



Introduction
A limited understanding of immune regulatory mechanisms hinders the implementation of immune-based protocols in cancer treatment. Currently, immunotherapy is recognized as one of the most effective ways to treat cancer patients, and its promising effects bring us closer to a future where this disease can be successfully controlled through the human life cycle. In practice, immunotherapeutic approaches to cancer have revolutionized this decade owing to the exponential growth in the number of clinical studies started in the last years. Figure 1 summarises immunotherapeutic clinical trials for breast, lung, colorectal and prostate cancer, the first-four most common cancer types worldwide according to the International Agency for Research on Cancer (IARC) (Ref. Reference Bray1).
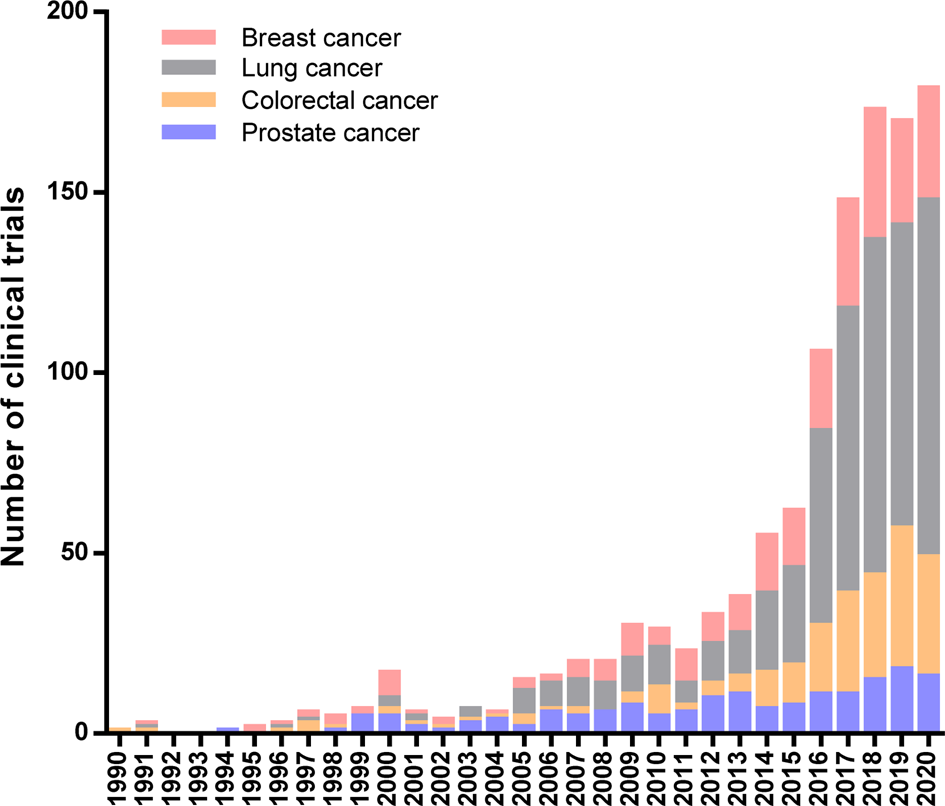
Fig. 1. Immunotherapeutic Clinical Trials started during 20th century worldwide. Number of clinical trials per year for the four most common cancer types: breast, lung, colorectal and prostate cancer. Data extracted from the ClinicalTrials.gov database according to the following search strategy: (a) study type: interventional studies, (b) status of study results: all studies, (c) additional criteria: study starts from 01/Jan/1990 to 30/Nov/2020 (data updated until 1/Dec/2020).
The effectiveness of immunotherapeutic protocols in cancer control is attributed to their high specificity since prescribed treatments are conditioned to the molecular characteristics of each kind of tumour. Unlike conventional therapies that just focused on wiping out cells with a high division rate, cancer immunotherapy seeks to address and counter tumour immune evasion strategies, and/or uses the fingerprints acquired by malignant cells to differentiate them from healthy ones in the body (Refs Reference Hilf2, Reference Karapetyan3). Through these mechanisms, immunotherapy is able to enhance the patient's immune response to act specifically on tumour cells. Although specificity portraits the greatest advantage of immunotherapy, at the same time, it turns out to be the main cause that prevents the massive use of these therapeutic approaches.
For this reason, it is necessary to develop better and more rigorous methodologies for tumour taxonomy as part of cancer diagnosis, in order to improve treatment decisions. For a long time, cancer was diagnosed and treated according to itslocation and histopathological characteristics. However, the long-standing experience in treating patients based on this methodology has demonstrated a broad response spectrum for equal treatments, including people with null or poor treatment effects. This heterogeneous response from patients evidences the existence of cancer subtypes and the lack of an accurate taxonomic system (Ref. Reference Jordan4). Multiple bioinformatics studies performed in the last years have shown better classification strategies for patient diagnosis and treatment prognosis based on molecular markers (Ref. Reference Pajtler5). More recently, it has been proposed to consider a tumour immune classification for the diagnosis, treatment and prognosis of cancer patients because it has already been demonstrated that immune characteristics of tumour microenvironment play a decisive clinical role in the therapeutic outcome (Refs Reference Binnewies6, Reference Zhou7).
The molecular and immune diversity from each cancer intricates the design of a single and equally functional treatment for all cases. Additionally, tumour genetic heterogeneity may lead to tumour resistance to treatments (Refs Reference Andor8, Reference Stewart9). Inter- and intra-tumour heterogeneity produces variable therapeutic results depending on the type of neoplasm and patient's genetic composition. For this reason and despite all of the advancements, cancer remains one of the leading causes of death worldwide (Ref. Reference Soerjomataram, Bray, Wild, Weiderpass and Stewart10). Hence, much effort is currently devoted to producing more specific treatments, improving response rates and identifying biomarkers to predict those patients who would benefit from receiving a specific targeted therapy. Throughout this article, we will make a conceptual and historical review of the main advances in our understanding of the role of the immune system in cancer, while describing methodological details successfully implemented on cancer treatments.
Antigen presentation by cancer cells
In the last decade, 10 hallmarks of cancer were described related to tumour progression in patients (Ref. Reference Hanahan and Weinberg11). Most of these characteristics explain cancer cell mechanisms to survive, proliferate, migrate and colonize different organs and systems. Nevertheless, all of these mechanisms seem to converge at genome instability and gene mutations that initiate and allow the disruption of cellular functions, promoting the acquisition of tumour features.
Even though not all somatic mutations induce changes in protein function, those that involve modifications of amino acid sequence or variations in the stop codon may allow structural and chemical alterations which, subsequently, may lead to dysregulation of normal cell functions. For instance, cell cycle alterations including death inhibition, survival and proliferative induction are responsible for tumour development. However, most of the mutations found in tumour cells have been described as passengers because they do not contribute to cancer development. Meanwhile, a minority of mutations, known as driver mutations, are responsible for cancer cell survival, growth and thus tumour progression (Ref. Reference Dietlein12).
