Introduction
Autism spectrum disorders (ASDs) are a group of lifelong and devastating neurodevelopmental disabilities that affect 1 in 88 children in the USA (Autism, Developmental Disabilities Monitoring Network Surveillance Year Principal I, Centers for Disease C, & Prevention, 2012). Multiple lines of evidence show that ASDs have a strong genetic basis. Currently, there are no effective treatments available for ASDs. Deciphering the genetic code of the ASDs would benefit the affected children through early diagnosis and intervention (Rogers et al., Reference Rogers, Estes, Lord, Vismara, Winter, Fitzpatrick, Guo and Dawson2012), and identify new biomarkers for drug development and new therapy (Silverman et al., Reference Silverman, Smith, Rizzo, Karras, Turner, Tolu, Bryce, Smith, Fonseca, Ring and Crawley2012). After decades of efforts, many ASDs-related syndromic genes, rare penetrant genes, rare chromosome abnormalities and structural variations have been identified, but the heritability was still missed largely (>70%) (Devlin & Scherer, Reference Devlin and Scherer2012). The state-of-the-art high-throughput sequencing (HTS) technique already showed promise in disease-causing genes discovery not only for monogenic disorders but also for genetic diseases with complex inheritance (Rehm, Reference Rehm2013). This review will focus on the ASDs genome studies using HTS.
ASDs, problems and hope
The alarmingly high prevalence of ASDs poses a serious public health challenge globally and brings a high burden to the ASDs families. Multiple lines of evidence suggested a strong genetic factor in the aetiology. However, the considerable clinical and genetic heterogeneity have made the identification of genetic aetiology studies of ASDs more complex than expected. Recent studies have identified several de novo events, that is single nucleotide variants (SNVs), insertion and deletions (InDels), copy-number variations (CNVs) or structural variations (SVs), with high frequency among ASDs families (Veltman & Brunner, Reference Veltman and Brunner2012). In addition, new clues also revealed some new environmental risks such as prenatal nutritional factors to ASDs (Grabrucker, Reference Grabrucker2012), including microbial infection (Heijtz et al., Reference Heijtz, Wang, Anuar, Qian, Bjorkholm, Samuelsson, Hibberd, Forssberg and Pettersson2011) and epigenetic regulation (Shulha et al., Reference Shulha, Cheung, Whittle, Wang, Virgil, Lin, Guo, Lessard, Akbarian and Weng2012).
In a clinical setting, the diagnosis is mostly made based on behavioural evaluation. Recently, the application of copy number variant by chromosome microarray has become the standard of care in clinical evaluation of ASDs, and is widely used for clinical diagnosis of ASDs at an early stage (Miller et al., Reference Miller, Adam, Aradhya, Biesecker, Brothman, Carter, Church, Crolla, Eichler, Epstein, Faucett and Feuk2010). The gene panel based on syndromic gene and non-syndromic gene with high penetrance holds great prospects. Behavioural treatment such as the Early Start Denver Model (ESDM), has been reported to be effective if used early (Rogers et al., Reference Rogers, Estes, Lord, Vismara, Winter, Fitzpatrick, Guo and Dawson2012).
