


1. Introduction
Difference in recombination rates between the sexes were observed in early cytological studies as different chiasma frequencies in female and male samples (Burt et al., Reference Burt, Bell and Harvey1991), and heterochiasmy has been subsequently noted in a diverse array of taxa (Lenormand & Dutheil, Reference Lenormand and Dutheil2005). Initial explanations for this wide-spread phenomenon stemmed from the observation that in some species, the heterogametic sex is achiasmate, or completely lacking any recombination, and it was suggested that recombination may be suppressed to varying degrees in the heterogametic sex in all organisms in order to prevent crossing over between the different sex chromosomes (Haldane, Reference Haldane1922; Huxley, Reference Huxley1928). While this hypothesis, named the Haldane–Huxley Rule, has been carried forward and is often assumed to be true in the current literature, it only applies to the small fraction of species where one sex is achiasmate; for the majority of species where both sexes recombine, but at different rates, the Haldane–Huxley Rule is clearly untenable as many species with both male (Burt et al., Reference Burt, Bell and Harvey1991) and female (Hansson et al., Reference Hansson, Åkesson, Slate and Pemberton2005; Åkesson et al., Reference Åkesson, Hansson, Hasselquist and Bensch2007) heterogametic systems fail to conform to the predicted pattern.
Further fine-scale work indicates that not only overall recombination rate but also the location of recombination within the genome varies greatly between females and males (Perry & Jones, Reference Perry and Jones1974; Shifman et al., Reference Shifman, Bell, Copley, Taylor, Williams, Mott and Flint2006). Based on this variety, it would be reasonable to speculate that recombination is not subject to selection, but rather the patterns we observe are the result of biological differences between females and males in meiosis. Specifically, the long-term torpor of vertebrate ova in the penultimate stages of arrested meiosis may produce fundamental differences between male and female recombination rate due simply to genomic and biochemical affinities rather than evolutionary mechanisms. However, the lack of a sex-specific pattern in the data makes this explanation also somewhat unsatisfying.
Studies have shown that recombination differs not only between the sexes but also among individuals (Broman et al., Reference Broman, Murray, Sheffield, White and Weber1998), populations (McVean et al., Reference McVean, Myers, Hunt, Deloukas, Bentley and Donnelly2004; Hernandez et al., Reference Hernandez, Hubisz, Wheeler, Smith, Ferguson, Rogers, Nazareth, Indap, Bourquin, McPherson, Muzny, Gibbs, Nielsen and Bustamante2007), and closely related species (Winckler et al., Reference Winckler, Myers, Richter, Onofrio, McDonald, Bontrop, McVean, Gabriel, Reich, Donnelly and Altshuler2005), suggesting that if these differences are heritable, sufficient variation exists to provide a target for selection pressure. Recent re-examinations of the heterochiasmy phenomenon with this in mind have resulted in evolutionary theory linking recombination rate differences between males and females to differences in sexually antagonistic selection pressures. One suggestion is that, as sexual selection is typically more powerful for males, this selective regime would produce reduced recombination in males in order to preserve the favourable gene combinations that confer male reproductive success (Trivers, Reference Trivers, Michod and Levin1988). There is some circumstantial evidence for this, as successful male tends to produce successful sons (Wedell & Tregenza, Reference Wedell and Tregenza1990); however, mathematical treatments failed to support the conjecture (Burt et al., Reference Burt, Bell and Harvey1991). Additionally, comparative phylogenetic work across plants and animals did not support the link between sexual selection and male recombination, although this may be due to vast differences in the sexual selection regimes of plants and animals resulting from differences in haploid versus diploid selection, parental provisioning and factors contributing to fertilization success.
More recently it was suggested that the pattern of heterochiasmy is a function of sex-specific selection during the haploid phase (Lenormand & Dutheil, Reference Lenormand and Dutheil2005), and results from the difference in strength of gametic selection for females and males. For vertebrates, female meiosis is typically completed at the point of fertilization, effectively eliminating any haploid selection for ova. However, haploid selection in males could theoretically result from sperm competition regimes, presumably with stronger sperm competition resulting in higher levels of heterochiasmy. Several studies have indicated that genes that function in sperm production or function are under exceptionally strong selective regimes (Aguade, Reference Aguade1999; Meiklejohn et al., Reference Meiklejohn, Parsch, Ranz and Hartl2003; Begun & Lindfors, Reference Begun and Lindfors2005). Additionally, Drosophila recombination, which is limited to females, was shown to be more common in regions of male-biased gene expression, suggesting higher recombination in females for genes related to sperm function (Zhang & Parsch, Reference Zhang and Parsch2005). Therefore, this hypothesis shows some promise in explaining heterochiasmy patterns.
