


1. Introduction
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability (ID) and co-morbid autism. It is characterized by an abnormal amplification of CGG repeats (>200) in the 5′UTR of the FMR1 gene, located on chromosome Xq27·3 (Verkerk et al., Reference Verkerk, Pieretti, Sutcliffe, Fu, Kuhl, Pizzuti, Reiner, Richards, Victoria and Zhang1991; Hagerman et al., Reference Hagerman, Berry-Kravis, Kaufmann, Ono, Tartaglia, Lachiewicz, Kronk, Delahunty, Hessl, Visootsak, Picker, Gane and Tranfaglia2009). However, loss of function mutations (large deletions and point mutations) have also been associated with FXS, although they are far less documented (De Boulle et al., Reference De Boulle, Verkerk, Reyniers, Vits, Hendrickx, Van Roy, Van den Bos, de Graaff, Oostra and Willems1993; Coffee et al., Reference Coffee, Ikeda, Budimirovic, Hjelm, Kaufmann and Warren2008; Luo et al., Reference Luo, Huang, Xia, Du, Wu and Duan2014). FXS affects individuals of both sexes with a wide spectrum of neurobehavioural phenotypes. Of these, 30–60% of boys, depending on the study, have autism spectrum disorders (Harris et al., Reference Harris, Hessl, Goodlin-Jones, Ferranti, Bacalman, Barbato, Tassone, Hagerman, Herman and Hagerman2008; Budimirovic & Kaufmann, Reference Budimirovic and Kaufmann2011; Abbeduto et al., Reference Abbeduto, McDuffie and Thurman2014).
A variety of genetic conditions related to the FMR1 gene occur as a result of unstable CGG repeats, which are responsible for fragile X-associated genetic disorders (FXD), including FXS, fragile X-associated tremor/ataxia syndrome (FXTAS) and fragile X-associated primary ovarian insufficiency (FXPOI) (Hagerman & Hagerman, Reference Hagerman and Hagerman2013). A combination of molecular techniques including PCR and Southern blot (SB) are required to accurately determine the number of repeats as well as the methylation status of the promoter associated with expanded alleles in FXD (Hagerman, Reference Hagerman2008).
Clinically, FXS is associated with a wide spectrum of neurodevelopmental phenotypes with cognitive functioning ranging from mild learning difficulties to severe ID (Harris et al., Reference Harris, Hessl, Goodlin-Jones, Ferranti, Bacalman, Barbato, Tassone, Hagerman, Herman and Hagerman2008; Hagerman, Reference Hagerman2013). The most common cognitive and behavioural characteristics identified in fragile X individuals are deficits in language and mathematics, social phobia, attention deficit, aggressive behaviour and hyperactivity. Physical features include large and prominent ears, elongated face with a prominent chin, hypermobility of the joints of the fingers, smooth skin and flat feet (Pieretti et al., Reference Pieretti, Zhang, Fu, Warren, Oostra, Caskey and Nelson1991).
Full mutation (FM) alleles exceeding 200 CGG repeats are associated with hypermethylation in the promoter region causing an absence of mRNA expression and subsequent deficit of the Fragile X Mental Retardation Protein (FMRP), essential for normal neurodevelopment (Pieretti et al., Reference Pieretti, Zhang, Fu, Warren, Oostra, Caskey and Nelson1991). Prevalence of FM in the general population has been reported to be higher in males than females, affecting 1 in 4000 males and 1 in 8000 females (Hagerman et al., Reference Hagerman, Berry-Kravis, Kaufmann, Ono, Tartaglia, Lachiewicz, Kronk, Delahunty, Hessl, Visootsak, Picker, Gane and Tranfaglia2009; Song et al., Reference Song, Barton, Sleightholme, Yao and Fry-Smith2003; Hunter et al., Reference Hunter, Rivero-Arias, Angelov, Kim, Fotheringham and Leal2014).