A complex genetic architecture of ASDs
Four types of genetic defects have been identified in ASDs (Devlin & Scherer, Reference Devlin and Scherer2012): (i) single genes linked to synromic ASDs (Betancur, Reference Betancur2011), such as FMR1 that causes fragile X syndrome (OMIM: 309548) associated with ASDs with high frequency. These genes can explain ∼10% of individuals with ASDs; (ii) rare chromosome abnormalities (Marshall et al., Reference Marshall, Noor, Vincent, Lionel, Feuk, Skaug, Shago, Moessner, Pinto, Ren, Thiruvahindrapduram and Fiebig2008) that were observed in ∼5% ASDs cases; (iii) rare CNVs that contribute to ∼5% cases (Malhotra & Sebat, Reference Malhotra and Sebat2012); (iv) rare penetrant genes that account for ∼5% cases (Banerjee-Basu & Packer, Reference Banerjee-Basu and Packer2010; State, Reference State2010; Xu et al., Reference Xu, Li, Huang, Zhao, Tang and Wei2012). As we still miss the molecular basis for the rest ∼70% cases, the studies focusing on common variants have not identified any significant sites associated with ASDs (Anney et al., Reference Anney, Klei, Pinto, Almeida, Bacchelli, Baird, Bolshakova, Bolte, Bolton, Bourgeron, Brennan, Brian and Casey2012). Recently, state-of-the-art HTS technology has shown promise in disease gene discovery and was applied to detect rare de novo or inherited variants linked to ASDs.
The progress of HTS-based ASDs studies
The recent advance in target-enrichment strategies (Mamanova et al., Reference Mamanova, Coffey, Scott, Kozarewa, Turner, Kumar, Howard, Shendure and Turner2010) and HTS has made the detection of rare variants linked to ASDs feasible. Nowadays, one can focus on single genes, multiple genes, important pathway genes, chromosome X genes, or use whole exome sequencing (WES) and whole genome sequencing (WGS) to study genes at genome width.
Candidate gene sequencing on ASDs
For candidate gene sequencing strategy, one can focus on a single reported ASDs gene and try to find new risk variants, centre on multiple ASDs candidate genes for validation of previous results, focus on important pathway genes and try to identify new susceptible genes, or study the X-linked susceptible genes by targeting the whole X chromosome. AFF2 is a known fragile X syndrome gene, duplications or deletions at the AFF2 locus have also been reported in cases with moderate intellectual disability and ASDs. Mondal et al. (Reference Mondal, Ramachandran, Patel, Hagen, Bose, Cutler and Zwick2012) sequenced the AFF2 genomic region in 202 male ASDs probands. They found that 2·5% of males had missense mutations at highly conserved evolutionary sites and identified rare AFF2 3′UTR variants at conserved sites, which alter gene expression in a luciferase assay. These new identified rare variants in AFF2 may be new ASDs susceptibility locus. A large amount of ASDs candidate genes identified by large-scale sequencing project provide a source to further study the aetiology of the ASDs. O'Roak et al. (Reference O'Roak, Vives, Fu, Egertson, Stanaway, Phelps, Carvill, Kumar, Lee, Ankenman, Munson, Hiatt, Turner, Levy, O'Day, Krumm, Coe, Martin, Borenstein, Nickerson, Mefford, Doherty, Akey, Bernier, Eichler and Shendure2012a) used an ultra-low-cost molecular inversion probe to capture and sequence 44 candidate genes, which were found in the previous WES study (O'Roak et al., Reference O'Roak, Deriziotis, Lee, Vives, Schwartz, Girirajan, Karakoc, Mackenzie, Ng, Baker, Rieder, Nickerson, Bernier, Fisher, Shendure and Eichler2011, Reference O'Roak, Vives, Girirajan, Karakoc, Krumm, Coe, Levy, Ko, Lee, Smith, Turner, Stanaway, Vernot, Malig, Baker, Reilly, Akey, Borenstein, Rieder, Nickerson, Bernier, Shendure and Eichler2012b), in 2446 ASDs probands, the results show several new clues: (i) 27 de novo events in 16 genes, 59% of which are predicted to truncate proteins or disrupt splicing; (ii) recurrent disruptive mutations are observed in six genes – CHD8, DYRK1A, GRIN2B, TBR1, PTEN and TBL1XR1; (iii) these de novo events may contribute to 1% of sporadic ASDs; and (iv) associations between specific genes and reciprocal subphenotypes (CHD8-macrocephaly, DYRK1A-microcephaly). This new discovery may highlight the importance of a β-catenin/chromatin remodelling network to ASDs aetiology (Talkowski et al., Reference Talkowski, Rosenfeld, Blumenthal, Pillalamarri, Chiang, Heilbut, Ernst, Hanscom, Rossin, Lindgren, Pereira and Ruderfer2012).