Beyond the functional significance of these mutations in disease progression, the alterations in gene expression products on cancer cells could be also used to differentiate them from normal cells. The identification of cancer cells as a target by the immune system may lead to the subsequent specific-elimination of these malignant cells. In this way, the presentation of intracellular antigens to immune cells is performed through Major Histocompatibility Complex class I (MHC-I). The antigen presentation process starts with peptidases and proteasome protein complexes located in the cytoplasm, which mediate protein degradation to peptides. After degradation, short sequence products are translocated to the endoplasmic reticulum, where they are charged on the MHC-I complex and relocated to the extracellular membrane to expose intracellular peptides to immune cells, more specifically to CD8+ T cells (Ref. Reference Dersh, Hollý and Yewdell13). Hence, through this machinery, tumour mutant antigens – also referred to as neoantigens – are exposed to be recognized by the immune system (Ref. Reference McGranahan14). Based on the possibility of specific identification of tumour cells by antigen presentation mechanisms, the study of neoantigens vaccine and their possibility to activate the immune system against cancer has been investigated for some time (Refs Reference Monach15, Reference Duperret16).
Although several non-silent mutations were identified through tumour DNA sequence analysis, a reduced fraction of these were capable of activating the antitumor immune response in preclinical studies (Ref. Reference Hundal17). The immunogenicity of neoantigens will depend on several factors, such as (1) the degradation pathway for the mutated proteins, (2) their interaction with molecules of the antigenic presentation pathway, (3) their capacity to produce 8–11 amino acids sequence needed to interact with the MHC-I complex, (4) the affinity of the mutated peptides to be loaded in the MHC-I molecules, and (5) their ability to be exposed outwards the MHC-I/peptide complexes allowing their recognition by T lymphocytes. Owing to the complexity of this system, neoantigens immunogenicity is poorly predictable through standard bioinformatics methods (Ref. Reference Koşaloğlu-Yalçın18).
In this regard, new techniques were proposed to search for effective neoantigens. Whole-exon sequencing technology is a current methodology used to predict with great efficacy tumour antigens capable of CD8+ T-cell activation. Although this methodology may be effective when designing personalized vaccines, there is a risk of finding tumour subpopulations that do not express these neoantigens owing to tumour intrinsic heterogeneity (Ref. Reference Abécassis19). The survival of tumour subpopulations after treatment, which usually cannot be detected by current medical examinations, leads to cancer relapse. Beyond neoantigen discovery, the high variety of MHC-I molecules found in human population (product of a combination up to six different alleles per individual) (Ref. Reference Marty20), besides the ability of tumour cells to prevail by reducing both immune cell recruitment and effector immune response on tumour microenvironment, are facts that hinder the effectiveness of clinical trials of neoantigen-based cancer immunotherapy.
Immune response and cancer progression
The first work that relates immune response to cancer was developed by William Bradley Colley at the end of the 19th century (Ref. Reference Coley21). Based on previously documented cases of about 50 hospitalized patients with cancer who improved their health upon contracting a bacterial infection, Coley prepared a safe mixed vaccine using both heat-inactivated streptococcal bacteria and its products (i.e. Streptococcus pyogenes and Serratia marcescens). After several years using these bacterial toxins, he reported improved medical outcomes and tumour regression in treated patients with bone and soft sarcoma. Even though his scientific contribution was not recognized at that time, nowadays Coley is considered the father of immunotherapy because of this contribution (Ref. Reference Decker and Safdar22).
At the beginning of the 20th century, Paul Ehrlich proposed that malignant cells emerge continuously in the organisms and, similarly, the permanent surveillance carried out by immune cells would be involved in controlling tumour growth at early stages (Ref. Reference Ribatti23). Decades later, thanks to new knowledge acquired about the role of the immune response in transplant rejection, Burnet (1957) and Thomas (1959) brought back the hypothesis of immune-surveillance in cancer (Ref. Reference Dunn24). These events marked important pillars in the recognition of the essential role played by the immune system against cancer, leading to the implementation of immunotherapeutic approaches against this disease.
Currently, it is largely known that cancer cells could be eliminated by the immune system as a natural result from immune-surveillance, continuous supervision mediated by our immune cells to detect and eliminate tumours at the beginning, avoiding tumour progression (Ref. Reference Mittal25). Immune surveillance is mediated by innate and adaptive immune mechanisms (Refs Reference Bottos26–Reference Anton and Yewdell28) and is activated by different factors, such as cellular stress and cellular alteration signals, that is dysregulated proliferation, DNA damage and senescence (Refs Reference Senovilla29, Reference Galluzzi30), besides neoantigen recognition (Ref. Reference McGranahan14).
Several innate immune cells participate in the recognition and elimination of tumour cells. One of the best-studied innate immune populations in cancer is NK cells. This cell population is responsible for the recognition of no-MHC expressing cancer cells, eliminating them by cytotoxic induction mainly through the release of granzymes and perforin; also by receptor-mediated death pathways such as TRAIL and FasL, and antibody-dependent cellular cytotoxic (ADCC) mechanism (Ref. Reference Wagner31). Other minor innate populations also display their cytotoxic effects against the tumour, such as NKT type I (iNKT) and gamma/delta T cells (ɣδT cells). iNKT cells recognize glycolipid neoantigens presented on CD1d molecules, such as tumour-derived ganglioside GD3 (Ref. Reference Paget32). Meanwhile, ɣδT cells are activated by recognition of stress-induced molecules, such as MICA, MICB, ULBP 1–4, RAET1 (Ref. Reference Siegers33). Thus, these early cytotoxic effector functions contribute to the recognition of tumour antigens and allow the stimulation of other immune cell populations.
Antigen-presenting cells (macrophages and DCs) can recognize dying cells because of the ability of their membrane receptors to differentiate specific intracellular products displayed on the extracellular surface owing to cell damage. After recognition, macrophages and DC phagocyte the apoptotic tumour cells and become activated. In this state, DCs are able to migrate to the nearest lymph nodes and induce T-cell activation through antigen presentation, serving as a link between innate and adaptive immune responses (Ref. Reference Zhang34). Antigens carried on MHC-II molecules allow the activation of specific CD4+ T lymphocytes (T-helper lymphocytes), whereas antigens carried on MHC-I molecules (by cross-presentation) mediates the activation of CD8+ T lymphocytes (cytotoxic T cells) (Ref. Reference Veglia35). Once activated, CD4+ T cells release cytokines that could mediate B-cell maturity and antigen-specific antibody production. Also, CD4+ T lymphocytes help on CD8+ T-cell activation. Activated CD8+ T cells work mainly on tumour cell elimination (Refs Reference Ahrends36, Reference Chevalier37). All these events take place simultaneously, thus adaptive immune mechanisms also contribute to the effector response mediated by the innate cells. For example, antibodies released by B cells bind to specific neoantigens on tumour cells, whereas allow their interaction with innate immune cells by Fcɣ receptor leading to phagocyte activation (Ref. Reference Gallo, Gonçalves and Mosser38). Similarly, CD8+ T cells contribute positively to feedback activation of NLRP3 on innate cells by perforin-dependent mechanism (Ref. Reference Yao39).