Several sex-specific linkage maps and cytological screens are now available for the eutherian (placental) mammals, making it possible to quantify the degree of heterochiasmy for these lineages (Table 1). These data make tenable a rigorous test of the role of sex-specific selection in the diploid (Trivers, Reference Trivers, Michod and Levin1988) versus haploid (Lenormand & Dutheil, Reference Lenormand and Dutheil2005) phases of the life cycle within a clade that shares many reproductive aspects. Additionally, eutherian mammals all share placental gestation, which is a major component of female reproductive fitness. Confining analysis to this clade therefore controls, at least to some extent, for variance in female-specific selection regimes, making it possible to focus on male fitness components and their relationship to recombination.
Table 1. Eutherian taxa included in this analysis, along with information on sperm competition and sexual dimorphism
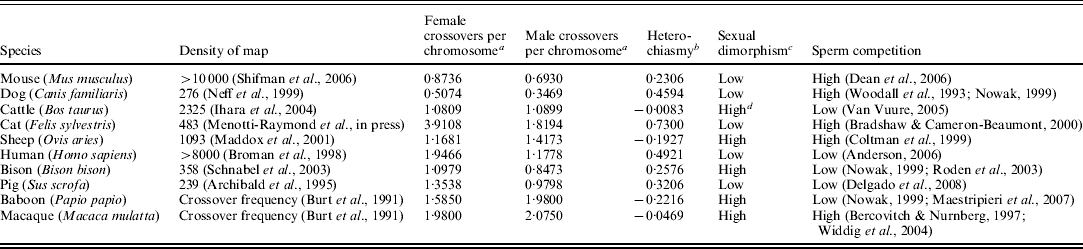
a Corrected for variation in marker density (Chakravarti et al., Reference Chakravarti, Lasher and Reefer1991) and undetected crossover events in unmapped regions (Hall & Willis, Reference Hall and Willis2005).
b (Female recombination per chromosome-male recombination per chromosome)/average recombination per chromosome.
c High levels of sexual dimorphism is defined as difference in body weight >30%, or the presence of a sex-specific sexual ornament, such as horns or antlers. All estimates taken from (Nowak, Reference Nowak1999) unless otherwise noted.
d (Van Vuure, Reference Van Vuure2005)
If sexually antagonistic selection, either in the diploid or haploid phase, is linked to recombination rate differences between the sexes, there should be a clear pattern in the data. Specifically if differences in sex-specific selection on the diploid phase of the life cycle resulting from sexual selection are ultimately causing the heterochiasmy under the model presented by Trivers (Reference Trivers, Michod and Levin1988), we would expect lineages with greater variance in male mating success to have less recombination in males, therefore sexually selected lineages would show greater heterochiasmy in recombination as a function of suppression of recombination in males.
If heterochiasmy is the product of differences between females and males in haploid selection in the gametic phase, somewhat different patterns are expected. In mammals, the haploid phase of the life cycle is relatively limited in both sexes, though most so in females, where meiosis is completed at fertilization, and therefore is nearly non-existent. Haploid selection, if it is a substantive evolutionary force at all, would be observed primarily for sperm. Therefore, if heterochiasmy is a function of sexually antagonistic haploid selection, those species with more intense sperm competition should show greater differences in female and male recombination.
2. Materials and methods
I found sex-specific recombination data, either from linkage mapping studies or recombination nodule counts, for ten eutherian mammal species. Although many more eutherian mammals have been the subject of linkage mapping, the vast majority of these studies computed only a sex-averaged map. Where multiple studies existed for a single species, I selected those based on linkage mapping rather than cytology, and where multiple linkage maps existed, I chose those with greater marker density and genome coverage. The species included in this analysis (Table 1) are human (Broman et al., Reference Broman, Murray, Sheffield, White and Weber1998), baboon (Burt et al., Reference Burt, Bell and Harvey1991), macaque (Burt et al., Reference Burt, Bell and Harvey1991), sheep (Maddox et al., Reference Maddox, Davies, Crawford, Hulme, Vaiman, Cribiu, Freking, Beh, Cockett, Kang, Riffkin, Drinkwater, Moore, Dodds, Lumsden, van Stijn, Phua, Adelson, Burkin, Broom, Buitkamp, Cambridge, Cushwa, Gerard, Galloway, Harrison, Hawken, Hiendleder, Henry, Medrano, Paterson, Schibler, Stone and van Hest2001), pig (Archibald et al., Reference