Many important biological pathways have been found associated with ASDs (Sakai et al., Reference Sakai, Shaw, Dawson, Dugas, Al-Mohtaseb, Hill and Zoghbi2011). Study of the gene implicated in an important pathway became a good choice for studying potential risk genes or variants. The metabotropic glutamate-receptors (mGluRs) signalling pathway is one of the important pathways that has been linked to the pathophysiology of the ASDs. Several syndromic and non-syndromic genes, such as TSC1, TSC2 and SHANK3, have been linked to ASDs. Most preclinical drugs, though just on the horizon, are based on mGluRs. To study most of the potential rare mGluRs variants linked to ASDs, Kelleher 3rd et al. (2012) analysed 16 genes encoding proteins in mGluRs-related pathways from pooled samples of 290 non-syndromic autism cases and 300 ethnically matched controls by using two independent next generation platforms. They (i) identified HOMER1, as a novel ASDs-risk gene, which encodes a scaffolding protein at the post-synaptic density-localized scaffolding protein that interacts with SHANK3 to regulate mGluRs activity; and (ii) found rare ASDs-associated coding variants predicted to have damaging effects on components of the Ras/MAPK cascade. These discoveries will benefit the mGluRs-based ASDs drug development activity.
The observed 1/4 (female/male) ratio among ASDs suggested that X-linked genes may be implicated although there is no evidence from the genetic linkage studies so far. Followed by enriching the whole chromosome X exome using a custom target region capture array, Nava et al. (Reference Nava, Lamari, Heron, Mignot, Rastetter, Keren, Cohen, Faudet, Bouteiller, Gilleron, Jacquette and Whalen2012) sequenced 12 unrelated families with two males affected with ASDs and identified (i) 36 possibly deleterious variants in 33 candidate genes; (ii) damaging mutations in PHF8 and HUWE1 that were previously implicated in intellectual disability (ID); (iii) a non sense mutation in TMLHE in two brothers with autism and ID; and (iv) TMLHE linked to ASDs by a large-scale Sanger sequencing-based screening and functional analysis.