In addition to the direct action on tumour cells, immune cells have other mechanisms for tumour elimination at work. This role is carried out through the cytokines secretion which may contribute to the activation of anti-tumour function on surrounding immune cells. IFNɣ is an important pro-inflammatory cytokine released by different cell types such as macrophages, NK, iNKT and ɣδT cells, and even by T cells. Several studies have demonstrated that this cytokine contributes to activating NK and CD8+ T cells. Also, IFNɣ plays an important role during macrophage differentiation; leading them to acquire the anti-tumour M1 profile. At the same time, M1 macrophage activation induces CD4+ T-cell polarisation into the Th1 proinflammatory profile and stimulates CD8+ T cell activation towards an antitumor effector-cell profile (Ref. Reference Alspach, Lussier and Schreiber40).
During the execution of the anti-tumour response, the immune system exerts selective pressure on tumour cells. Since most tumours are made of heterogeneous populations of cells, known as tumour subpopulations (Refs Reference Welch41, Reference Lawson42); immune response activation against certain neoantigens may not be enough to successfully eliminate the whole tumour mass. Instead, subpopulations not expressing target-neoantigens may survive. Thus, cells with different phenotypes may escape the immune response, being responsible for tumour progression despite treatments. This is evidenced by disease recurrence after long periods of remission, leading to assume that an undetectable number of cancer cells remained after treatment. The remaining tumour cells could stay in the body in a silent form for a long time via a process described as tumour equilibrium with the potential to form stem cell-like hierarchies (Ref. Reference Espinoza-Portocarrero43). Little is known about the equilibrium phase; a balance between effector immune response and tumour tolerogenic activity is assumed, where the immune system is able to control and retain tumour growth, while tumour cells adopt favourable phenotypes to achieve their survival. During equilibrium, continued cycles of tumour elimination and immune escape take place (Ref. Reference Mittal25). This stage ends when the selective pressure imposed by the immune system finally lets the generation and progression of tumour subpopulations being able to evade anti-tumour response, producing a way to escape. All of these processes (tumour elimination, equilibrium and scape) are part of a more general event called cancer immune-editing. In this regard, immune-editing explains how the immune response contributes to both tumour cell elimination and the selection of competent tumour subpopulations (Ref. Reference Mittal25).
Owing to the important role of T lymphocytes in tumour cell elimination, many treatments aim to recruit these immune cells towards the tumour microenvironment. Thus, the absence of T cells on tumour tissue represents the incapacity of immune cells to arrive at the tumour microenvironment, making it unlikely to fulfill their antitumor role. Therefore, tumour immunogenicity is measured in relation to its ability to recruit large numbers of tumour-infiltrating lymphocytes (TILs). Histological studies report major immunogenic differences between tumours (Ref. Reference van der Woude44). Inflamed tumours, also known as hot tumours, present TILs. Meanwhile, cold tumours are devoid of TILs and are unable to activate or recruit immune cells. Cold tumours unable to both activate and recruit immune cells are also known as ignored tumours. On the other hand, cold tumours with the ability to activate immune cells but fail to recruit them inside the tumour (immune cells located in the periphery) are called excluded tumours (Ref. Reference Binnewies6). These differences in tumour-infiltrating immune cells are caused by evasion mechanisms developed by tumour cells. To survive, cancer cells take advantage of negative regulatory mechanisms of the immune system, which are important checkpoints to prevent autoimmunity and usually help to restore body homeostasis after the immune response deployed against an infection.
Inhibition of negative immune regulators as a cancer therapy strategy
Oncogenesis is a complex process that produces cell homeostasis dysregulation that is caused by the incidence of mutations in the cell genome. Although the accumulation of mutations increases tumour genetic diversity and contributes to generating cancer cells with an improved evolutionary fitness for survival, divergence from a healthy cell increases the likelihood of being recognized for elimination by the immune system. However, tumour cells may mask their genetic divergence by suppression of immune responses via activation of negative regulatory pathways, or mutate to actively escape immune detection. The interplay at the core of this balancing act may explain that the TMB contributes to immune recognition of cancer which may determine responses to cancer immunotherapy (Ref. Reference Liu45).
In 1987, Brunet et al. discovered a new molecule member of the immunoglobulin superfamily called cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), which was found expressed on the surface of activated T lymphocytes (Ref. Reference Brunet46). Some years later, James Allison's group in the USA studied the activity of this molecule, finding that it was related to the inhibition of the immune activity discovering the first immune checkpoint molecule. Then, through the use of anti-CTLA-4-specific antibodies in murine models, Allison and coworkers bore out experimentally that blocking this molecule provides better control of tumour progression (Ref. Reference Leach, Krummel and Allison47). Simultaneously, Tasuku Honjo's group discovered in Japan the programmed cell death protein 1 (PD-1), a macromolecule expressed by T cells upon its activation. Moreover, Honjo described the negative regulatory function exerted by PD-1 molecules on the immune response (Ref. Reference Nishimura48). The works performed by Allison and Honjo were recognized as the basis for a new approach in cancer therapy, awarding them with the Nobel Prize in 2018. Unlike other cancer immunotherapies, therapies based on immune checkpoint inhibitors can act on multiple types of cancer, independently of its heterogeneity (Ref. Reference Xin Yu49).
Currently, CTLA-4 and PD-1 are the most studied immune checkpoint pathways. It has been proposed that CTLA-4 expression is induced on conventional CD4+ T cells after activation and remains constitutively expressed on FoxP3 + Treg cells. According to that, CTLA-4 may control T-cell activation through both (a) the competition with co-stimulatory macromolecule CD28 to binds with its ligands B7.1 (CD80) and B7.2 (CD86) found in antigen-presenting cells or (b) by blocking CD28 expression on the outer membrane (Refs Reference de Vos50, Reference Ovcinnikovs51). Likewise, it was demonstrated that PD-1 regulates immune cells binding to its ligand PD-L1 (B7-H1/CD274) and PD-L2 (B7-DC/CD273) which are differentially expressed on specific immune cell subsets. After the interaction, PD-1 elicits an inhibitory signal to attenuate T-cell activity, preventing cell activation, IL-2 production besides cell proliferation (Ref. Reference Kim and Chen52). These immune checkpoints are part of the peripheral tolerance mechanisms developed by the immune system. Regularly, they prevent the development of autoimmune responses by inactivation of autoreactive lymphocytes, as well as allow to maintain body homeostasis during immune response execution by preventing an exacerbated proliferation of immune cells that may trigger death (Ref. Reference Wei, Duffy and Allison53).
