


1. Introduction
miRNAs are single-stranded, small, non-coding RNAs composed of approximately 18–25 nucleotides (Zhong et al., Reference Zhong, Coukos and Zhang2012). miRNAs are derived from pri-miRNA, which has a hairpin-like structure and undergoes subsequent cleaving processes to make mature miRNA by Drosha and Dicer (Lee et al., Reference Lee, Han, Yeom, Jin and Kim2006). miRNAs have inhibitory functions for modulating the post-transcriptional regulation of gene expression by partial or complete binding to the 3′-untranslated region (UTR) of their target mRNAs, thereby mediating mRNA translational inhibition or degradation (Ha & Kim, Reference Ha and Kim2014). On the basis of bioinformatics analyses, individual miRNAs are able to bind up to several hundred genes, and miRNAs potentially regulate a third of human mRNAs (John et al., Reference John, Enright, Aravin, Tuschl, Sander and Marks2004; Lewis et al., Reference Lewis, Burge and Bartel2005), implying that miRNAs may impact all aspects of cell biology. Indeed, miRNAs play important roles in various biological processes, such as development, proliferation, differentiation, cell fate determination, apoptosis, signal transduction, host–viral interaction and tumorigenesis (Stern-Ginossar et al., Reference Stern-Ginossar, Elefant, Zimmermann, Wolf, Saleh and Biton2007; Welch et al., Reference Welch, Chen and Stallings2007; Choy et al., Reference Choy, Siu, Kok, Lung, Tsang and To2008; Ivey & Srivastava, Reference Ivey and Srivastava2010, Baumjohann & Ansel, Reference Baumjohann and Ansel2013). Dysregulation of miRNA–target gene interaction can result in debilitating diseases, including cancer development and metastasis (Tavazoie et al., Reference Tavazoie, Alarcón, Oskarsson, Padua, Wang and Bos2008; Hayes et al., Reference Hayes, Peruzzi and Lawler2014).
Despite multiple computational tools being useful in predicting miRNA target genes, candidate genes should be experimentally validated in order to obtain full legitimacy. Conventional molecular techniques such as northern blot, reverse transcription polymerase chain reaction, microarrays, sequencing of RNA, western blot and enzyme-linked immunosorbent assay have been used to validate target genes. Nevertheless, these methods are not appropriate for validating the entire lists of candidate genes predicted from computational algorithms due to the limitations of these techniques in terms of revealing direct correlations between an miRNA and a candidate gene.
A 3′-UTR reporter assay has been commonly used in order to experimentally validate direct miRNA targets (Aldred et al., Reference Aldred, Collins and Trinklein2011). This method is based on the idea that miRNAs function by mediating translational repression of their target mRNAs. Many studies have been performed using in vitro 3′-UTR reporter assays in order to validate the target genes of specific miRNAs (Kuhn et al., Reference Kuhn, Martin, Feldman, Terry, Nuovo and Elton2008; Thomson et al., Reference Thomson, Bracken and Goodall2011). Furthermore, a luciferase reporter system has been recently utilized in order to study whether miRNAs bind to the 3′-UTR regions of target genes (Oh & Do Won Hwang, Reference Oh and Do Won Hwang2013).
To apply the luciferase reporter system, the 3′-UTR regions of potential miRNA target genes have to be cloned into a reporter vector. Traditionally, the whole 3′-UTR region of a target gene was cloned for use in the luciferase reporter system (Fig. 1). Even though the whole 3′-UTR sequence is ideal for studying an miRNA and its targets, it can take time and effort to clone it. Recently, the use of only a surrounding region of the miRNA regulatory element (MRE) (~60 bp) was successful at validating the target genes (Jin et al., Reference Jin, Chen, Liu and Zhou2013). This study used a chemically synthesized ‘linker’ instead of amplifying the whole 3′-UTR region in order to validate candidate genes. The ‘linker’ has a duplex form and contains the predicted MRE from computational algorithms and restriction sites at both ends. Even though this study proved that the surrounding region of the MRE is sufficient for validating candidate genes, the linker took a relatively long time to synthesize and the cloning efficiency was low because the linker had the same restriction enzyme at both ends.
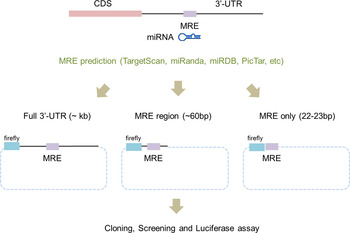
Fig. 1. Scheme of a modified linker reporter assay. According to the target prediction algorithms, including TargetScan, miRanda and PicTar, the full sequences of 3′-untranslated regions (UTR) were analysed in order to find miRNA regulatory element (MRE) sequences that putatively bind to miR-708. For the experimental validation, either the whole 3′-UTR (left), the MRE with a partial sequence of the 3′-UTR (middle) or only MRE sequences (right) were incorporated into the reporter vector. Two cases – whole 3′-UTR (left) and only MRE sequences (right) – were tested in this study.
