


Introduction
Inherited optic neuropathies are a heterogeneous group of disorders characterized by mild to severe visual loss, colour vision deficit (dyschromatopsia), central or paracentral visual field defects and optic disc pallor. Optic atrophy can be isolated or associated with multi-systemic involvement (syndromic; Allen et al., Reference Allen, Gaier and Wiggs2015) such as in Costeff's syndrome (methyl-glutaconic aciduria type 3; OMIM 258501; Ho et al., Reference Ho, Walter and Christodoulou2008), Behr's syndrome (OMIM 210000; Pyle et al., Reference Pyle, Ramesh, Bartsakoulia, Boczonadi, Gomez-Duran, Herczegfalvi, Blakely, Smertenko, Duff, Eglon, Moore, Man, Douroudis, Santibanez-Koref, Griffin, Lochmüller, Karcagi, Taylor, Chinnery and Horvath2014) or Wolfram syndrome (OMIM 598500; Safarpour et al., Reference Safarpour Lima, Ghaedi, Daftarian, Ahmadieh, Jamshidi, Khorrami, Noroozi, Sohrabifar, Assarzadegan, Hesami, Taghavi, Ahmadifard, Atakhorrami, Rahimi-Aliabadi, Shahmohammadibeni, Alehabib, Andarva, Darvish and Emamalizadeh2016). While multiple modes of inheritance have been reported, autosomal dominant optic atrophy (ADOA) and mitochondrial inherited Leber's hereditary optic neuropathy (LHON) are the most common forms (Pfeffer et al., Reference Pfeffer, Burke, Yu-Wai-Man, Compston and Chinnery2013; Skidd et al., Reference Skidd, Lessell and Cestari2013). Optic atrophy type 1, caused by mutations in the OPA1 gene is believed to be the most common hereditary optic neuropathy, and most patients inherit a mutation from an affected parent. Both ADOA and LHON are characterized by selective involvement of the retinal ganglion cells. Optic atrophy type 1 is characterized by bilateral and symmetric optic nerve pallor, associated with a childhood-onset decrease in visual acuity, visual field defects and colour vision defect (often tritanopia, blue–yellow loss). The estimated prevalence of OPA1-associated optic atrophy is 1:10 000–1:50 000. Up to 10% of individuals with OPA1 mutations have additional extra-ophthalmlogic abnormalities such as sensorineural hearing loss, ataxia and myopathy, termed ‘OPA1 plus’ (Amati-Bonneau et al., Reference Amati-Bonneau, Guichet, Olichon, Chevrollier, Viala, Miot, Ayuso, Odent, Arrouet, Verny, Calmels, Simard, Belenguer, Wang, Puel, Hamel, Malthièry, Bonneau, Lenaers and Reynier2005; Yu-Wai-Man et al., Reference Yu-Wai-Man, Griffiths, Burke, Sellar, Clarke, Gnanaraj, Ah-Kine, Hudson, Czermin, Taylor, Horvath and Chinnery2010; Skidd et al., Reference Skidd, Lessell and Cestari2013). In this study we used whole-exome sequencing to investigate the genetic aetiology in a patient affected with isolated optic atrophy.
Patients and methods
We applied whole-exome sequencing to a consanguineous family in which the proband presented with isolated optic atrophy. Local approval for this study was provided by the Rabin Medical Center Institutional Review Board, and all participants signed an informed consent.