In an attempt to evade the immune response, tumour cells may express ligands for CTLA-4 and PD-1 receptors. Clinical trials using monoclonal antibodies to hinder the interaction of CTLA-4 and PD-1 to its ligand have shown strong evidence of a long-term anticancer response in cancer patients, with minimal toxic effects (Refs Reference Kim and Chen52–Reference Prieto54). For this reason, some immune checkpoint inhibitor therapies have been approved for clinical use by the Food and Drug Administration (FDA). Ipilimumab, CTLA-4-specific monoclonal antibody therapy, was approved by the FDA for the first time in 2011, being indicated for patients with late-stage melanoma. Later, its use was extended for the treatment of unresectable or metastatic melanoma, intermediate- and poor-risk advanced renal cell carcinoma, and metastatic colorectal cancer. Unlike CTLA-4 therapy, Nivolumab, a PD-1-specific monoclonal antibody, was approved for the first time in 2014. Currently, nivolumab is indicated for the treatment of advanced melanoma, lung cancer, advanced lung cancer, metastatic renal cell carcinoma, Hodgkin lymphoma, head and neck cancer, advanced or metastatic urothelial carcinoma, metastatic colorectal cancer, hepatocellular carcinoma, completely resected or metastatic melanoma, intermediate- and poor-risk advanced renal cell carcinoma. Furthermore, since 2015 Ipilimumab and Nivolumab are used in combination as a complementary treatment in several types of cancer leading to a better clinical response than expected from single-agent therapy (Ref. Reference Gao and McDermott55).
Immunological strategies applied in cancer immunotherapy
Several immunotherapeutic strategies were proposed over the last 30 years against cancer, as previously shown in Figure 1. However, during the translation of preclinical tests to clinical trials, the protocols do not always show the promising results observed during their early experimental stages. Therefore, over the years, the FDA has approved those immunotherapeutic methodologies that managed to replicate their beneficial results in cancer treatment, with the least toxic effect. Those approved immunotherapeutic methods are described in Table 1. It is important to note that all of these immunotherapeutic strategies have been approved to be applied as a complementary therapy to conventional treatments (chemotherapy and radiotherapy).
Table 1. Immunotherapeutic drugs approved by FDA (1997–2019)

Record of immunotherapeutic drugs approved by the FDA (www.fda.gov) for the treatment of several cancer types and their mechanism of action (data updated until 31/Dec/2019).
The first immunotherapeutic treatment against cancer, Rituximab, was approved in 1997 (Ref. Reference James and Dubs56). Rituximab is a monoclonal antibody medicine that recognizes the CD20 receptor expressed exclusively in the extracellular membrane of B lymphocytes and malignant B cells (Ref. Reference Casak57). In this regard, Rituximab was initially indicated for patients with B-cell non-Hodgkin's lymphomas to recognize and eliminate of malignant B cells. Currently, Rituximab is approved to treat also chronic lymphocytic leukaemia and follicular lymphoma. It has been shown to have a great effect on patients, reducing the number of circulating tumour cells (Ref. Reference Salles58).
The upgrade of this therapeutic tool was introduced through protein engineering in 2002 when the FDA approved Ibritumomab tiuxetan. Its improvement lies in the coupling of yttrium-90 radionuclide that allows tumour-specific radiation therapy (radio-immunotherapy) with fewer side effects for patients. Eight years later, the FDA approved the commercialization of Ofatumumab, a human monoclonal antibody directed against the CD20 antigen. The absence of murine regions in its composition increased drug tolerance, avoiding the treatment rejection observed in some patients undergoing the aforementioned drugs. A few years later, another anti-CD20 medication was approved by the FDA, called Obinutuzumab. This new drug is a type 2 antibody and links to CD20 in a different way than performed by type 1 antibodies (e.g. rituximab, ofatumumab), increasing the drug's performance. Thus, Obinutuzumab is capable of inducing direct cell death, unlike type 1 antibodies whose cytotoxic effect depends on the complement system. Type 1 antibodies may induce its internalization and subsequent drug destruction by the lysosomal pathway, whereas type 2 antibodies prevent internalization and avoid drug loss. Because of these features, Obinutuzumab has strong B-cell depleting activity which represents improvements in therapeutic effect (Refs Reference Beers59, Reference Elias60).
The biological treatments mentioned above use the CD20 receptor as a tumour-associated antigen present on malignant B lymphocytes where the uncontrolled proliferation of cells leads to myeloid lymphoma or leukaemia. Antibodies linked to tumour cell surfaces allow both the recognition of tumour cells by the antigen-presenting cells for phagocytosis and the ADCC for tumour cell elimination. Similarly, other tumour antigens associated with diverse types of cancer have been discovered over the years. These neoantigens are usually found to be overexpressed in tumour cells, whereas in normal tissues they are absent or scarcely expressed. Some immunotherapies based on monoclonal antibodies against these neoantigens are listed in Table 1. For example, GD2 ganglioside is a neoantigen overexpressed on neuroblastoma. Unlike other gangliosides also expressed in neuroblastoma, the presence of GD2 in healthy tissue is almost null making it the best candidate for the immunotherapeutic treatment of this disease. Dinutuximab is a monoclonal antibody drug designed to target the GD2 receptor to induce tumour cell death by ADCC but also by the activation of intrinsic apoptotic pathway (Refs Reference Voeller and Sondel61–Reference Erbe63).
Another example of immunotherapies recognizing tumour antigens is provided by the CD38 and SLAMF7 molecules. These are membrane receptors prominently overexpressed in multiple myeloma cells, whose activation is associated with tumour progression. Expressed in normal tissues, CD38 is related to lymphocyte proliferation, whereas SLAMF7 is responsible for NK cell activation. Daratumumab and Elotuzumab are monoclonal antibodies that target CD38 and SLAMF7 neoantigens on myeloma cells, respectively. In addition to target tumour cells and inducing cytotoxicity by ADCC, elotuzumab is able to bind to the SLAMF7 receptor on NK cells, activating them to secret IFNɣ, regulating immune response against the tumour. The implementation of these drugs in multiple myeloma treatment allowed a considerable improvement in the progression-free survival of patients (Refs Reference Janmaat, Fischer, Klein and Childers64, Reference Malaer and Mathew65).