Archibald, Haley, Brown, Couperwhite, McQueen, Nicholson, Coppieters, Vandeweghe, Stratil, Wintero, Fredholm, Larsen, Nielsen, Milan, Woloszyn, Robic, Dalens, Riquet, Gellin, Caritez, Burgaud, Ollivier, Bidanel, Vaiman, Renard, Geldermann, Davoli, Ruyter, Verstege, Groenen, Davies, Hoyheim, Keiserud, Andersson, Ellegren, Johansson, Marklund, Miller, Dear, Signer, Jeffreys, Moran, Letissier, Muladno, Tuggle, Vaske, Helm, Liu, Rahman, Yu, Larson and Schmitz1995), dog (Neff et al., Reference Neff, Broman, Mellersh, Ray, Acland, Aguirre, Ziegle, Ostrander and Rine1999), cat (Menotti-Raymond et al., Reference Menotti-Raymond, David, Schaffer, Tomlin, Eizirik, Phillip, Wells, Pontius, Hannah and O'Brien2009), cow (Ihara et al., Reference Ihara, Takasuga, Mizoshita, Takeda, Sugimoto, Mizoguchi, Hirano, Itoh, Watanabe, Reed, Snelling, Kappes, Beattie, Bennett and Sugimoto2004), bison (Schnabel et al., Reference Schnabel, Taylor and Derrm2003) and mouse (Shifman et al., Reference Shifman, Bell, Copley, Taylor, Williams, Mott and Flint2006). Although sex-specific maps exist for marsupial species (Zenger et al., Reference Zenger, McKenzie and Coop2002; Samollow et al., Reference Samollow, Kammerer, Mahaney, Schneider, Westenberger, VendeBerg and Robinson2004), the reproductive differences resulting from the evolution of extended internal gestation and the placenta in the eutherians vastly alter the female-specific selection regime from that of the non-placental clades, therefore these linkage maps were not included in this analysis.
For each of these species, I also searched the current literature for information about mating system in order to assign the degree of dimorphism and sperm competition. The effort and expense of linkage map analysis means that, until recently, most efforts were either confined to those animals of medical (mouse and macaque), commensal (dog and cat) or agricultural (sheep, pig and cattle) importance, and for these species I either searched for mating system information on the wild (macaque and mouse), ancestral (cattle) or feral (cat, dog, sheep and pig) populations. Based on mating system assessments (Woodall et al., Reference Woodall, Pavlo and Tolley1993; Nowak, Reference Nowak1999; Bradshaw & Cameron-Beaumont, Reference Bradshaw and Cameron-Beaumont2000; Maestripieri et al., Reference Maestripieri, Mayhew, Carlson, Hoffman and Radtke2007) and genetic paternity analyses (Bercovitch & Nurnberg, Reference Bercovitch and Nurnberg1997; Coltman et al., Reference Coltman, Bancroft, Robertson, Smith, Clutton-Brock and Pemberton1999; Nowak, Reference Nowak1999; Roden et al., Reference Roden, Vervaecke, Mommens and van Elsacker2003; Widdig et al., Reference Widdig, Bercovitch, Streich, Sauermann, Nurnberg and Krawczak2004; Anderson, Reference Anderson2006; Dean et al., Reference Dean, Ardlie and Nachman2006; Delgado et al., Reference Delgado, Fernandez-Llario, Azevedo, Beja-Pereira and Santos2008), I determined whether lineages were likely to experience relatively high or low sperm competition. It is important to note that a certain degree of sperm competition is possible in virtually every type of mating system, given sufficient time and scrutiny; however, some mating systems are far more likely to contain higher levels of sperm competition than others. Specifically mate guarding in conjunction with pair-bonded and haremic mating systems will generally result in less sperm competition than more promiscuous behavioural ecologies.
Sexual dimorphism can be used to assess the degree to which pre-copulatory sexual selection defines the majority of variance in male mating success, which is predicted to correlate with heterochiasmy under diploid models (Trivers, Reference Trivers, Michod and Levin1988). Sexual dimorphism is present to some extent in all eutherians due to the physiological and anatomic requirements of internal gestation and lactation, therefore the strict presence or absence of sexual dimorphism alone is not helpful in this analysis. However, the degree of sexual dimorphism can be used as an indicator, therefore, I assessed whether species were strongly sexually dimorphic, exhibiting either large body size differences (>30%) or sexually selected traits such as horns in sheep (Nowak, Reference Nowak1999).