WES or WGS studies on ASDs
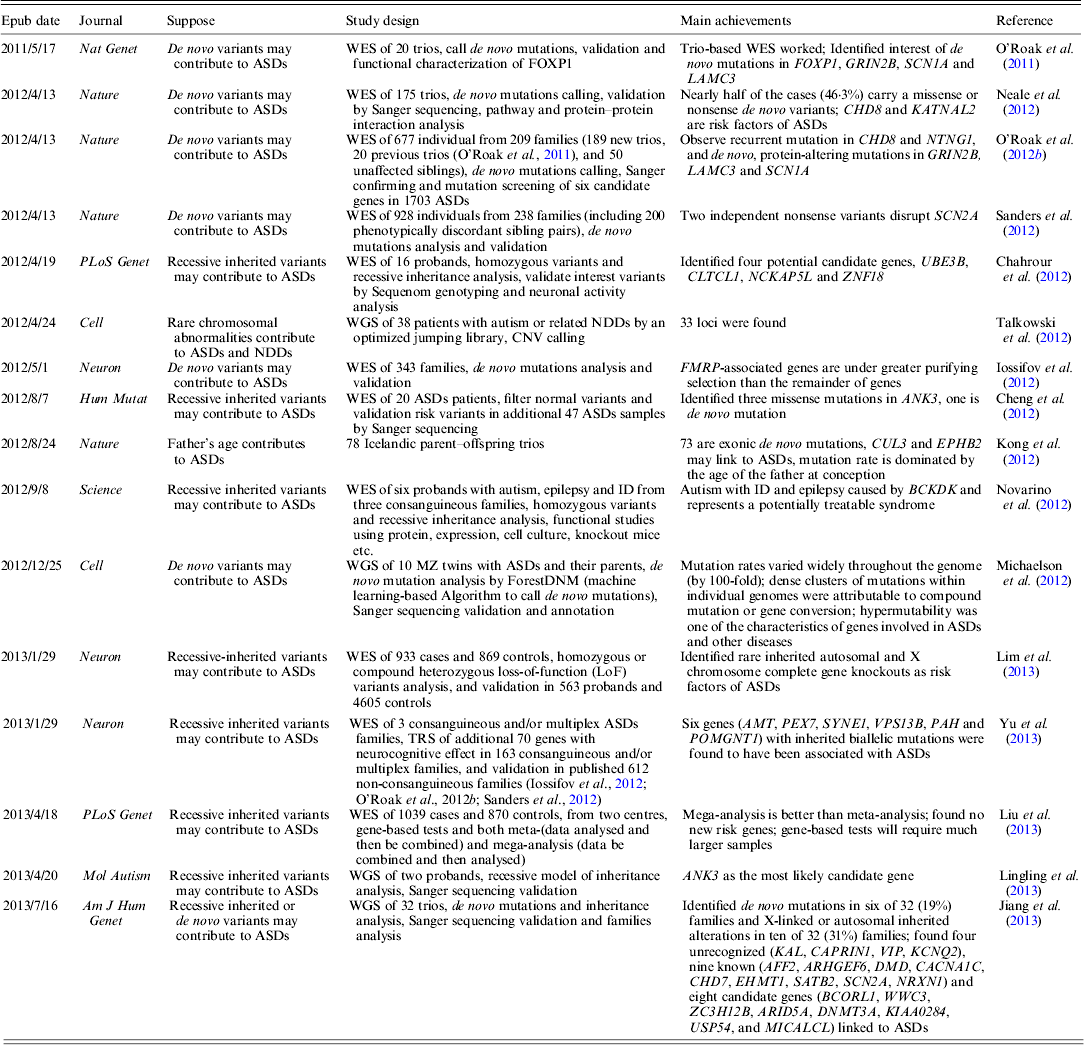
WES has been widely used to target disease gene of unknown human disorder (Gilissen et al., Reference Gilissen, Hoischen, Brunner and Veltman2012). As the cost for human WGS is going down very rapidly, one can afford to study the whole genome of a common disorder (Anney et al., Reference Anney, Klei, Pinto, Almeida, Bacchelli, Baird, Bolshakova, Bolte, Bolton, Bourgeron, Brennan, Brian and Casey2012). To date, several large-scale ASDs WES studies have been published; a brief summary of these studies is shown in Table 1.
Table 1. Summary of current WES and WGS studies on ASDs

The achievements of HTS studies on ASDs
Identified amount of rare de novo point mutations linked to ASDs
De novo mutations have been found in many human genetic diseases (Veltman & Brunner, Reference Veltman and Brunner2012); to study de novo mutations linked to ASDs, one can use WES of several or large-scale trios. Many rare de novo damaging mutations linked to ASDs have been identified to date.
O'Roak et al. (Reference O'Roak, Deriziotis, Lee, Vives, Schwartz, Girirajan, Karakoc, Mackenzie, Ng, Baker, Rieder, Nickerson, Bernier, Fisher, Shendure and Eichler2011) performed the first trios-based WES on ASDs, after excluding CNVs by the CGH array, they sequenced 20 trios and (i) detected a total of 21 de novo variants and 11 were protein alterations, which occurred on different genes and were predicted to be damaging; (ii) suggested that the probands harboured more highly conserved and disruptive amino acid mutations than controls; and (iii) identified four potentially causative de novo mutations, GRIN2B(IVS9-2A > G), SCN1A(p.Pro1894Leu), LAMC3(p.Asp399Gly) and FOXP1(p.Ala339Ser fsX4), as new candidates linked to ASDs.