On the other hand, epidermal growth factor receptor 2 (Her2) is a proto-oncogene related to cellular proliferation and survival. In breast and gastric cancer, several mutations on the Her2 sequence induce the overexpression of this cellular receptor, favouring tumorigenic processes (Ref. Reference Pahuja66). Trastuzumab and pertuzumab are anti-Her2 monoclonal antibodies approved by the FDA with a synergistic treatment effect. Trastuzumab targets Her2 subdomain IV, meanwhile pertuzumab was designed to bind Her2 subdomain II. These drug treatments induce ADCC and prevent the formation of Her2-protein complexes (homodimerization and heterodimerization) necessary for Her2 intervention on cell cycle. Hence, using both drugs together inhibits Her2 canonical and non-canonical activation pathways, improving results for patients (Ref. Reference Hao67). Immediately, a variation of trastuzumab was approved for commercialization by the FDA. Trastuzumab emtansine is an antibody conjugated to the chemotherapeutic DM1 molecule, a microtubule polymerization inhibitor. This modification of trastuzumab reduces the side effects caused by conventional DM1 chemotherapy to healthy tissues, transporting the DM1 cytotoxic compound to interact directly and specifically with the tumour cells overexpressing Her2 receptor (Ref. Reference Perez68).
Other drugs based on conjugated antibodies are Gemtuzumab ozogamicin and Brentuximab vedotin, which are covalently linked to a cytotoxic chemotherapeutic agent. Gemtuzumab ozogamicin is a monoclonal antibody targeting the CD33 receptor highly expressed in myeloid cell lines, and conjugated with a calicheamicin synthetic molecule. After CD33 recognition, the antibody is interiorized and taken to the lysosomes where the acid medium allows calicheamicin activation. Then, calicheamicin leaves the lysosome and induces DNA breaking and tumour death (Ref. Reference Godwin, Gale and Walter69). Meanwhile, Brentuximab vedotin is a monoclonal antibody directed against CD30 receptor, overexpressed on systemic anaplastic large-cell lymphoma and Hodgkin lymphoma. This antibody is linked with an anti-tubulin agent named monomethyl auristatin E. In patients, Brentuximab is internalized by tumour cells where, similarly, it is activated after its arrival to the lysosome. Once released from the digestive vesicle, the molecule reaches the microtubules, from where it inhibits its polymerization avoiding cell division and tumour progression (Ref. Reference Schönberger70). Both conjugated antibodies allow a precise and stable chemotherapeutic delivery system since they travel through the body as an inactive molecule and acquire an activated state right after antigen-specific recognition.
Although the recognition of tumour cells is an important process in immunotherapeutic approaches, in several cases, this methodology is not enough to mitigate tumour progression. Owing to immune evasion mechanisms developed into the tumour microenvironment, therapies focused on the modulation of the immune checkpoints are being used as promising alternatives. In this way, some therapies such as Pembrolizumab, Nivolumab and Cemiplimab target cell death receptor PD-1 located on the surface of TILs. This antibody recognition avoids the interaction of PD-1 with its ligand on tumour cells (PD-L1), preventing lymphocyte inhibition (Ref. Reference Gao and McDermott55). In the same regulation pathway, Atezolizumab, Avelumab and Durvalumab are monoclonal antibody drugs that recognize and bind to PD-L1 molecules, avoiding contact and subsequent activation of the death receptor on immune cells (Ref. Reference Kim and Chen52). Another immune regulatory molecule is CTLA-4, expressed on activated T lymphocytes. The interaction of this molecule with its ligand on tumour cells inhibits the antitumor immune response. Ipilimumab and Tremelimumab are monoclonal antibodies that target CTLA-4 preventing contact with its ligand. This treatment contributes by regulating positively the immune response against cancer (Ref. Reference Arce Vargas71).
Further development on the use of monoclonal antibodies for immunotherapy is the production of antibodies with double specificity since they are able to bind to two different therapeutic targets. Blinatumomab was the first bispecific antibody approved by the FDA. Its double specificity has been addressed to interact with B and T lymphocytes, simultaneously. Therefore, Blinatumomab is able to bind and stimulate T cells through its CD3 receptor and, at the same time, it binds to the CD19 receptor expressed in B-cell lymphomas. This double specificity induces a spatial approach between T and B cells and allows a direct effector function of T lymphocytes against CD19-expressing B cells. Owing to its proven efficacy in clinical trials, Blinatumomab was approved for the treatment of relapsed or refractory precursor B-cell acute lymphoblastic leukaemia (Ref. Reference Przepiorka72).
Beyond the innovation already happening in monoclonal antibody technology, new strategies are in the pipeline for cancer immunotherapies with vastly promising results. Genetic engineering techniques applied to adoptive T-cell transfer strategy resulted in the production of T lymphocytes with a chimeric antigen receptor (CAR), also known as CAR-T cells. Through this technology, the T-cell receptor (TCR) was modified by adding the B-cell receptor variable regions to replace the conventional recognition site. This CAR allows the recognition of three-dimensional protein structures by T cells, whereas TCR recognizes just linear peptide sequences carried on the MHC molecule. Thereby, CAR incorporation allows direct recognition and binding of T cells to tumour surface antigens. Thus, CAR-T cells managed to appropriately associate both the wide recognition range of B lymphocytes receptor and the cytotoxic functions of T lymphocytes (Ref. Reference Ying73). In 2017, two treatments based on specific CAR-T cells against CD19 were approved by the FDA, Tisagenlecleucel and Axicabtagene ciloleucel. It has been observed that patients diagnosed with diffuse large B-cell lymphoma treated with Axicabtagene ciloleucel show a high remission rate (complete remission = CR) reaching 58% of patients (Refs Reference Schuster74, Reference Abramson75). Meanwhile, those patients treated with anti-CD20 monoclonal antibodies reach only 7% of CR (Ref. Reference Crump76). Despite the great results obtained with immunotherapeutic strategies based on monoclonal antibodies, clinical trials with CAR-T cells have shown higher promising results for cancer patients.
Limitations of current biomarkers for immunotherapy
For the assignment of a correct immunotherapeutic approach, the expression of certain biomarkers is previously evaluated in patients. However, the biomarkers currently used fail to have great precision as a prognostic factor of treatment. Despite the wide use of PD-L1 and TMB, up-to-date, there is no perfect biomarker for immunotherapy. Although PD-L1 expression by immunohistochemistry is the most widely accepted test to select patients who will receive immune checkpoint inhibitors, issues that still need to be addressed are antibody clone-dependent thresholds and inter-laboratory variability. FDA-approval of different immune checkpoint inhibitors under PD-L1 evaluation with a companion diagnostic assay and the interchangeability between antibodies from different clones is still controversial.
In addition, sensitivity to immunotherapy is driven by the intrinsic characteristics of the tumour cell and the tumour microenvironment. The TMB is known to be directly related to the efficacy of immune checkpoint inhibitors, where a higher TMB indicates a higher presence of neoantigen and therefore an improved immune response. However, this assumption may not always be correct for reasons yet to be fully understood (Ref. Reference Bardhan, Anagnostou and Boussiotis77).