Here, we describe the improved luciferase 3′-UTR reporter assay containing only a short MRE sequence (~22 bp) and demonstrate that our new, improved technique provides a simplified platform for validating target genes compared with the conventional luciferase reporter assay.
2. Materials and methods
2.1. Primer and linker design
Lists of putative target genes are obtained from miRNA target prediction tools such as miRanda, TargetRank and TargetScan (Ryu et al., Reference Ryu, McDonnell, Choi, Gao, Hahn and Joshi2013). MRE sequences are gained from the 3′-UTR sequences of putative target genes by analysing predicted miR-708 binding sequences. Linker sequences are designed to have computationally predicted MRE sequences that can complementally bind to miRNA (Fig. 2(b)). Two short, single-stranded primers were chemically synthesized and solubilized in 10 mM Tris (Qiagen, Venlo, The Netherlands) as 100 μM stock (Table 1). Two paired primers were self-ligated to form a double-stranded linker. Simply put, each primer was mixed in a ratio of 1:1 and boiled at 95 °C for 5 minutes and slowly cooled to room temperature.
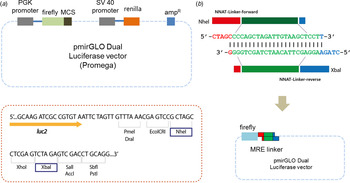
Fig. 2. Designing a linker. (a) Either a miRNA regulatory element linker or whole 3′-untranslated regions of target genes regulated by miR-708 were incorporated into the pmirGLO Dual Luciferase vector. Two different enzymes – NheI and XbaI – are used for cloning. (b) The ‘linker’ is derived from self-ligation of two synthetically designed, single-stranded primers. When two primers form a duplex, both ends become sticky ends automatically (5′-NheI and 3′-XbaI) and bind directly to digested vectors without further enzyme digestion.
Table 1. Linker sequence information of putative miR-708 target genes.

To ligate with a reporter vector, a linker should have restriction enzyme sites at both ends. In our method, the linker has two different restriction enzyme sites: NheI and XbaI at the 5′ and 3′ ends of the MRE sequence, respectively (Fig. 2(b)). Simply put, when two primers form a duplex, both ends are sticky and automatically mimic enzyme-digested inserts. There is no extra restriction enzyme digestion step. Sequence information is shown in Table 1 and Table 2. All linkers were mixed in a single tube and ligated with a vector at the same time. Vectors and linkers were mixed in a ratio of 1:3 for ligation. T4 DNA ligase enzyme was used for the ligation. Three-fold greater numbers of colonies than numbers of linkers were selected in order to determine the type of linker.
Table 2. Primer sequence information for cloning whole 3′-untranslated regions of putative miR-708 target genes.

2.2. Construction of the linker reporter vector
pmirGLO Dual Luciferase vector was purchased (Promega, Fitchburg, WI) and used to make the modified luciferase reporter assay system. This vector contains firefly luciferase (Fluc), which can be used to measure the interactivity of miR-708 as described previously (Ko et al., Reference Ko, Lee and Kim2009). Renilla luciferase (Rluc) is used to normalize the efficiency of the transfection of the DNA vector (Ko et al., Reference Ko, Lee and Kim2009). Either the ‘linker’ or the whole 3′-UTR region of the target genes that are regulated by miR-708 were incorporated into the pmirGLO vector (Fig. 2(a)).
2.3. DNA transfection and luciferase activity assay
Transfection of the reporter vector was carried out using a mixture of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) with reduced serum medium, Opti-MEM (Thermo Scientific, Waltham, MA) in 293 T cells according to the manufacturers’ instructions. Either ‘linker reporter plasmid vector’ or empty vectors with or without miR-708 were transfected into 293 T cells. A total of 1 μg of each MRE linker reporter plasmid and empty or miR-708-bearing vector was used for transfection into 293 T cells with 4 μl of Lipofectamine 2000. After 24-hour incubations, 293 T cells were lysed with 1X passive lysis buffer and luciferase activity was measured following manufacturer's instructions (Fig. 1).
3. Results
We previously reported the characteristic functions of miR-708 and revealed its biological role in cancer metastasis by regulating NNAT expression (Ryu et al., Reference Ryu, McDonnell, Choi, Gao, Hahn and Joshi2013). NNAT was identified as a true target gene using a reporter vector with a whole 3′-UTR region of NNAT, and this was confirmed by using a reporter vector with a mutated binding site (Ryu et al., Reference Ryu, McDonnell, Choi, Gao, Hahn and Joshi2013). In this study, we have utilized NNAT as a confirmed target gene of miR-708 in order to test our newly developed technique.