O'Roak et al. (Reference O'Roak, Vives, Girirajan, Karakoc, Krumm, Coe, Levy, Ko, Lee, Smith, Turner, Stanaway, Vernot, Malig, Baker, Reilly, Akey, Borenstein, Rieder, Nickerson, Bernier, Shendure and Eichler2012b) selected a total of 677 individual exomes, including 189 new trios and 20 trios that were previously reported (O'Roak et al., Reference O'Roak, Deriziotis, Lee, Vives, Schwartz, Girirajan, Karakoc, Mackenzie, Ng, Baker, Rieder, Nickerson, Bernier, Fisher, Shendure and Eichler2011), for WES. They identified that (i) de novo point mutations were overwhelmingly paternal in origin (4:1 bias) and positively correlated with paternal age; (ii) 39% (49 of 126) of the most severe or disruptive de novo mutations mapped to highly interconnected b-catenin/chromatin remodelling protein network genes; (iii) two genes, CHD8 and NTNG1, were found with recurrent damaging mutations in probands; and (iv) mutation screening of six candidate genes (four reported previously (O'Roak et al., Reference O'Roak, Deriziotis, Lee, Vives, Schwartz, Girirajan, Karakoc, Mackenzie, Ng, Baker, Rieder, Nickerson, Bernier, Fisher, Shendure and Eichler2011)) in 1703 ASDs probands identified additional de novo and protein-altering mutations in GRIN2B, LAMC3 and SCN1A.
Sanders et al. (Reference Sanders, Murtha, Gupta, Murdoch, Raubeson, Willsey, Ercan-Sencicek, DiLullo, Parikshak, Stein, Walker, Ober and Teran2012) performed whole-exome sequencing of 928 individuals in 238 families, including 200 discordant sibling pairs, and observed highly disruptive (nonsense and splice-site) de novo mutations in brain-expressed genes that are associated with ASDs and a total of 279 de novo coding mutations. Although there is only one single instance in the probands and none in the siblings, SCN2A which harboured two independent nonsense variants in probands may linked to ASDs.
Neale et al. (Reference Neale, Kou, Liu, Ma'ayan, Samocha, Sabo, Lin, Stevens, Wang, Makarov, Polak and Yoon2012) sequenced the exomes of 175 trios and found that: (i) nearly half of the cases (46·3%) carry missense or nonsense de novo variants; (ii) the overall rate of mutation is ∼2 × 10−8 per base per generation and only modestly higher than expected; (iii) the disrupted genes were found to be connected with previously reported ASDs genes via a protein–protein interaction analysis; and (iv) CHD8 and KATNAL2 may link to ASDs which need further validation.
Iossifov et al. (Reference Iossifov, Ronemus, Levy, Wang, Hakker, Rosenbaum, Yamrom, Lee, Narzisi, Leotta, Kendall and Grabowska2012) sequenced 343 family ‘quads’ (the parents of a single child on the autism spectrum and its unaffected sibling), although the numbers of de novo missense mutations between affected and unaffected children are similar, gene-disrupting mutations (nonsense, splice site and frame shifts) are found twice as frequently, 59 to 28, in ASDs children. These mutations were mostly from the paternal line and in an age-dependent manner. Many of these disrupted genes are associated with the fragile X protein, FMRP.
Identified multiples of rare inherited point mutations, may lead to cure of ASDs
To study rare inherited mutations linked to ASDs, WES of several probands or large-scale case–control samples can be applied.
Chahrour et al. (Reference Chahrour, Yu, Lim, Ataman, Coulter, Hill, Stevens, Schubert, Collaboration, Greenberg, Gabriel and Walsh2012) sequenced the whole exomes of 16 probands from 16 non-consanguineous families that showed evidence of distant shared ancestry. By using the homozygosity analysis and multidimensional strategy for filtering whole-exome sequence data to find candidate recessive mutations, four candidate genes (e.g. UBE3B, CLTCL1, NCKAP5L and ZNF18) were identified, and the expression of these genes were highly responsive to neuronal activity.