Future immunotherapies
Despite the great progress that has been made in recent years regarding cancer treatment, immunotherapy protocols still show a wide spectrum response on patients treated. This fact suggests that a better tumour taxonomic system is needed since a specific diagnosis is linked to suitable treatment decisions and improvement in patient prognosis. Therefore, the discovery of predictive biomarkers must progress hand-over-hand with therapeutic approaches. Thereby, these tools will allow proposing the most appropriate therapy according to each cancer-specific profile.
In the last decade, tumour-infiltrating immune cells were associated with cancer prognosis, even relating them to early diagnosis (Ref. Reference Zhou7). Hence, the presence/absence of infiltrating cells is currently being proposed as a biomarker for cancer treatment. Some of the immune cell lines reported in tumour infiltrate are DCs (Ref. Reference Truxova78), macrophages (Ref. Reference Cassetta79), B cells (Ref. Reference Liudahl and Coussens80) and mainly T lymphocytes (Refs Reference Uryvaev81, Reference D'Arena82). Despite the large number of studies carried out on this topic, no pattern that allows establishing a relationship between tumour infiltrate characteristics and patient prognosis has been found yet. Similarly, some studies focused on the characterization of the T lymphocyte subpopulations (i.e. CD8+, Th1, Th2, Th17, Treg), in an attempt to relate the profile of the immune response with the evolution of the patients. However, a strong association between the specific T-cell population present in the tumour microenvironment and the clinical outcome of cancer patients has not yet been established. In consequence, in-depth studies that consider the interaction among immune cell populations, its location in the tumour microenvironment, the type of tissue where cancer appears, as well as the places where they metastasized are needed (Ref. Reference Fridman83). Accordingly, it is expected to obtain a vast database of larger heterogeneity, whose diversity will not be restricted to cancer types but from one patient to another. For this reason, in recent years, the idea of a predictive method based on the characteristics of infiltrating immune cells has been developed and supported by powerful bioinformatics tools that allow the analysis of all the factors. This method is known as immune-score and is expected to help further personalize cancer treatment (Ref. Reference Hao84).
Furthermore, beyond the description of tumour-infiltrating immune populations, the characterization of gene expression profiles could help to unveil new factors responsible for immune evasion mechanisms in the tumour microenvironment. For instance, sequencing of immune cells is helping to simultaneously study several metabolic pathways, while allowing to identify both alterations in gene expression profile and mutations on immune checkpoint sequences (Refs Reference Liu45, Reference Guo85). This information may be used to propose relevant and personalized therapeutic targets. Currently, specific therapies have been proposed to neutralize tumour immune suppression, for the purpose of helping to activate anti-tumour immune response. Some of these immunomodulatory molecules are currently under study in clinical trials, those involved in breast, lung, colorectal and prostate cancer treatment are summarised in Figure 2 and Table S1.
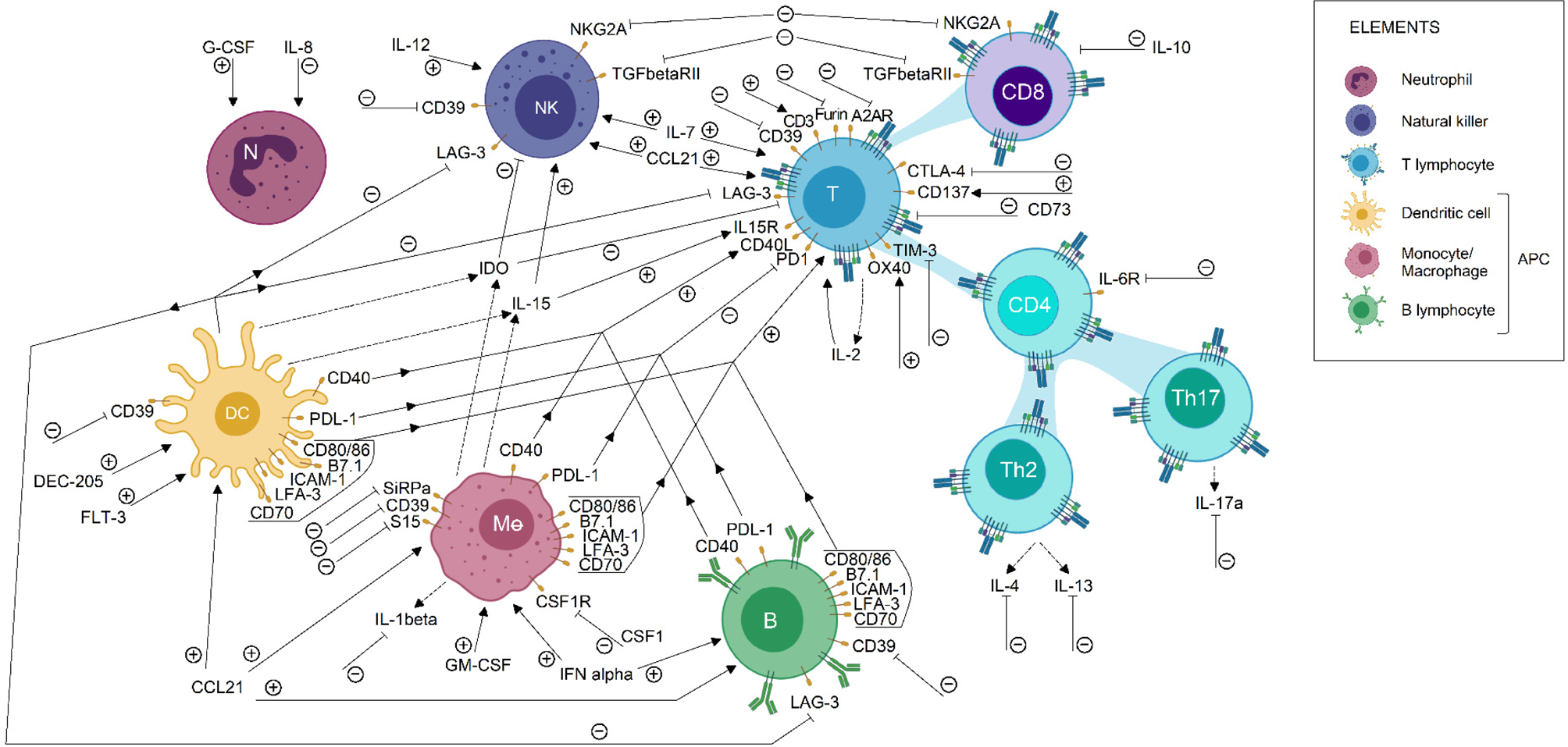
Fig. 2. Immunomodulatory targets for cancer treatment. Some immunomodulatory agents currently in clinical trial status for breast, lung, colorectal and prostate cancer were shown in this picture. Its effect on the immune cell population is described briefly. Each arrow symbolizes a stimulating/potentiating effect on immune cell effector functions, whereas the truncated lines represent an inhibitory effect on effector response related to the molecule. Immunotherapeutic protocols are designed to induce or blockade the activation of the pathways related to the molecules shown. Therefore, the action of immunotherapeutic approaches on each molecule is represented by (+) or (−), according to its activating or inactivating role, respectively. Also, dotted lines indicate a secreted compound. These data were extracted from the ClinicalTrials.gov database, according to the following parameters: (1) study type: interventional studies, (2) recruitment status: not stopped studies (i.e. not yet recruiting, recruiting, enrolling by invitation, active not recruiting and completed), (3) study results: all studies, (4) study start: from 01/Jan/2010 to 04/Dec/2019. APC, antigen-presenting cells; DC, dendritic cell; NK, natural killer.