We obtained a list of predicted target genes, including NNAT, by using several miRNA target prediction algorithms such as miRanda, TargetRank and TargetScan (Ryu et al., Reference Ryu, McDonnell, Choi, Gao, Hahn and Joshi2013). NNAT and HOXB3 are predicted by all three distinct algorithms (Ryu et al., Reference Ryu, McDonnell, Choi, Gao, Hahn and Joshi2013). In addition, SSRP1, HNRNPK, EPDR1 and FLT1 have been predicted by more than two algorithms in our previous research (Ryu et al., Reference Ryu, McDonnell, Choi, Gao, Hahn and Joshi2013).
Based on the results, we designed the appropriate primers to amplify the whole 3′-UTR and to synthesize the ‘linker’ for a given list of candidate target genes (Fig. 1). The lengths of the 3′-UTR regions varied from several hundred to nearly 1000 bp (Table 2). We then cloned them into the luciferase reporter vector, the pmirGLO Dual Luciferase vector. This vector contains two independent luciferases: firefly luciferase (Fluc) for the translational suppression and Renilla luciferase (Rluc) for the normalization (Fig. 2).
To make the linkers, we chemically synthesized two short sequences of ‘linker’ in duplex form, having restriction enzyme sites at both ends of the MRE sequence, as is shown in Table 1. In this study, we used different restriction enzymes – NheI and XbaI – for each end in order to prevent self-ligation of the vector without a dephosphorylation step (Fig. 2(b)), which was used in the previous method employing a surrounding MRE region (Jin et al., Reference Jin, Chen, Liu and Zhou2013).
First, we tested whether our newly developed technique was accurate enough to suppress luminescence signals by miRNAs in comparison with the conventional luciferase reporter assay. We subsequently measured luciferase activity after the transfection of either empty or miR-708-expressing vectors in 293 T cells. Importantly, the luciferase activity linked to the NNAT linker was dramatically decreased after transfection of miR-708-expressing vector compared to empty vector (Fig. 3(a)). Critically, we demonstrated the validated target gene NNAT as being directly regulated by miR-708 using a linker reporter assay.
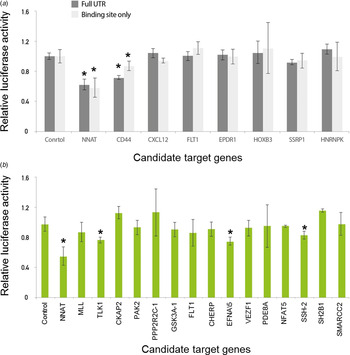
Fig. 3. Modified linker reporter assays are comparable to the whole 3′-untranslated region (UTR) reporter assay. Various candidate genes, including NNAT and CD44, were predicted by several target prediction algorithms and selected for 3′-UTR cloning. (a) Either empty or miR-708-bearings vector were co-transfected with a reporter vector into 293 T cells. After 24 hours of incubation, the activity of luciferase was measured. The luciferase signals from the whole 3′-UTR were compared with the signals from a miRNA regulatory element (MRE) linker. (b) Many linkers were selected for high-throughput miRNA target assay. The MRE sequences were designed by the Probability of Interaction by Target Accessibility (PITA) target prediction algorithm and cloned into the reporter vector. Either empty or miR-708-bearing vectors were co-transfected with a reporter vector into 293 T cells. After 24 hours of incubation, luciferase activity was measured. *p-value < 0·05.
We then compared the luciferase activity between the full lengths of the 3′-UTR regions and a MRE linker for the various candidate target genes of miR-708. However, the other genes were not heavily affected by the transfection of miR-708, which is consistent with the previous conventional luciferase reporter assay (Fig. 3(a)). The results of both the conventional 3′-UTR and the linker luciferase assay consistently discriminated genuine targets (e.g., NNAT and CD44) from a list of candidate target genes of miR-708. Taken together, these data suggest that our newly developed technique is reliable for validating target genes relative to conventional methods.
We found that NNAT and CD44 are the true targets of miR-708 by both conventional 3′-UTR and linker luciferase assays, as previous studies have shown (Saini et al., Reference Saini, Majid, Shahryari, Arora, Yamamura and Chang2012; Ryu et al., Reference Ryu, McDonnell, Choi, Gao, Hahn and Joshi2013). To find novel, genuine targets of miR-708, we further investigated the candidate target genes of miR-708 using another prediction algorithm, Probability of Interaction by Target Accessibility (PITA). This algorithm had been developed to predict the target genes of miRNAs based on target gene accessibility and similarity (Kertesz et al., Reference Kertesz, Iovino, Unnerstall, Gaul and Segal2007).