Cheng et al. (Reference Cheng2012) performed whole-exome sequence and extensive bioinformatic analysis of a cohort of 20 ASDs patients, identified novel mutations in seven genes that are implicated in synaptic function and neurodevelopment. After sequencing an additional 47 ASDs samples, they identified three different missense mutations in ANK3 in four unrelated ASDs patients, one of which, c.4705T > G (p.S1569A), is a de novo mutation. There is shared molecular pathophysiology between the ASDs and other neuropsychiatric disorders considering that ANK3 was previously reported to be linked to Schizophrenia and Bipolar Disorder (Schizophrenia Psychiatric Genome-Wide Association Study C, 2011). The ANK3 was replicated by Lingling et al. (Reference Lingling2013) through a WGS study of two probands from a large pedigree (including two parents and eight children). They (i) identified a list of 59 candidate variants that may increase susceptibility to autism; (ii) manual examination of this list identified ANK3 as the most likely candidate gene; and (iii) identified 33 prioritized non-coding variants such as those near SMG6 and COQ5, based on evolutionary constraint and experimental evidence from ENCODE.
Yu et al. (2013) firstly apply WES to a cohort of three consanguineous and/or multiplex ASDs families which shared ancestry between the parents, typically as cousins; then they used TRS to screen for mutations in additional 70 genes with neurocognitive effect in a total of 163 consanguineous and/or multiplex families. Lastly, they used 612 published non-consanguineous families data for validation, finally six genes (e.g., AMT, PEX7, SYNE1, VPS13B, PAH and POMGNT1) with inherited biallelic mutations were found to have been associated with ASDs.
The identified rare variants may lead to better cure of ASDs. To develop a better cure for ASDs, Novarino et al. (Reference Novarino, El-Fishawy, Kayserili, Meguid, Scott, Schroth, Silhavy, Kara, Khalil, Ben-Omran, Ercan-Sencicek, Hashish, Sanders, Gupta, Hashem, Matern, Gabriel, Sweetman, Rahimi, Harris, State and Gleeson2012) performed whole-exome sequencing from two consanguineous families, one of Turkish descent and a second of Egyptian ancestry, and identified inactivating mutations in the gene Branched Chain Ketoacid Dehydrogenase Kinase (BCKDK) in consanguineous families with autism, epilepsy and ID. The patients with homozygous BCKDK mutations display reductions in BCKDK mRNA and protein, and BCKDK knockout mice show abnormal brain amino acid profiles and neurobehavioural deficit. Nevertheless, the deficit is reversible using dietary supplementation. This may represent a potentially treatable syndrome in ASDs.
De novo mutations varied widely in ASDs and were associated with father's age
Kong et al. (Reference Kong, Frigge, Masson, Besenbacher, Sulem, Magnusson, Gudjonsson, Sigurdsson, Jonasdottir, Jonasdottir, Wong, Sigurdsson, Walters, Steinberg, Helgason, Thorleifsson, Gudbjartsson, Helgason, Magnusson, Thorsteinsdottir and Stefansson2012) conducted a study of genome-wide mutation rates by WGS of 78 Icelandic parent–offspring trios at high coverage (30X average coverage). They show that (i) with an average father's age of 29·7, the average de novo mutation rate is 1·2 × 10−8 per nucleotide per generation; (ii) 73 are exonic de novo mutations, among which CUL3 and EPHB2 may link to ASDs and (iii) the diversity in mutation rate of single nucleotide polymorphisms is dominated by the age of the father at conception of the child and father's age.