(i) Recombination data
Linkage mapping data must be corrected mathematically for variation in marker density and coverage in order to make comparable mapping efforts made with different panels of genetic markers. I first corrected the linkage mapping data for variance in marker density using the equations in Chakravarti et al. (Reference Chakravarti, Lasher and Reefer1991), as greater marker density is more likely to recover double crossing over. I then corrected for undetected crossover events on unmapped portions according to Hall & Willis (Reference Hall and Willis2005). Once these corrections were made, the sex-averaged cytological data from macaque and baboon matched well with the sex-averaged linkage maps of these and closely related species (Rogers et al., Reference Rogers, Mahaney, Witte, Nair, Newman, Wedel, Rodriguez, Rice, Slifer, Perelygin, Slifer, Palladino-Negro, Newman, Chambers, Joslyn, Parry and Morin2000, Reference Rogers, Garcia, Shelledy, Kaplan, Arya, Johnson, Bergstrom, Novakowski, Nair, Vinson, Newman, Heckman and Cameron2006; Cox et al., Reference Cox, Mahaney, VandeBerg and Rogers2006), therefore recombination nodule information was included in further analyses. For all species, I only analysed the autosomal portions of the genome, as the sex chromosomes do not recombine along the majority of their length in the heterogametic sex.
The size and number of chromosomes can influence recombination (de Villena & Sapienza, Reference de Villena and Sapienza2001), therefore I corrected for this by calculating the number of crossover events per chromosome, defined here as homologous autosomes for females, males and averaged across sexes. I also calculated a measure of heterochiasmy, or the difference in recombination between the sexes, defined as (recombinationfemale−recombinationmale)/recombinationaverage.
(ii) Comparative analysis
Recombination hotspots change rapidly among related species (Winckler et al., Reference Winckler, Myers, Richter, Onofrio, McDonald, Bontrop, McVean, Gabriel, Reich, Donnelly and Altshuler2005) and within populations of the same species (McVean et al., Reference McVean, Myers, Hunt, Deloukas, Bentley and Donnelly2004; Hernandez et al., Reference Hernandez, Hubisz, Wheeler, Smith, Ferguson, Rogers, Nazareth, Indap, Bourquin, McPherson, Muzny, Gibbs, Nielsen and Bustamante2007), and a strong case can be made that the millions of years that separate the species in this study would obliterate any phylogenetic signal in recombination rate. Therefore I first examined the data regardless of shared ancestry by computing the average female, male and sex-averaged crossovers per chromosome for species with high and low sexual dimorphism, and high and low sperm competition. I also computed the average degree of heterochiasmy and assessed whether this was related to dimorphism or sperm competition. Significant difference was assessed with a two-tailed t test in each case.
Despite the potential for a quickly evolving trait to change more rapidly than can be mapped onto a sparse topology, it is important to test for phylogenetic signal as shared ancestry can create spurious associations. I therefore used the complete mitochondrial coding content of the species in this analysis to construct a neighbour-joining phylogeny in MEGA (Tamura et al., Reference Tamura, Dudley, Nei and Kumar2007), using the PAM Dayhoff Matrix of amino acid substitution, and assuming a difference rate of substitution among sites. Significance was assessed with 1000 bootstrap replicates. The branch lengths and topology from this tree were used to identify whether the continuous characters of female crossovers per chromosome and male crossovers per chromosome evolve in accordance with the phylogenetic history, or whether shared ancestry is not relevant to the distribution of the trait. This was done with the CoMET module (Lee et al., Reference Lee, Blay, Mooers, Singh and Oakley2006) in Mesquite 2.6 (Maddison & Maddison, Reference Maddison and Maddison2009). Both male and female chiasmata per chromosome fit best to the non-phylogenetic, equal matrix model, suggesting that phylogenetic history could be ignored for these characters.
Interestingly, the heterochiasmy score did follow a phylogenetic distribution (pure phylogenetic model). I therefore divided the heterochiasmy data into two categories, those taxa with higher recombination in females and those with higher recombination in males, and then tested the relationship between direction of heterochiasmy (male or female) and sexual dimorphism (high or low) and sperm competition (high or low) using the MESQUITE implementation of Pagel's maximum likelihood estimation of discrete character evolution (Pagel, Reference Pagel1994), using 1000 simulations to assess significance in each case.
3. Results
Recombination rates varied widely over the taxa assessed here, and the sex-averaged recombination rate per chromosome was lowest in dog (0·5371), where the long stretches of linkage disequilibrium (Karlsson & Lindblad-Toh, Reference Karlsson and Lindblad-Toh2008) have proved ideal for haplotype mapping of breed-specific traits and diseases (Drogemuller et al., Reference Drogemuller, Karlsson, Hytonen, Perloski, Dolf, Sainio, Lohi, Lindblad-Toh and Leeb2008; Wiik et al., Reference Wiik, Wade, Biagi, Ropstad, Bjerkas, Lindblad-Toh and Lingaas2008). Average recombination was highest in cat (2·8651).