Whole-exome sequencing
Genomic DNA extraction, exome enrichment, sequencing and analysis have been completed as described before (Reinstein et al., Reference Reinstein, Orvin, Tayeb-Fligelman, Stiebel-Kalish, Tzur, Pimienta, Bazak, Bengal, Cohen, Gaton, Bormans, Landau, Kornowski, Shohat and Behar2015). Genomic DNA was extracted from peripheral leukocytes following standard protocols. Exome enrichment was achieved by the Nextera Rapid Capture Expanded Exome Kit (FC-140-1006) following manufacturer's guidelines. Sequencing was performed using the Illumina HiSeq 2500 machinery to generate paired end reads of 150 bp with average coverage of 80X. The analytic pipeline available at the genetics and genomic medicine laboratory (Gene by Gene, Houston, Texas) was used to map the obtained fragments to the human reference genome (hg19) with the Burrows–Wheeler Alignment (BWA MEM) algorithm and genetic variants were identified using SAM tools. The variants were annotated using SnpEff (Li et al., Reference Li, Handsaker, Wysoker, Fennell, Ruan, Homer, Marth, Abecasis and Durbin2009; Cingolani et al., Reference Cingolani, Platts, Wang le, Coon, Nguyen, Wang, Land, Lu and Ruden2012). The results were analysed based on all modes of inheritance. Variants were filtered to generate a final list of rare functional variants only (missense, nonsense, splice site variants and indels). Variants with minor allele frequency >0·01 in the Exome Variant Server (release ESP6500) or that have allele count >150 in the ExAC database of Europeans (NFE) were removed. For validation and segregation analyses PCR primers were designed to amplify the regions flanking the two mutations. PCR products were purified using magnetic-particle technology (Seradyn, Inc.). After purification, all fragments were sequenced using forward and backward internal primers to determine the noted regions. Sequencing was performed on a 3730xl DNA Analyser (Life Technologies), and the resulting sequences were analysed with the Sequencher software (Gene Codes Corporation). The Illumina HumanOmniExpress-24 v1·0 BeadChip was used to detect copy number variations and for homozygosity mapping.
Clinical report
The nuclear family is of Arab Muslim ancestry, and consists of parents and four children. The parents are first-degree cousins and report no medical problems. The proband has three healthy siblings, aged 13, 12 and 4 years, with normal vision. The proband is a 7 year old male, with no significant past medical history. He was admitted to the pediatric ward because of headache, nausea, vomiting and decrease in visual acuity (VA) in both eyes over the past few months. The parents reported regression in his learning ability as well as in his fine motor skills recently and he was diagnosed with attention deficit disorder by a child neurologist. Physical examination revealed normal findings with no facial dysmorphism. Ophthalmology examination revealed reduced VA, 6/30 both eyes on first exam, with mild deterioration to 6/60 both eyes (right eye [RE] 6/60 and left eye [LE] 6/90) 2 years later. Near vision was Jeger 16 for both eyes together at the time of diagnosis and remained stable until the end of the follow up. Refraction was +2·25 RE and +2·50 LE with no improvement in vision with this correction. No relative afferent pupillary defect (normal pupillary response to rapid shift of light from one eye to the other indicates equal optic nerve function) was detected. He had normal colour vision (Ishihara colour plate), 12/12 for each eye at diagnosis, and 8/12 for each eye on last exam. Orthoptic evaluation revealed exotropia of -30 prism diopters (PD) for distance and -10 PD for near. Fundoscopic examination showed no papilloedema or other abnormal findings except for diffuse optic disc pallor. Visual field test was considered as unreliable. Additional work up including full-field rod–cone electro-retinogram and flash visual evoked potential were normal. Optical coherence tomography measured severe reduction in average nerve fibre layer thickness around the optic disc. Initial metabolic work up including plasma amino acids, very long chain fatty acids, carnitine, acylcarnitine and urinary organic acids was normal. Serum vitamin E and vitamin A levels were within normal limits. Serologic test results were negative for aquaporin-4 (AQP4) antibodies (detected in neuromyelitis optica), anti-proteinase 3 and anti-myeloperoxidase (detected in vasculitides-associated optic neuropathies). Serology for Bartonella henselae (cat-scratch disease) was also negative. Levels of C3 and C4 were normal. Cerebrospinal fluid oligo-clonal bands (detected in optic neuritis) were negative. Lumbar puncture showed normal opening pressure. Brain MRI scan was normal except for bilateral thickening of the optic nerves with increased fluid around the optic nerve sheaths. Based on these findings, a diagnosis of pseudotumour cerebri was suspected and he was treated with oral acetazolamide and steroids with no improvement in his symptoms.