Michaelson et al. (Reference Michaelson, Shi, Gujral, Zheng, Malhotra, Jin, Jian, Liu, Greer, Bhandari, Wu and Corominas2012) firstly developed a machine-learning based de novo mutation calling pipeline, ForestDNM, by which they analysed de novo point mutations in ten identical twin pairs and their parents. Results showed that: (i) mutation rates varied widely throughout the genome (by 100-fold) and could be explained by the intrinsic characteristics of the DNA sequence and chromatin structure; (ii) dense clusters of mutations within individual genomes were attributable to compound mutation or gene conversion; and (iii) hypermutability was one of the characteristics of genes involved in ASDs and other diseases.
Failed to identify common variants linked to ASDs
Although Genome Wide Association Studies (GWAS) have not made a significant contribution to ASDs gene discoveries, large-scale WES-based case-control study shows promise on common diseases as they can focus on sites with MAF < 5%. Liu et al. (Reference Liu, Sabo, Neale, Nagaswamy, Stevens, Lim, Bodea, Muzny, Reid, Banks, Coon and Depristo2013) evaluated the association of rare variants and ASDs in 1039 cases and 870 controls with similar ancestry, about half of the samples were sequenced on the Solid platform (Baylor: 505 cases, 491 controls) and the remainder were sequenced on the Illumina platform (Broad: 534 cases, 379 controls). Gene-based association analyses were conducted but no gene showed exome-wide significant association. This may suggest that rare risk variants are scattered across these many genes, and larger samples would be required.
WGS holds promise to identify rare chromosome abnormalities
To study rare chromosome abnormalities associated with ASDs, especially balanced chromosomal abnormalities (BCAs), which represent a relatively untapped reservoir of single gene disruptions in neurodevelopmental disorders (NDDs), a cost-effective pipeline was developed to call genomic rearrangements and structural variations at base pair resolution (Talkowski et al., Reference Talkowski, Ernst, Heilbut, Chiang, Hanscom, Lindgren, Kirby, Liu, Muddukrishna, Ohsumi, Shen, Borowsky, Daly, Morton and Gusella2011). Talkowski et al. (Reference Talkowski, Rosenfeld, Blumenthal, Pillalamarri, Chiang, Heilbut, Ernst, Hanscom, Rossin, Lindgren, Pereira and Ruderfer2012) sequenced BCAs in 38 patients with autism or related NDDs by using an optimized jumping library protocol (21 subjects), a targeted DNA capture protocol (10 subjects), a standard paired-end (PE) protocol (two subjects), and an Illumina mate-pair (MP) protocol (five subjects). As a consequence, disruptions of 33 loci were found in four general categories: (1) genes previously associated with abnormal neurodevelopment (e.g. AUTS2, FOXP1 and CDKL5); (2) single-gene contributed to microdeletion syndromes (MBD5, SATB2, EHMT1 and SNURF-SNRPN); (3) novel risk loci (e.g. CHD8, KIRREL3 and ZNF507); and (4) genes associated with later-onset psychiatric disorders (e.g. TCF4, ZNF804A, PDE10A, GRIN2B and ANK3). By evaluation of CNVs in independent subjects, significantly increased burdens of CNVs from these 33 loci were found. By performing gene-set enrichment analysis using published GWAS data and network analysis, significant enrichments of polygenic risk alleles in these 33 loci were also found.
WGS shows better performance to identify rare variants linked to ASDs
Jiang et al. (Reference Jiang, Yuen, Jin, Wang, Chen, Wu, Ju, Mei, Shi, He, Wang, Liang and Wang2013) used WGS to study 32 ASDs trios; after de novo mutations and inheritance analysis, they (i) identified that deleterious de novo mutations in six of 32 (19%) families and X-linked or autosomal inherited alterations in 10 of 32 (31%) families, may be due to the comprehensive and uniform coverage afforded by WGS; (ii) deleterious variants were found in four unrecognized (e.g. KAL, CAPRIN1, VIP and KCNQ2), nine known ASDs genes (e.g. AFF2, ARHGEF6, DMD, CACNA1C, CHD7, EHMT1, SATB2, SCN2A and NRXN1) and eight candidate ASDs risk genes (e.g. BCORL1, WWC3, ZC3H12B, ARID5A, DNMT3A, KIAA0284, USP54 and MICALCL).