Male and female recombination also varied widely, both within and between sexes. Heterochiasmy estimates show that while many species have higher recombination in females, cattle have slightly more recombination in males, and sheep, baboons and macaques have significantly male-biased recombination rates. Therefore, heterogamety cannot explain heterochiasmy as all eutherian mammals share a homologous system of male heterogamety, and the Haldane–Huxley Rule fails to explain 30–40% of the data.
Assessing all data irrespective of phylogenetic history showed a strong overall relationship between heterochiasmy and sexual dimorphism (P<0·005, Fig. 1), though neither male nor female crossovers per chromosome differed significantly by themselves by sexual dimorphism class (P>0·13 in both cases). Sperm competition was not significantly associated with sex-specific recombination or heterochiasmy rates (P>0·5, Fig. 2).
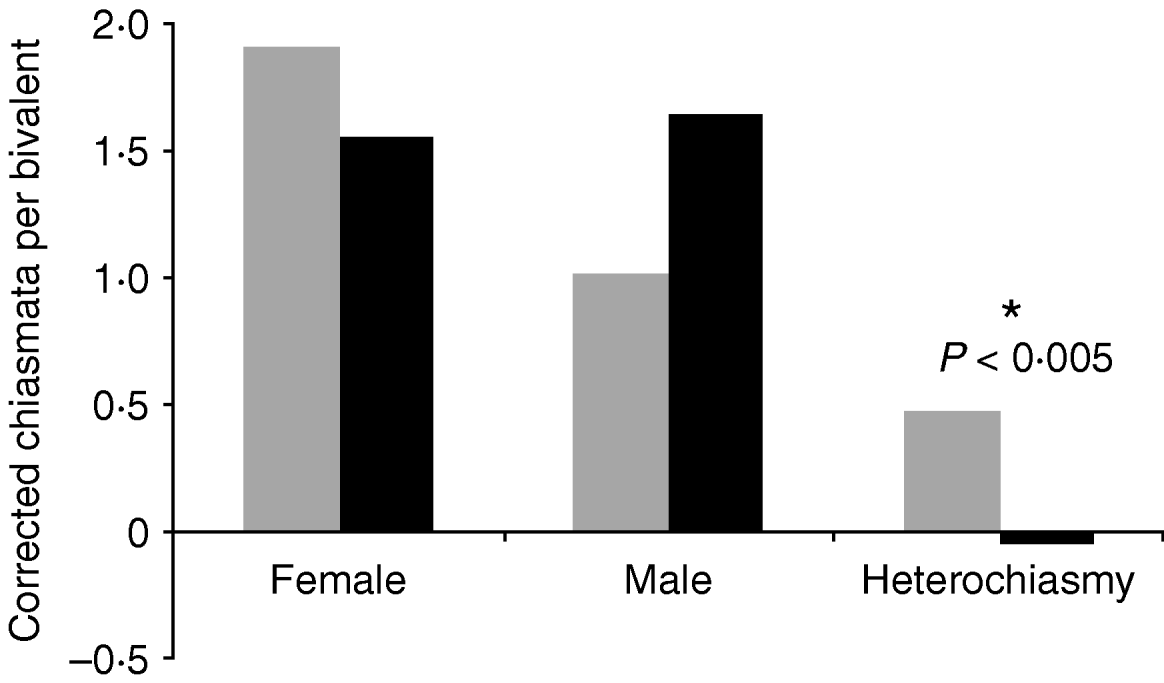
Fig. 1. Average number of crossovers per chromosome for taxa with high and low levels of sexual dimorphism. The comparison of overall heterochiasmy was significantly different (two-tailed t test). Grey bars indicate taxa with low sexual dimorphism, black bars indicate high sexual dimorphism.

Fig. 2. Average number of crossovers per chromosome for taxa with low (grey) and high (black) levels of sperm competition, based on mating system and/or genetic parentage analysis. No comparison was significant (two-tailed t test).
The mitochondrial coding content was used to derive a phylogenetic reconstruction of these taxa, shown in Fig. 3. This phylogeny is topologically identical to recent mitochondrial (Arnason et al., Reference Arnason, Adegoke, Gullberg, Harley, Janke and Kullberg2008) and genomic (Prasad et al., Reference Prasad, Allard, Green and Program2008) phylogenies of the mammals. As all branches were supported by at values >95% (based on 1000 bootstrap replicates), there was no need to collapse any branches into polytomies to indicate phylogenetic uncertainty.