Premutation (PM) is a medium size expansion (55–200 CGG repeats) mostly identified in unaffected carrier parents. Studies indicate a prevalence of 1 in 209 and 1 in 430 for females and males, respectively (Song et al., Reference Song, Barton, Sleightholme, Yao and Fry-Smith2003; Tassone et al., Reference Tassone, Iong, Tong, Lo, Gane, Berry-Kravis, Nguyen, Mu, Laffin, Bailey and Hagerman2012). In contrast to FM, PM alleles remain unmethylated and FMR1 mRNA increases by up to five times the normal level, leading to mild to moderate FMRP production (Tassone et al., Reference Tassone, Hagerman, Chamberlain and Hagerman2000; Leehey et al., Reference Leehey, Munhoz, Lang, Brunberg, Grigsby, Greco, Jacquemont, Tassone, Lozano, Hagerman and Hagerman2003). Presence of high levels of mRNA produces a toxic effect on brain cells linked to late-onset disorders including FXTAS (Tassone et al., Reference Tassone, Iwahashi and Hagerman2004) usually affecting males >50 years of age (Rousseau et al., Reference Rousseau, Labelle, Bussieres and Lindsay2011; Santa Maria et al., Reference Santa Maria, Pugin, Alliende, Aliaga, Curotto, Aravena, Tang, Mendoza-Morales, Hagerman and Tassone2014). Females carrying a PM are at an increased risk (~20%) of developing FXPOI, which usually occurs before 40 years of age (Schwartz et al., Reference Schwartz, Dean, Howard-Peebles, Bugge, Mikkelsen, Tommerup, Hull, Hagerman, Holden and Stevenson1994; Sherman et al., Reference Sherman2000). Moreover, the risk of expanding from PM to FM in subsequent generations is determined by the CGG number, alleles >90 repeats have a 100% risk of expansion to a FM (Fisch et al., Reference Fisch, Snow, Thibodeau, Chalifaux, Holden, Nelson, Howard-Peebles and Maddalena1995; Nolin et al., Reference Nolin, Brown, Glicksman, Houck, Gargano, Sullivan, Biancalana, Brondum-Nielsen, Hjalgrim, Holinski-Feder, Kooy, Longshore, Macpherson, Mandel, Matthijs, Rousseau, Steinbach, Vaisanen, von Koskull and Sherman2003). On the other hand, intermediate or grey zone (GZ) alleles between 45 to 54 repeats overlap between PM and normal (<45) CGG size ranges, with a frequency of 1 in 112 males and 1 in 66 females (Tassone et al., Reference Tassone, Iong, Tong, Lo, Gane, Berry-Kravis, Nguyen, Mu, Laffin, Bailey and Hagerman2012). Expansions of the GZ are not clearly associated with a particular phenotype, but studies have suggested that carriers of these alleles have slightly increased risk of the FMR1-related disorders FXTAS and FXPOI compared with normal alleles (Bennett et al., Reference Bennett, Conway, Macpherson, Jacobs and Murray2010).
It is not uncommon that individuals with FXS may carry different CGG repeat allele sizes: some cells carry FM alleles while others carry PM alleles (Tassone et al., Reference Tassone, Longshore, Zunich, Steinbach, Salat and Taylor1999; Lokanga et al., Reference Lokanga, Entezam, Kumari, Yudkin, Qin, Smith and Usdin2013). This instability of the CGG repeats within the FMR1 gene is termed ‘size’ mosaicism and has been reported in 41% of males affected with FXS (Nolin et al., Reference Nolin, Glicksman, Houck, Brown and Dobkin1994). In addition, some individuals exhibit ‘methylation’ mosaicism, in which some cells have fully methylated FM alleles while other cells possess unmethylated FM alleles (Merenstein et al., Reference Merenstein, Sobesky, Taylor, Riddle, Tran and Hagerman1996; Loesch et al., Reference Loesch, Sherwell, Kinsella, Tassone, Taylor, Amor, Sung and Evans2012; Pretto et al., Reference Pretto, Hunsaker, Cunningham, Greco, Hagerman, Noctor, Hall, Hagerman and Tassone2013).