Future direction
To study the epigenetic risk factor linked to ASDs
MZ twins discordant for ASDs provide an ideal model to study epigenetics risk factors of ASDs since the twins share most of their genetic background. Wong et al. (Reference Wong, Meaburn, Ronald, Price, Jeffries, Schalkwyk, Plomin and Mill2013) performed a genome-wide analysis of DNA methylation in a sample of 50 MZ twin pairs (100 individuals), included twins discordant and concordant for ASDs, ASDs-associated traits and no autistic phenotype. Within-twin and between-group analyses identified numerous differentially methylated regions associated with ASDs.
To study the gene expression of ASDs genes
Study of the gene expression of ASDs holds promise in deciphering the DNA mutation effect of ASDs. As brain tissue is not available from most samples, study of the gene expression of peripheral blood mononuclear cell (PBMC) provides an easy way. Luo et al. (Reference Luo, Sanders, Tian, Voineagu, Huang, Chu, Klei, Cai, Ou, Lowe, Hurles, Devlin, State and Geschwind2012) interrogated gene expression in lymphoblasts from 244 families with discordant siblings by microarray.
To study the gut microbes linked to ASDs
Gut microbiomes are a new frontier in autism research (Mulle et al., Reference Mulle, Sharp and Cubells2013); although no direct evidence exists, there are clues that gut microbiota modulate brain development and behaviour (Heijtz et al., Reference Heijtz, Wang, Anuar, Qian, Bjorkholm, Samuelsson, Hibberd, Forssberg and Pettersson2011). Ming et al. (Reference Ming, Stein, Barnes, Rhodes and Guo2012) performed liquid- and gas-chromatography-based mass spectrometry to study metabolomics in urinary specimens from 48 children with ASDs and 53 age-matched controls. They detected abnormal amino acid metabolism, increased oxidative stress, and altered gut microbiomes in ASDs.
Prospective
The problem of ASDs has been exposed to the spotlight; several large-scale WES have led to the identification of multiple ASDs candidate genes. The WGS showed better detection rate not only on coding and non-coding variants (Jiang et al., Reference Jiang, Yuen, Jin, Wang, Chen, Wu, Ju, Mei, Shi, He, Wang, Liang and Wang2013), but also on CNVs and SVs. The state-of-the-art HTS will enable us to decode the missed heritability for ASDs. The challenge is how to analyse the de novo or inherited event of rare variations and understand their functions.
Recently, several large-scale WGS projects have begun, one of which is called the Autism Genome 10 K project (http://www.autismgenome10k.org/) initiated by BGI, the world's largest genomic organization, and Autism Speaks, the world's largest autism science and advocacy organization. The project aims to: (i) sequence the genomes from 10 000 individuals belonging to 2000 ASDs families from the Autism Speaks Autism Genetic Resource Exchange (AGRE) and 1000 Chinese ASDs families; (ii) analyse de novo events or inherited SNVs, InDels, CNVs, SVs etc.; (iii) identify ASDs genes or variants; and (iv) screen targets for drug development and therapy. The pilot results are published (Jiang et al., Reference Jiang, Yuen, Jin, Wang, Chen, Wu, Ju, Mei, Shi, He, Wang, Liang and Wang2013) and the planned transomics study of ASDs, which include transcriptome, epigenome, proteome, metabolome and metagenome study, will focus on the ASDs families which lack explanation by the genetic studies. The project may bring together scientists, ASDs families, funding agencies from governments, non-government organizations which provide autism intervention services and drug companies from all over the world.
We thank Dr Yong-hui Jiang and Dr Zhongsheng Sun's suggestions for preparing and revising this manuscript.