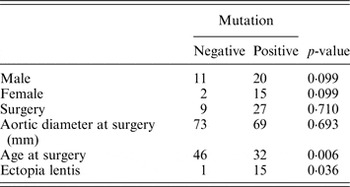
Next, statistical analysis for possible genotype/phenotype correlations was performed (Table 3). We found a significant difference between the FBN1 mutation positive and mutation negative patients for the age at surgery (Mann–Whitney U, p = 0·006) and the presence of ectopia lentis (Fisher's exact test, p = 0·036). Patients with a mutation in FBN1 had aortic surgery at an earlier age (32 years on average) than mutation negative patients (46 years on average). This emphasizes the importance of genetic screening for the identification of patients that are at higher risk for developing aortic aneurysms and dissection. According to the literature, a higher frequency of truncating and splicing variants in FBN1 can be observed in patients with an aortic event (Baudhuin et al., Reference Baudhuin, Kotzer and Lagerstedt2015). In our cohort, no significant difference could be observed between truncating or splice variants and missense variants in patients with an aortic event (Fisher's exact test, p = 0·282). Of course, our study was not sufficiently powered to detect such differences.
Table 3. Clinical data of 38 MFS probands and their family members.
4. Conclusion
In conclusion, we identified FBN1 gene mutations in Ukrainian MFS patients for the first time. Since the clinical picture of these patients is not always clear, genetic screening can help to establish a diagnosis and to identify patients at high risk for developing life-threatening complications such as aortic aneurysm and dissection.
We are grateful to all patients who participated in this study. B. L. Loeys is senior clinical investigator and J. A. N. Meester is a predoctoral researcher of the Fund for Scientific Research, Flanders (FWO, Belgium). This research was supported by funding from the University of Antwerp (Lanceringsproject), the Fund for Scientific Research, Flanders (FWO, Belgium) (G.0221·12), an ERC starting grant to B. L. Loeys and the Hercules Foundation.
Declaration of interest
None.
Supplementary material
The online supplementary material can be found available at http://dx.doi.org/10.1017/S0016672316000112