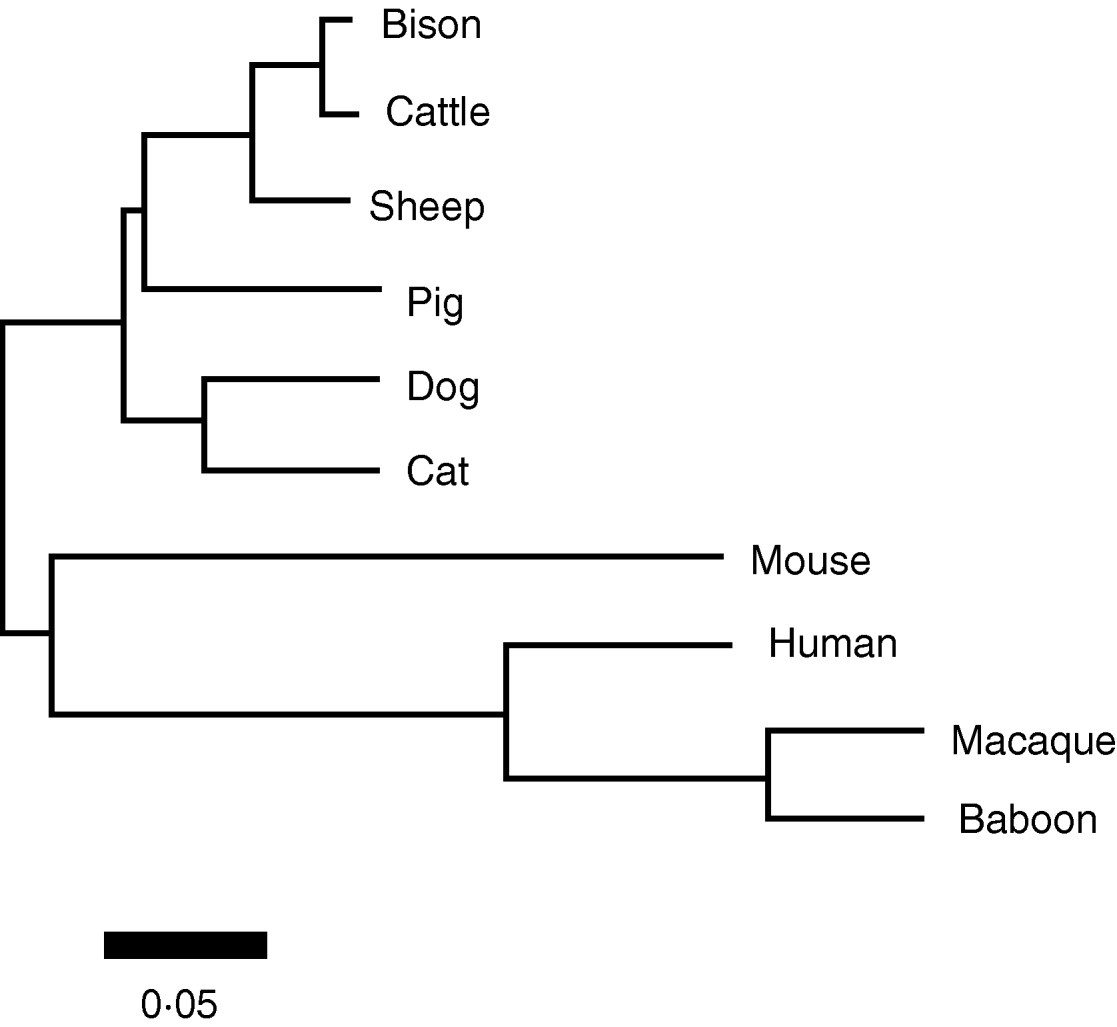
Fig. 3. Neighbour-joining reconstruction of the phylogenetic relationships among the taxa in this study based on the full protein-coding complement of the mitochondrial genome. In all cases, bootstrap values of significance exceeded 95%. Relative branch lengths are indicated, indicated by the scale bar.
I used this phylogeny to determine whether the recombination traits assessed here show an associated due to shared ancestry, or whether phylogenetic history can be safely ignored. Model testing (Lee et al., Reference Lee, Blay, Mooers, Singh and Oakley2006) indicated that while sex-specific estimates of chiasmata per chromosome did not show a phylogenetic distribution greater than that expected by chance (for reasons discussed later), the direction of heterochiasmy did show some phylogenetic signal in the continuous data.
Due to this phylogenetic signal, I also performed assessments of the relationship between heterochiasmy and sexual dimorphism and sperm competition using maximum likelihood models of discrete character evolution (Pagel, Reference Pagel1994), incorporating the topology and branch-length estimates from the phylogeny in Fig. 1. For this analysis, I assigned the heterochiasmy data into two states; taxa either showed greater recombination in females or males. The results from this analysis are convergent with those counting species independently. Specifically, lineages with high levels of sexual dimorphism were more likely to experience greater relative recombination in males (1000 simulations, P=0·02). There was no relationship between heterochiasmy and sperm competition (1000 simulations, P=0·45).