The previous approach supports the crucial role of tumour-infiltrating immune cells during immunotherapy protocols (Ref. Reference Trujillo86). In addition, a poor anti-tumour response after the immunotherapeutic intervention has been reported on patients with ignored tumours that fail to recruit immune cells (Ref. Reference Ariyan87). Therefore, the efforts to transform a non-inflammatory tumour into an inflammatory one led to a new approach to conventional cancer treatment tools such as chemotherapy and radiotherapy, by deep analysis of their role as immune-stimulating agents for the antitumor response, beyond its well-described cytotoxic effect on cells with high division rate. For instance, over the last years, we have learned that there is indeed a relationship between radiotherapy and the immune system (Ref. Reference Jiang88). The use of radiotherapy has synergistic effects with immune checkpoint therapy on both local and systemic control of the tumour. Locally, the exposure to radiation increases the tumour mutational load and enhances MHC-I expression on tumour cells leading to neoantigens presentation. Because of radiation, the death receptor Fas increases its expression on the tumour membrane, allowing the activation of a cell death pathway after binding to its ligand. In addition, damage signals produced during radiation induce the activation of DC (professional antigen-presenting cells) leading to increased TILs (Ref. Reference Sharabi89). In addition, failures in DNA mismatch repair enzymes increase the number of neoantigens triggering immune system responses (Ref. Reference Germano90). A similar mechanism has been described for non-small cell lung cancer, where radiotherapy potentiates the beneficial effects provided by immune checkpoint therapeutic protocols, showing significant improvement in overall survival (Ref. Reference Shaverdian91). On the other hand, systemic control is conducted by the abscopal effect induced by radiation. After radio- and immune-therapy combined protocols on a primary tumour, distant anti-tumour effects were described which cause metastatic tumours regression. In the clinic, this abscopal effect is not observed in patients treated only with immunotherapeutic protocols (Ref. Reference Chandra92). In the same way, chemotherapy has demonstrated to improve response to immunotherapy in patients with solid tumours by increasing activated lymphocytes in the tumour microenvironment (Refs Reference Ariyan87, Reference Heinhuis93).
Another approach studied for cancer treatment is patient sensitization against tumour antigens. Until now, just a few neoantigen vaccines were approved by the FDA (Table 1). However, several vaccine candidates are still being analyzed in clinical trials. Some of these candidates are exemplified in Figure 3 and described in more detail in Table S2. Most tumour-associated antigens are proposed for the treatment of more than one type of cancer. Highlight cases of brachyury, CEA, MUC-1 and NY-ESO-1 antigens, which were proposed as therapeutic methods for the four types of cancer, are analyzed in this work. In normal conditions, brachyury, CEA and NY-ESO-1 are expressed in the early stages of human development (e.g. germline cells or embryonic tissues) but high TMB produces sequence genetic alterations, allowing the re-expression of these genes in adulthood. Despite the possibility to be used in the treatment of different cancer types, a variable response has been observed in patients treated with those neoantigen vaccines. For example, NY-ESO-1 expression was identified in more than 10 different tumour types. Nevertheless, a non-homogeneous expression of the antigen was reported in tumours, whose presence may vary between 20 and 100% of tumour cells depending on the cancer type (Ref. Reference Thomas94). Therefore, notwithstanding the improved effectiveness observed in comparison to conventional therapies, neoantigen vaccines have certain restrictions. One of the most worrisome effects is the selection of tumour clones that do not express the vaccine antigen. Thus, these tumour cells could acquire a selective advantage that allows them to continue proliferating despite treatment. For this reason, new therapeutic approaches seek to customise these treatments, sensitizing patients against multiple tumour-specific neoantigens in an attempt to cope with the heterogeneity of tumour cell subpopulations (Refs Reference Hilf2, Reference Karapetyan3). Hence, several clinical trials using personalized anti-cancer vaccines are currently proposed for FDA approval.
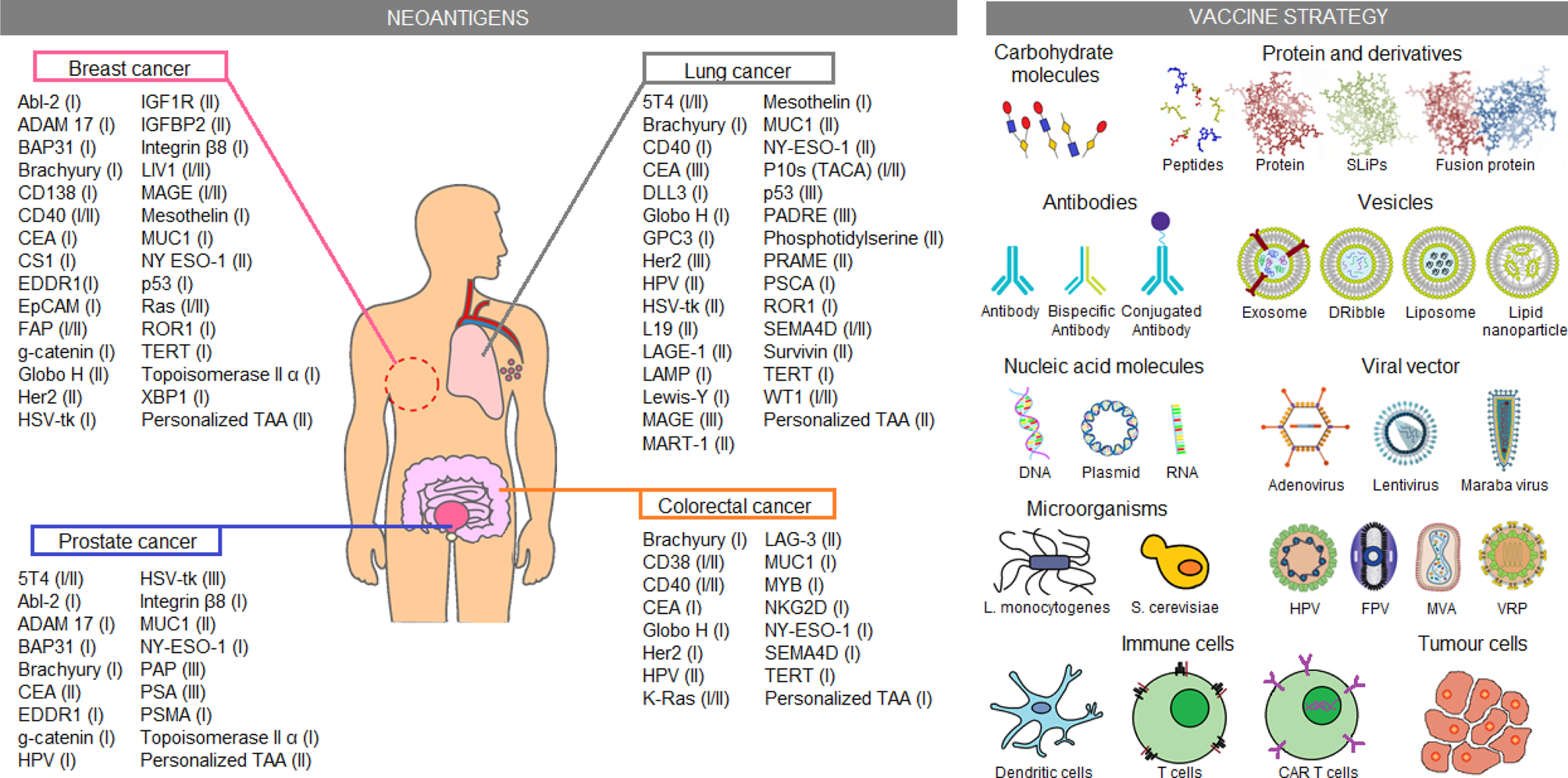