Results
Screening for the three common LHON-associated mtDNA mutations (m.11778G > A, m.3460G > A and m.14484 T > C) revealed normal results. Subsequently, a SNP-based microarray was completed and showed no copy number variations. We did not identify known optic-atrophy associated genes within the homozygous regions, which encompassed 4% of the DNA (~1300 OMIM morbid genes). Next, whole-exome sequencing was carried out on the proband, his 13 year old healthy brother, and their parents. Exome results were analysed for all modes of inheritance, and revealed a heterozygous variant in the OPA1 gene in the proband: OPA1 c.1189A > G, p.Lys397Glu (chr3:193361347A > G). This variant has never been reported in any public domains. The lysine residue in this position is highly conserved in evolution (Fig. 1), and prediction algorithms (SIFT, Polyphen) showed high functional effect. The variant was validated by Sanger sequencing and segregation analysis showed that the proband's sibling and parents do not carry the identified OPA1 variant.

Fig. 1. Evolutionary conservation analysis of the OPA1 mutated domain. Lys 397 is marked.
Discussion
We report a novel de novo OPA1 variant in a child presenting with isolated optic atrophy. The vast majority of individuals diagnosed with optic atrophy type 1 have an affected parent. Hence, since the proband was the only affected individual in his extended family, and was a product of consanguineous marriage, a recessive inheritance was suspected and homozygosity mapping followed by whole-exome sequencing were pursued, rather than a targeted OPA1 gene test. De novo OPA1 mutations have been reported in the literature before. In one report, OPA1 testing in 20 patients with ADOA and no family history revealed two cases (10%) with de novo OPA1 mutations (Baris et al., Reference Baris, Delettre, Amati-Bonneau, Surget, Charlin, Catier, Derieux, Guyomard, Dollfus, Jonveaux, Ayuso, Maumenee, Lorenz, Mohammed, Tourmen, Bonneau, Malthièry, Hamel and Reynier2003). In a larger study, mutation screening for LHON, OPA1 and OPA3 genes was performed in 980 patients with suspected hereditary optic neuropathy (Ferré et al., Reference Ferré, Bonneau, Milea, Chevrollier, Verny, Dollfus, Ayuso, Defoort, Vignal, Zanlonghi, Charlin, Kaplan, Odent, Hamel, Procaccio, Reynier and Amati-Bonneau2009). Molecular diagnosis was established in 440 (45%) patients, out of which 295 patients (67%) had an OPA1 mutation and half of them (157/295) were apparently ‘sporadic’ cases without known family history. However, since parental samples were not available for testing, de novo mutations were confirmed in only 12 (4%) of the apparently sporadic cases.
Non-syndromic recessive optic atrophy is a rare form of optic neuropathy. To date, only two genes (TMEM126A and ACO2) and one locus (8q) have been identified (Allen et al., Reference Allen, Gaier and Wiggs2015). The main forms of non-syndromic optic atrophy are depicted in Table 1.
Table 1. Main forms of non-syndromic optic atrophy.