Previously, cases have shown that although mosaic individuals can have milder cognitive involvement, they can be at risk for developing FXTAS if their FMR1 mRNA levels are elevated (de Vries et al., Reference de Vries, Jansen, Duits, Verheij, Willemsen, van Hemel, van den Ouweland, Niermeijer, Oostra and Halley1996; Lokanga et al., Reference Lokanga, Entezam, Kumari, Yudkin, Qin, Smith and Usdin2013). In post pubescent males methylation and size mosaicism has also been associated with higher IQ scores and lower phenotypic presentation, when compared to males with fully methylated FM (Tassone et al., Reference Tassone, Longshore, Zunich, Steinbach, Salat and Taylor1999; Lokanga et al., Reference Lokanga, Entezam, Kumari, Yudkin, Qin, Smith and Usdin2013; Pretto et al., Reference Pretto, Mendoza-Morales, Lo, Cao, Hadd, Latham, Durbin-Johnson, Hagerman and Tassone2014 a ). Recently it has been shown that, FMR1 mRNA and FMRP expression are directly correlated with percentage methylation of the FMR1 allele. In addition, IQ scores were inversely correlated with percentage methylation and positively correlated with FMRP expression (Pretto et al., Reference Pretto, Yrigollen, Tang, Williamson, Espinal, Iwahashi, Durbin-Johnson, Hagerman, Hagerman and Tassone2014 b ).
Chile has 16 million inhabitants, composed predominantly of a mixture of Amerindian and Spanish populations (Cruz-Coke, Reference Cruz-Coke1976). As in many other countries, the real number of individuals with CGG expansions remains unknown (Tejada et al., Reference Tejada, Glover, Martinez, Guitart, de Diego-Otero, Fernandez-Carvajal, Ramos, Hernandez-Chico, Pintado, Rosell, Calvo, Ayuso, Ramos-Arroyo, Maortua and Mila2014). The first case of FXS was reported in 1983 (Lacassie et al., Reference Lacassie, Moreno, de la Barra, Bianca Curotto, Angelica Alliende, Anriquez, Barahona and Muzzo1983). Since then, two small studies have reported 2 and 3% of FXS among individuals attending special schools (Aspillaga et al., Reference Aspillaga, Jara, Avendano and Lopez1998; Alliende et al., Reference Alliende, Campora, Curotto, Toro, Valiente, Castillo, Cortes, Trigo, Alvarado, Silva and Caru2008). Although there are several large-scale studies reporting prevalence of FXS in different populations (Rousseau et al., Reference Rousseau, Labelle, Bussieres and Lindsay2011; Tassone et al., Reference Tassone, Iong, Tong, Lo, Gane, Berry-Kravis, Nguyen, Mu, Laffin, Bailey and Hagerman2012), few analyses have been published in Latin America, underscoring the importance of our report. Our aim was to present 30 years of laboratory experience diagnosing FXS and its related disorders among individuals with ID referred for fragile X testing as well as in their family members.
2. Materials and methods
(i) Sample
A retrospective review was performed on 2321 cases referred for FMR1 molecular testing conducted at the Cytogenetics and Molecular Laboratory at the Institute of Nutrition and Food Technology (INTA), University of Chile from 1983 to 2013.
(ii) Methods
FMR1 CGG-repeat size alleles were defined as normal size when between 5–44 repeats; intermediate or GZ when between 45–54 repeats; premutation when between 55–200 repeats and FM when >200 repeats. Fragile X Molecular Testing Guidelines of the American College of Medical Genetics, Standards and Guidelines for Clinical Genetics Laboratories (Monaghan et al., Reference Monaghan, Lyon and Spector2013) were used.
(a) Cytogenetics
Initially (in 1983), fragile X diagnosis was based only on clinical and cytogenetic analyses (Lacassie et al., Reference Lacassie, Moreno, de la Barra, Bianca Curotto, Angelica Alliende, Anriquez, Barahona and Muzzo1983). However, by 1997 the Southern blot was introduced and positive cases were subsequently confirmed.