4. Discussion
In this study, I tested two alternative hypotheses related to the difference in recombination between the sexes. Previous studies of this phenomenon (Burt et al., Reference Burt, Bell and Harvey1991; Lenormand & Dutheil, Reference Lenormand and Dutheil2005) attempted to circumvent the paucity of data by pooling across diverse clades with vastly different fertilization and maternal provisioning characteristics. By employing information solely from the eutherian mammals, a clade defined by internal fertilization and placental gestation, this analysis controls for variation in female-specific fitness related to maternal–foetal conflict and internal fertilization, both of which can significantly influence the female fitness topology.
I found sex-specific recombination data for ten eutherian species distributed throughout the mammalian phylogeny, including eight molecular linkage maps that I corrected for marker density (Chakravarti et al., Reference Chakravarti, Lasher and Reefer1991) and genome coverage (Hall & Willis, Reference Hall and Willis2005), and two visual cytological scans for recombination nodules. All of these assessments were corrected for variation in chromosome number, as this can affect rates of recombination (de Villena & Sapienza, Reference de Villena and Sapienza2001). The inclusion of these latter karyotypic studies is not expected to be problematic for two primary reasons. Most importantly, the majority of this study is concerned with relative within-species differences between the sexes, and while there may be differences in overall measurement between the different types of analysis used here, there is no reason to suspect a sex-specific bias, therefore the use of a heterochiasmy ratio would not be affected by the use of different types of recombination data. Additionally, the karyotypic studies showed similar numbers of chiasmata per chromosome to corrected sex-averaged linkage maps for these and closely related taxa (Rogers et al., Reference Rogers, Mahaney, Witte, Nair, Newman, Wedel, Rodriguez, Rice, Slifer, Perelygin, Slifer, Palladino-Negro, Newman, Chambers, Joslyn, Parry and Morin2000, Reference Rogers, Garcia, Shelledy, Kaplan, Arya, Johnson, Bergstrom, Novakowski, Nair, Vinson, Newman, Heckman and Cameron2006; Cox et al., Reference Cox, Mahaney, VandeBerg and Rogers2006).
Interesting, female- and male-specific estimates of crossovers per chromosome did not show a phylogenetic signal in this study, which differs from work done on a similar set of taxa (Dumont & Payseur, Reference Dumont and Payseur2007). This may reflect the difference in marker density in the data of Dumont & Payseur (Reference Dumont and Payseur2007) and that employed here. While these studies used overlapping taxon samples, the requirement of sex-specific maps in this analysis necessitated the inclusion of some maps constructed from far fewer markers (Archibald et al., Reference Archibald, Haley, Brown, Couperwhite, McQueen, Nicholson, Coppieters, Vandeweghe, Stratil, Wintero, Fredholm, Larsen, Nielsen, Milan, Woloszyn, Robic, Dalens, Riquet, Gellin, Caritez, Burgaud, Ollivier, Bidanel, Vaiman, Renard, Geldermann, Davoli, Ruyter, Verstege, Groenen, Davies, Hoyheim, Keiserud, Andersson, Ellegren, Johansson, Marklund, Miller, Dear, Signer, Jeffreys, Moran, Letissier, Muladno, Tuggle, Vaske, Helm, Liu, Rahman, Yu, Larson and Schmitz1995; Neff et al., Reference Neff, Broman, Mellersh, Ray, Acland, Aguirre, Ziegle, Ostrander and Rine1999; Maddox et al., Reference Maddox, Davies, Crawford, Hulme, Vaiman, Cribiu, Freking, Beh, Cockett, Kang, Riffkin, Drinkwater, Moore, Dodds, Lumsden, van Stijn, Phua, Adelson, Burkin, Broom, Buitkamp, Cambridge, Cushwa, Gerard, Galloway, Harrison, Hawken, Hiendleder, Henry, Medrano, Paterson, Schibler, Stone and van Hest2001). However, there is no reason to expect a sex-specific bias, and hence different linkage data is not a problem in this analysis as the majority of the study is concerned with the ratio between the sexes, rather than overall chiasmata numbers. This is supported by the recovery of a phylogenetic signal from the heterochiasmy ratio itself.
These data made it possible to test two alternative hypotheses proposed to explain the pattern of heterochiasmy. Lenormand & Dutheil (Reference Lenormand and Dutheil2005) showed mathematically that differences in sex-specific selection at the haploid level, presumably due to sperm competition, should predict recombination rate differences between the sexes, and I used information on mating system and genetic paternity analyses to infer the potential for sperm competition. However, there was no association in either the phylogenetically independent or controlled data between sperm competition and heterochiasmy. This may be because the haploid phase in males as well as females is functionally limited. While female haploid selection is very brief at best due to arrested meiosis, which is only completed at fertilization, the male haploid phase may also be limited due to limited gene expression in the spermatazoa.