Fig. 3. Immunotherapeutic clinical trials based on neoantigens. Current immunotherapeutic strategies based on tumour neoantigens were listed. In the picture, neoantigens proposed for clinical trials on breast, lung, colorectal and prostate cancer were summarised, the most advanced clinical trial phase (I–IV) is shown in parentheses. Similarly, several neoantigen vaccination strategies applied in the clinical trials are shown. These data were extracted from the ClinicalTrials.gov database, according to the following parameters: (1) study type: interventional studies, (2) recruitment status: not stopped studies (i.e. not yet recruiting, recruiting, enrolling by invitation, active not recruiting and completed), (3) study results: all studies, (4) study start: from 01/Jan/2010 to 04/Dec/2019. CAR T cell, chimeric antigen receptor T cells; DNA, deoxyribonucleic acid; Dribble, tumour-derived autophagosome vaccines; FPV, fowlpox virus; HPV, human papillomavirus; L. monocytogenes, Listeria monocytogenes; MVA, modified vaccinia Ankara; RNA, ribonucleic acid; S. cerevisiae, Saccharomyces cerevisiae; SLiPs, long-lived proteins; VRP, virus replicon particle.
In the search for personalized and more effective treatments, along with the great advances in genetic engineering, scientists have created ingenious therapeutic strategies. One of them is CAR-T cells. As described above, CAR molecule allows the recognition of more heterogeneous epitopes, as well as the direct binding to antigens expressed on tumour cells, independently of MHC molecules (Ref. Reference Ying73). These characteristics enabled a good therapeutic performance, reflected on the authorizations provided by the FDA for some CAR-T cell-based therapies (Refs Reference Schuster74–Reference Crump76). However, there are other types of cell-based therapies that are in cancer treatment clinical trials. An older strategy, but still in the early stages of clinical trial for cancer treatment is TCR engineered T cells (TCR-T). TCR-T cells are T-cell modified by viral vectors to express transgenic αβTCR chain sequences, with predefined antigen specificity (Refs Reference Heemskerk95, Reference Dossa96). This technology allows to control the affinity of TCR for a specific antigen. Unlike CAR-T cells, TCRs require antigen presentation by MHC molecules, and therefore are capable of recognizing intracellular antigens (Ref. Reference Xu97). At the same time, this characteristic limits its action in the tumour microenvironment with suppressed MHC expression. Also, TCR-T antigen recognition depends on the diversity of MHC molecules in patients, therefore it requires to be adapted for each person (Ref. Reference Zhao and Cao98). A less complex cell therapy protocol that does not involve genetic modifications is the adoptive transfer of TIL. For this procedure, TIL are isolated from the tumour microenvironment and cultured in the presence of irradiated tumour cells, together with costimulatory factors and proliferative cytokines which favour their activation and maturation (Ref. Reference van den Berg99). However, many doubts fall on the elimination processes of these adoptively transferred cells after treatment, as well as about the implications and risks for long-term health. On one hand, the high affinity for antigens in CAR-T cells has been associated with cytokine storm syndrome and consequent patient death (Ref. Reference Chen100). On the other hand, TCR-T cells have shown alterations because of incorrect positioning of inserted sequences, resulting in unwanted affinities (Refs Reference Provasi101–Reference van Loenen103).
Considering the various mechanisms applied by tumour cells to progress, it may be unrealistic to assume that just one kind of therapy will be enough to prevent tumour growth. For this reason, the integration of several therapeutic approaches seems to be the most reasonable solution. The great progress achieved in the last decade on immunotherapeutic tools (i.e. immune-modulatory therapies, immune checkpoint inhibitors, and individualized neoantigen vaccines), together with improvements in chemotherapy and radiotherapy specificity, represents important advancements in the search for the best treatment against cancer. It is imperative that these therapeutic advances are accompanied by integrated systems of tumour classification that include biomolecular markers, the location of surrounding immune populations and tumour taxonomy profiling; so that a specific diagnosis may lead to the best therapeutic decision for each patient.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/erm.2021.5.
Financial support
This research received no specific grant from any funding agency, commercial or not-for-profit sectors. Publication charges were defrayed by institutional internal funds.
Conflict of interest
None.
Ethical standards
The authors assert that this work does not involve human or animal experimental procedures.
Further reading, resources and contacts
• NCI Drug Dictionary
-
The NCI Dictionary of Cancer Terms stores the definition of the terms related to cancer and medicine allowing free access.
-
URL: https://www.cancer.gov/publications/dictionaries/cancer-drug
-
• NCI Dictionary of Cancer Terms
-
The NCI Drug Dictionary contains technical definitions for drugs or agents used to treat patients with cancer or conditions related to cancer, including synonyms, brand names and abbreviations related to the compounds.
-
URL: https://www.cancer.gov/publications/dictionaries/cancer-terms
-
• Clinical Trials
-
ClinicalTrials.gov is a database of privately and publicly funded clinical studies conducted around the world.
-







 Open access
Open access