(b) PCR
Initially PCR was performed as described by Haddad et al. (Reference Haddad, Mingroni-Netto, Vianna-Morgante and Pena1996) and applied only to males. PCR primers that flanked the trinucleotide repeats were used, with a product of 557 to 635 bp for normal size alleles visualized on silver-stained 6% polyacrylamide gel. Conditions were established such that FM that failed to amplify was then referred for SB analysis. PM alleles were not detected by this method. Since 2009, a new PCR assay was standardized, improving and refining the diagnostic testing by accurate amplification of fragments within the normal and PM range in both males and females (Saluto et al., Reference Saluto, Brussino, Tassone, Arduino, Cagnoli, Pappi, Hagerman, Migone and Brusco2005). PCR was performed with minor modifications to the protocol described by Saluto et al. (Reference Saluto, Brussino, Tassone, Arduino, Cagnoli, Pappi, Hagerman, Migone and Brusco2005). Briefly, PCR was based on 200 ng of gDNA to amplify the 5′UTR in the FMR1 gene using fluorescent started primers c (GCTCAGCTCCGTTTCGGTTTCACTTCCGGT) and f (TGGAGGAGCTGGTGGTGGAAGTGCGGGGCT). Expand Long Taq polymerase (Roche) and Betaine (Sigma) were used to provide successful amplification of longer PM alleles of up to 130 CGG repeats in males and females. The amplified fragments were analysed in agarose gel at 2%. Normal males obtained PCR products between ~300–350 bp. In GZ and PM males, PCR products extended beyond 350 bp. Nevertheless, PCR cannot amplify long fragments within the FM range (the target region). Therefore the absence of amplification product in males indicated that a FM allele would be present. In heterozygous females, two sizes of PCR bands were obtained. However, if samples gave a single PCR fragment in females or failed to amplify alleles in males, SB analysis was used to measure FM expansion and differentiate between females who were homozygous for a normal sized allele from females with a PM or FM heterozygous repeat expansion.
(c) CGG size amplification
Fragments greater than 350 bp long visualized by agarose gel (GZ and PM) were then sized using automated capillary electrophoresis ABI 3170xl (Applied Biosystems). Reactions were mixed with ROX 1000 size ladder and Hi-Di Formamide before being transferred to the Sequences Scan Analyzer. The fragment sizes were analysed using Gene Mapper 4·0.
(d) Southern blot
Direct analysis of the FMR1 gene was performed using methods previously described (Alliende et al., Reference Alliende, Campora, Curotto, Toro, Valiente, Castillo, Cortes, Trigo, Alvarado, Silva and Caru2008). A double digestion of 20 μg of gDNA was performed using restriction endonucleases EcoR1 and EagI overnight. After digestion and agarose gel electrophoresis, DNA was transferred to a nylon membrane Hybond N + (Amersham). The membrane was hybridized with 12·3 StB probe provided by J. L. Mandel (INSERM, France). Finally the membrane was marked with chemiluminescence (PCR DIG Probe Synthesis Kit, Roche) and visualized by autoradiography (Fu et al., Reference Fu, Kuhl, Pizzuti, Pieretti, Sutcliffe, Richards, Verkerk, Holden, Fenwick and Warren1991; Rousseau et al., Reference Rousseau, Heitz, Tarleton, MacPherson, Malmgren, Dahl, Barnicoat, Mathew, Mornet and Tejada1994; Alliende et al., Reference Alliende, Urzua, Valiente, Cortes, Curotto and Rojas1998).
(iii) Statistical analysis
Frequencies and percentages were used to describe overall findings. Logistic regression was used to predict the risk of having an affected child with FM. For that purpose, we firstly considered if the mother had a child with FM or not as a binomial outcome. In other words, if the mother had an affected child, then that was considered as ‘1’, if not, then we computed it as ‘0’. Subsequently, we took into account the maternal total CGG length as a continuous and independent variable to carry out the logistic regression using the IBM SPSS software version 22; a p-value <0·05 was considered as significant. Finally, we used the probability given for the CGG length to delineate that relationship.