According to the PITA algorithm, a total of 2069 genes were predicted as target candidates. A total of 45 genes were predicted by both PITA and the in-house algorithm, which only considers the maximum similarity value. We then selected 16 genes out of 45, including MLL and TLK1, for further validation (Fig. 3(b)). Even though several genes, including TLK1, EFNA5 and SSH-2, were suppressed (approximately 20% by miR-708), suppression was not as large compared with the suppression of NNAT, which was close to 50% (Fig. 3(b)). To confirm whether or not these signals were noise, we assessed another miRNA, miR-200a. As we expected, miR-200a does not suppress the luciferase activity of NNAT, SSH-2, EFAN5 and TLK1 linkers (Supplemental Figure 2(a) & 2(b)). By combining these data, we suggest that our new, modified linker reporter assay selectively suppresses the targets of miRNA dependent on their complementary binding.
4. Discussion
Bioinformatics tools facilitate the study of the characteristic functions of miRNAs by providing a list of potential target genes (Krek et al., Reference Krek, Grün, Poy, Wolf, Rosenberg and Epstein2005; Lewis et al., Reference Lewis, Burge and Bartel2005; Ryu et al., Reference Ryu, McDonnell, Choi, Gao, Hahn and Joshi2013). However, the currently available computational methods and techniques make it difficult to validate the entire list of hundreds to thousands of target genes obtained from target prediction algorithms due to their demands on time and effort. Recently, luciferase reporter assays have been widely used to monitor miRNA and validate target genes linked to the 3′-UTR (Kuhn et al., Reference Kuhn, Martin, Feldman, Terry, Nuovo and Elton2008; Thomson et al., Reference Thomson, Bracken and Goodall2011; Oh & Do Won Hwang, Reference Oh and Do Won Hwang2013). However, it is inefficient to clone each 3′-UTR region of hundreds of target genes.
In this study, we describe an improved linker reporter assay and have observed that this linker reporter assay produces data that are consistent with conventional reporter assays using whole 3′-UTR regions (Fig. 3(a)). These results indicate that our protocol is applicable for validating target genes. In addition, our improved linker reporter assay has economic advantages compared to previous reporter assays containing surrounding MRE regions (Jin et al., Reference Jin, Chen, Liu and Zhou2013). The linker is only 18–22 bp in length, corresponding to the predicted miRNA MRE sequence, in contrast to the previous protocols containing linkers of 60 bp in length for the surrounding MRE region (Jin et al., Reference Jin, Chen, Liu and Zhou2013). It is cost effective since shorter sequences can be chemically synthesized at lower costs.
We further tested several candidate genes, including NNAT, CD44, SSRP-1, TLK1, EFNA5, by using western blot analysis, but only NNAT showed suppressed signals (Supplemental Figure 2(c)). These data suggest that a further step using western blot analysis is necessary in order to validate the real target of an miRNA. It is possible that some MRE sequences are not accessible to miRNAs because a whole 3′-UTR sequence forms a complicated secondary structure, and so some MREs are not accessible to miRNAs. Nevertheless, our method can narrow the entire list of candidate targets from several hundreds to few strong candidates. These strong candidates will be further validated with western blot analysis at the protein level.
Previous protocols also performed an extra dephosphorylation step after cutting the vector in order to prevent self-ligations (Jin et al., Reference Jin, Chen, Liu and Zhou2013). However, our protocol does not require this step because the linker contains two different restriction enzyme sites at each end. Furthermore, our new, modified reporter assay also has outstanding advantages for designing mutated miRNA binding sites for the further validation of target genes. Taken together, our technique is a cost-effective and efficient method compared to the previous protocols that have been performed in a high-throughput manner.
Our technique will be applied not only for the validation of the target genes of miRNAs, but also for monitoring miRNA biogenesis in various experimental conditions in vivo. Thus, the study of miRNA biogenesis may allow us to understand more deeply the biological processes and cellular mechanisms of organogenesis and cell differentiation. Collectively, our approach is efficient for validating candidate miRNA target genes and may provide an opportunity to discover novel genes and cellular mechanisms as potential prognosis/diagnosis markers and therapeutic targets.
This research was supported by the Marine Biotechnology Program (PJT200620) funded by the Ministry of Oceans and Fisheries and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2014R1A1A1006650), Korea.
5. Declaration of interest
The authors have no commercial or financial conflicts of interest.
6. Authors’ contributions
J. Han, C. Choi and S. Ryu conceived and designed the experiments. J. Han, C. Choi, T. Nguyen, S Yoon and S. Ryu performed the experiments. S. Song and Y. Kim analysed the data. J. Han, C. Choi, Y. Kim and S. Ryu wrote the manuscript. All authors contributed to critically revising the manuscript for important intellectual content, gave final approval and agreed to be accountable for all aspects of the work.






 Open access
Open access