An alternative hypothesis does not require haploid selection, as Trivers (Reference Trivers, Michod and Levin1988) posited that sexual selection, in general, would select for reduced recombination in males in order to preserve the gene combinations that conferred high fitness in the father for his sons. I tested this by measuring the degree of heterochiasmy and relating it to the degree of sexual dimorphism, which is a measure of sexual selection and reproductive skew in males. Sexual dimorphism was significantly associated with heterogamety, both in the general analysis (P<0·005) and once controlled for phylogeny (P=0·02). However, the observed pattern did not fit the prediction, with male-biased recombination accompanying high levels of sexual dimorphism.
This raises the question of why males in systems with high levels of sex-specific fitness variance recombine more. The answer to this may reside in the prevalence in these mammals of male dispersal. Previous models assumed a static sex-specific fitness landscape (Trivers, Reference Trivers, Michod and Levin1988; Lenormand & Dutheil, Reference Lenormand and Dutheil2005), assuming the factors that predict reproductive fitness in one generation would also hold in subsequent generations. However, the tendency of males to disperse more often and farther than females in eutherian mammals (Pusey & Packer, Reference Pusey and Packer1986; Gates & Larter, Reference Gates and Larter1990; Thomson et al., Reference Thomson, Rose and Kok1992; Pal et al., Reference Pal, Ghosh and Roy1998; Turner & Bateson, Reference Turner and Bateson2000; Truve & Lemel, Reference Truve and Lemel2003) could produce a shifting male fitness topology.
In the absence of mutual mate choice, the main determinants of the female-specific fitness landscape at the diploid stage are linked to gestation and post-natal care, and these factors do not, in theory, change greatly over the generations. By contrast, the male-specific fitness landscape involves elements of female-choice and male–male competition. With the added component of male dispersal, this diploid landscape has the potential to shift among generations, and the genetic combinations that ensured the reproductive success of the father may no longer predict the reproductive success of his sons due to differences in female preference and adaptations of rival males. The temporal shifting of the male-specific fitness topology would potentially select for increased recombination in males, as changing ecologies have been shown to select for increased recombination (Charlesworth, Reference Charlesworth1976; Otto & Nuismer, Reference Otto and Nuismer2004). Therefore, differences in sex-specific selection pressures, in conjunction with changing fitness topologies for one sex alone, could very well exert a strong antagonistic evolutionary pressure to increase recombination in males and decrease it in females.
Additionally, theoretical studies have shown that higher rates of recombination may result from higher selection pressures (Felsenstein & Yokoyama, Reference Felsenstein and Yokoyama1976; Lenormand & Otto, Reference Lenormand and Otto2000; Otto & Barton, Reference Otto and Barton2001) in order to reduce the problems associated with the loss of genetic variation due to linkage disequilibrium. This is in accordance with the analysis presented here, as recombination rate in males under higher sex-specific selection pressures, show higher rates of recombination.
The increase in male recombination for lineages under greater levels of sexual selection provides an interesting potential solution to the lek paradox. Continuous and powerful selection for male traits theoretically depletes the genetic variability for male sexually selected traits, therefore reducing the heritability (Reynolds & Gross, Reference Reynolds and Gross1990; Ritchie, Reference Ritchie1996). Despite these predictions, systems with strong sexual selection often contain significant levels of heritability for male sexually selected traits (Norris, Reference Norris1993; Kruuk et al., Reference Kruuk, Slate, Pemberton, Brotherstone, Guinness and Clutton-Brock2002 ), and female choice for male traits persists in these lineages, creating a paradox that has proved difficult to explain in a population genetic context. The process of recombination has been suggested to be mutagenic (Lercher & Hurst, Reference Lercher and Hurst2002; Hellmann et al., Reference Hellmann, Ebersberger, Ptak, Pääbo and Przeworski2003), creating the possibility that the accelerated recombination in males provides a continuously renewing source of genetic variation to counter the depletions resulting from sexual selection.
The generation of additional high-density sex-specific linkage maps in the future will make it possible to test this hypothesis in several ways. Specifically, the inclusion of maps from a greater variety of eutherians will facilitate the study of the evolutionary forces exerted by mating system, including polyandry and monogamy, and female and male philopatry on recombination rate differences between the sexes. Additionally, maps of greater marker density will make it possible to pinpoint regions of heterochiasmy, and relate these to reproductive fitness components via gene function and expression.
This work was supported in part by a grant from the Royal Society. The helpful comments of two anonymous reviewers are gratefully acknowledged.