3. Results
A total of 2321 samples were referred to our laboratory for FXS testing over a 30-year period. Of these, 2202 (94%) were unrelated individuals with ID and clinical features of FXS whose ages ranged from 2 months to 25 years; 119 (6%) were family members of affected probands referred for cascade screening. The majority of patients were referred from a variety of medical specialists from other institutions in the country. The remaining patients were clinically examined by paediatricians, clinical geneticists and neurologists from the Centre for Diagnosis and Treatment of Fragile X Syndrome (CDTSXF) at INTA.
(i) FMR1 results among cases referred for fragile X testing
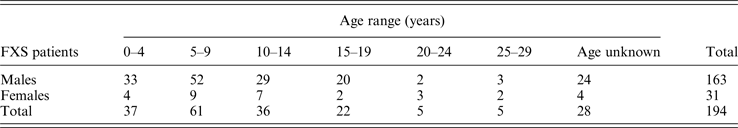
Among 2202 probands with ID and clinical features of FXS, 194 (8·8%) presented a FMR1 gene mutation, either a FM (n = 181), PM (n = 6) or GZ (n = 7). Table 1 shows the overall distribution of the different FMR1 mutations with >55 CGG repeats according to gender. The most frequent mutation was a FM/fully methylated allele identified in 165 (7·5%) of subjects, of them 135 (81·8%) were males and 30 (18·2%) were females. Whether all patients with any type of FM are considered, the overall prevalence of FMR1 mutations with >200 CGG repeats in the Chilean ID population is 8·2%.
Table 1. Distribution of FMR1 mutations with >55 CGG repeats detected in 194 FXS probands.

Considering family history, 86 (44%) had a family history of ID and 108 (56%) had no previous history of ID in the family. The 194 FXS patients corresponded to 152 different families and 42 corresponded to families where there were two or more ID affected children studied at the same time. The largest family registered comprised 22 individuals with a FMR1 expansion.
The average age at diagnosis for probands was 8·8 ± 5·4 years (mean calculated with the 166 cases in which the age at diagnosis was known). Table 2 shows that 77·7% of probands (129/166) were more than 5 years of age at diagnosis and only 22% (37/166) were diagnosed at <4 years of age.
Table 2. Distribution of FXS patients by age of diagnosis.
(ii) FMR1 results among cases referred for carrier detection
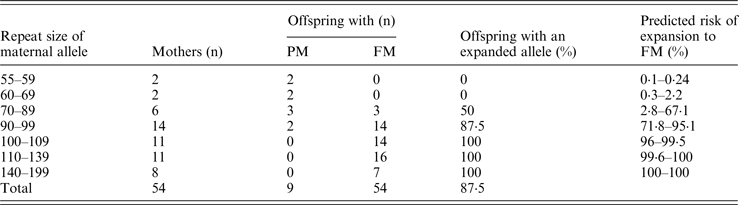
Among the 119 relatives of FXS patients referred for carrier detection or cascade screening, 72 (60%) were diagnosed with an expansion in the FMR1 gene; 61 were PM (54 females, seven males) and seven were FM females. Interestingly, two female obligate carriers presented different expansions in the FMR1 gene on both X chromosomes (compound heterozygous), they were daughters of non-consanguineous marriage, in which both parents were carriers of a PM. Additionally, an unmethylated FM male detected was part of a family study recently reported (Santa Maria et al., 2014). Table 3 shows the number of offspring with a FM and PM with respect to the range of maternal total CGG repeats and the predicted risk. As can be seen, the percentage of children with a FM is greater for those mothers with a longer expansion of the CGG repeats. Indeed, all females with >100 CGG repeats had only children with a FM. The logistic regression confidently predicted this situation (p = 0·011), which means that even if the mother transmitted the PM allele, the risk of having a child with FXS is directly related to the number of CGG repeats in that allele.
Table 3. Transmission of FMR1 premutation repeats from 54 females.