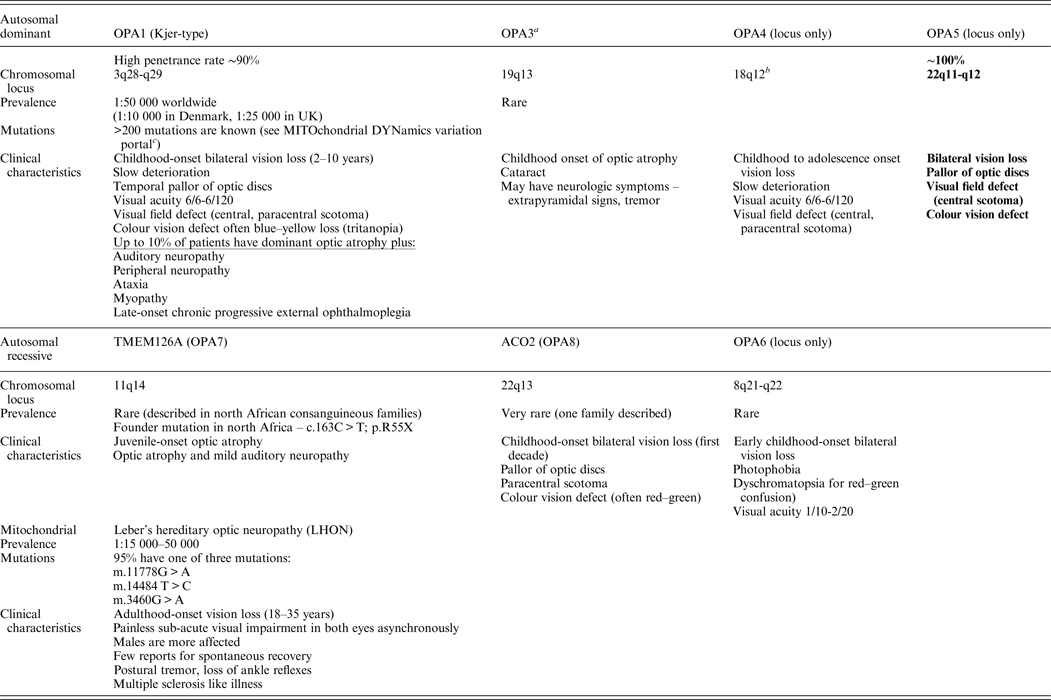
OPA1 and LHON are the most common forms of hereditary optic neuropathies.
a OPA3 mutations can also cause 3-methyl-glutaconic aciduria type 3 (Costeff syndrome), AR disease, early-onset bilateral optic atrophy. Later-onset spasticity, extrapyramidal dysfunction, cognitive impairment.
b Most pedigrees showed linkage to telomeric region of 3q28-q29.
c MITOchondrial DYNamics variation portal: http://mitodyn.org/home.php?select_db=OPA1.
Over 200 mutations in OPA1 have been reported hitherto (Ferré et al., Reference Ferré, Bonneau, Milea, Chevrollier, Verny, Dollfus, Ayuso, Defoort, Vignal, Zanlonghi, Charlin, Kaplan, Odent, Hamel, Procaccio, Reynier and Amati-Bonneau2009). Most of these mutations result in the loss of function of the mutated allele, supporting haploinsufficiency as the most likely mechanism (Pesch et al., Reference Pesch, Leo-Kottler, Mayer, Jurklies, Kellner, Apfelstedt-Sylla, Zrenner, Alexander and Wissinger2001). The estimated penetrence is ~90% in the familial forms of the disease (Cohn et al., Reference Cohn, Toomes, Potter, Towns, Hewitt, Inglehearn, Craig and Mackey2007). The OPA1 protein is composed of five regions: a mitochondrial leader sequence, a GTPase domain, a central dynamin domain and two coil-coiled regions (Alavi & Fuhrmann, Reference Alavi and Fuhrmann2013). Most reported mutations are located in the GTPase domain encoded by exons 8–16 or in the C-terminal coil-coiled domain encoded by exons 27–28. The identified mutation in this report also involves the GTPase domain of the OPA1 protein, which is highly conserved in evolution. Based on yeast studies showing that mutations in the yeast protein homologue, Mgm1, disrupt mitochondrial fusion, it was suggested that OPA1 mutations affect mitochondrial functions including fusion, energy metabolism, control of apoptosis and maintenance of mitochondrial genome integrity. Retinal ganglion cells are the most sensitive to apoptosis in optic atrophy type 1 patients and it still remains unclear why ADOA manifests with a restricted ocular phenotype, since the OPA1 gene is ubiquitously expressed.
In conclusion, although de novo OPA1 mutations are uncommon, testing of LHON associated mutations followed by OPA1 gene sequencing should be performed first in sporadic patients presenting with optic neuropathy, even when dominant inheritance is not apparent.
Declaration of interest
Doron M. Behar and Concetta Bormans are compensated and serve as the chief medical officer and the laboratory director of Gene by Gene, respectively.