4. Discussion
The present study is the first to estimate the prevalence of FMR1 expansions in the Chilean population. Although, a number of studies have been carried out reporting prevalence of FXS in different populations (Crawford et al., Reference Crawford, Acuna and Sherman2001; Puusepp et al., Reference Puusepp, Kahre, Sibul, Soo, Lind, Raukas and Ounap2008; Essop & Krause, Reference Essop and Krause2013; Tejada et al., Reference Tejada, Glover, Martinez, Guitart, de Diego-Otero, Fernandez-Carvajal, Ramos, Hernandez-Chico, Pintado, Rosell, Calvo, Ayuso, Ramos-Arroyo, Maortua and Mila2014), knowledge in Latin America is relatively scant (Lacassie et al., Reference Lacassie, Moreno, de la Barra, Bianca Curotto, Angelica Alliende, Anriquez, Barahona and Muzzo1983; Florencia et al., Reference Florencia, Irene and Veronica2006; Christofolini et al., Reference Christofolini, Abbud, Lipay, Costa, Vianna-Morgante, Bellucco, Nogueira, Kulikowski, Brunoni, Juliano, Ramos and Melaragno2009).
Overall, the diagnostic yield of FMR1 variant expansions (FM, PM, GZ, size and methylation mosaics) was 8·8% (194/2202) in unrelated individuals with ID, and specifically, 8·2% (181/2202) had an allele with >200 CGG repeats. Of them, 6·7% were males and 1·5% females. The overall frequency detected is relatively higher compared to other ID populations, which show overall ranges in males between 1 and 5·2% (Crawford et al., Reference Crawford, Acuna and Sherman2001; Essop & Krause, Reference Essop and Krause2013). This paper reports a high frequency of FMR1 mutations among Chilean ID patients, and it demonstrates the need for further testing in Latin American countries such as Chile. We cannot exclude a bias in the detection of the most affected patients and families with more than one member affected; due to economic and accessibility difficulties, only patients with a high suspicion of FXS are referred for molecular analysis.
Concerning mosaicism, the proportion of size mosaic detected having a PM and FM was low (15/194, 7·7% of FXS; Table 1) compared to 41% reported by Nolin et al. (Reference Nolin, Glicksman, Houck, Brown and Dobkin1994) and other studies (Pretto et al., Reference Pretto, Mendoza-Morales, Lo, Cao, Hadd, Latham, Durbin-Johnson, Hagerman and Tassone2014 a ). This could be explained by methodological differences. Standard protocols used in this study may not be sufficiently sensitive to detect expansions of more than 130 CGG repeats or methylation mosaics (McConkie-Rosell et al., Reference McConkie-Rosell, Lachiewicz, Spiridigliozzi, Tarleton, Schoenwald, Phelan, Goonewardena, Ding and Brown1993; Genc et al., Reference Genc, Muller-Hartmann, Zeschnigk, Deissler, Schmitz, Majewski, von Gontard and Doerfler2000). This result could also reflect a bias in the sample; mosaics that can produce some FMRP, depending on the methylation and the CGG allele size, are from less developmentally delayed patients and for economical and geographic reasons explained above, this population could be under-represented in this study (McConkie-Rosell et al., Reference McConkie-Rosell, Lachiewicz, Spiridigliozzi, Tarleton, Schoenwald, Phelan, Goonewardena, Ding and Brown1993; Nolin et al., Reference Nolin, Glicksman, Houck, Brown and Dobkin1994; Genc et al., Reference Genc, Muller-Hartmann, Zeschnigk, Deissler, Schmitz, Majewski, von Gontard and Doerfler2000; Tassone et al., Reference Tassone, Hagerman, Chamberlain and Hagerman2000; Loesch et al., Reference Loesch, Sherwell, Kinsella, Tassone, Taylor, Amor, Sung and Evans2012; Pretto et al., Reference Pretto, Mendoza-Morales, Lo, Cao, Hadd, Latham, Durbin-Johnson, Hagerman and Tassone2014 a ; Santa Maria et al., 2014).
As expected, a PM allele was detected in only 0·3% (6/2202) of individuals with ID (Table 1) supporting the notion that FM alleles are the most common cause of inherited ID. Moreover, as DNA testing was performed in blood and as these six PM patients showed signs of ID and behavioural problems, we cannot exclude that these patients correspond to a size mosaic pattern in other tissues (Pretto et al., Reference Pretto, Mendoza-Morales, Lo, Cao, Hadd, Latham, Durbin-Johnson, Hagerman and Tassone2014 a ).
The average age of diagnosis among probands was 8·8 ± 5·4 years, this estimation is considerably higher than the age reported in the USA, where it is around 3 years of age (Bailey et al., Reference Bailey, Raspa, Bishop and Holiday2009). One study in the US reported that 1 in 3 to 1 in 4 African–American children attending special education schools were diagnosed after 10 years of age. One possible explanation given was that social and cultural differences might be at play (Crawford et al., Reference Crawford, Meadows, Newman, Taft, Scott, Leslie, Shubek, Holmgreen, Yeargin-Allsopp, Boyle and Sherman2002). In the context of Chile, the delay in diagnosis might more likely be related to lack of opportunity for diagnosis because of difficulties for families and health care programs in accessing genetic testing in Chile.
Our findings demonstrate a considerable number of unrelated FXS families, including a non-consanguineous family with two female sibling compound heterozygous patients with expansions on both alleles, inherited from unrelated PM parents (data not shown). This condition has been very rarely reported, only in five other cases. This information could be an indicator that the prevalence of this condition is similar to other populations and as a result, further studies are needed to confirm the prevalence of PM in the whole Chilean population.
Cascade screening and carrier detection in affected families allowed for the detection of 72 individuals with PM, FM or intermediate allele, including mothers of affected children, siblings, grandparents and second or third degree relatives. Because the FMR1 mutation is rarely de novo, cascade screening from a proband is helpful to detect other cases in the family. In our study, the number of relatives tested by cascade screening was notably low indicating that many family members are not being tested or receiving appropriate genetic counselling. As a consequence, many young women will face a new pregnancy without the knowledge of a PM condition and will not be aware of the risk of developing an adult-onset disorder.
In this study PM alleles determined in 54 mothers were positively correlated with the risk of having FM offspring, confirming previous reports that an increasing likelihood for unstable transmissions in one generation with increasing repeat size (Table 3). It has been described that 55 CGG alleles can expand to FM in a single generation (Biancalana et al., Reference Biancalana, Beldjord, Taillandier, Szpiro-Tapia, Cusin, Gerson, Philippe and Mandel2004). Albeit two PM females with alleles in the 55–59 CGG range and two mothers with 60–69 CGG alleles did not have children with FM, this finding could be related to a sample size bias and the non-studied presence of AGG interruptions, which decrease the possibility of transmitting an expanded allele (Yrigollen et al., Reference Yrigollen, Durbin-Johnson, Gane, Nelson, Hagerman, Hagerman and Tassone2012). Hence, it would be important to determine the number of AGG interruptions and the length of uninterrupted CGG repeats at the 3′ end for better allele characterization and risk prediction.
The results presented in this study provide molecular information from a Latin American country about the complexity of a disorder with many molecular facets, including CGG repeat size and methylation, which can lead to the broad spectrum of clinical involvement in FXS. We emphasize that a detailed molecular diagnosis, particularly in those cases with mosaicism, could be used clinically to provide additional information for genetic counselling and expectations for the family. It could also guide clinicians and public health policy, specifically concerning early diagnosis.
5. Conclusion
This study evaluated the present status of fragile X diagnosis in Chile. Results indicate a need for education of health professionals from different disciplines regarding the importance of early detection, which could lead to minimizing or preventing the risk of transmission. Collaboration among professionals involved in the diagnosis of FXS in Chile has not been sufficient to break the chain of transmission.
There is a high probability of finding others affected or at risk of presenting FMR1-related disorders in a family when a FMR1 expansion is detected. Support is improved with a confirmed diagnosis and may allow for the detection of more carriers in a family. This issue is essential for therapeutic approaches in FXS children, both in medical and cognitive outcomes, especially in families with more than one affected child.
We thank the study participants for their contribution. Solange Aliaga is a PhD student supported by CONICYT, Chile's National Commission for Scientific and Technological Research, for an international doctoral scholarship.
Declaration of interest
None.



