In Europe, a new medicine can be marketed, prescribed and utilized by European citizens only after a careful scientific evaluation of its quality, safety and efficacy. This scientific evaluation is carried out by an agency of the European Union called European Medicine Agency (EMA) (http://www.ema.europa.eu). On the basis of the EMA's assessments, only medicines that have a positive risk–benefit profile are granted a marketing authorization and may reach the marketplace. Medicines approved by the EMA are automatically marketable in all the European Member States. EMA's decisions on new or old medicines relating to changes in therapeutic indications, approval, suspension or withdrawal of a product have to be accepted by all Member States. So far, the Agency has approved eight psychotropic drugs for 15 psychiatric indications: agomelatine, aripiprazole, duloxetine, melatonin, olanzapine, paliperidone, pregabalin and zaleplon (Barbui et al. Reference Barbui, Baschirotto and Cipriani2011).
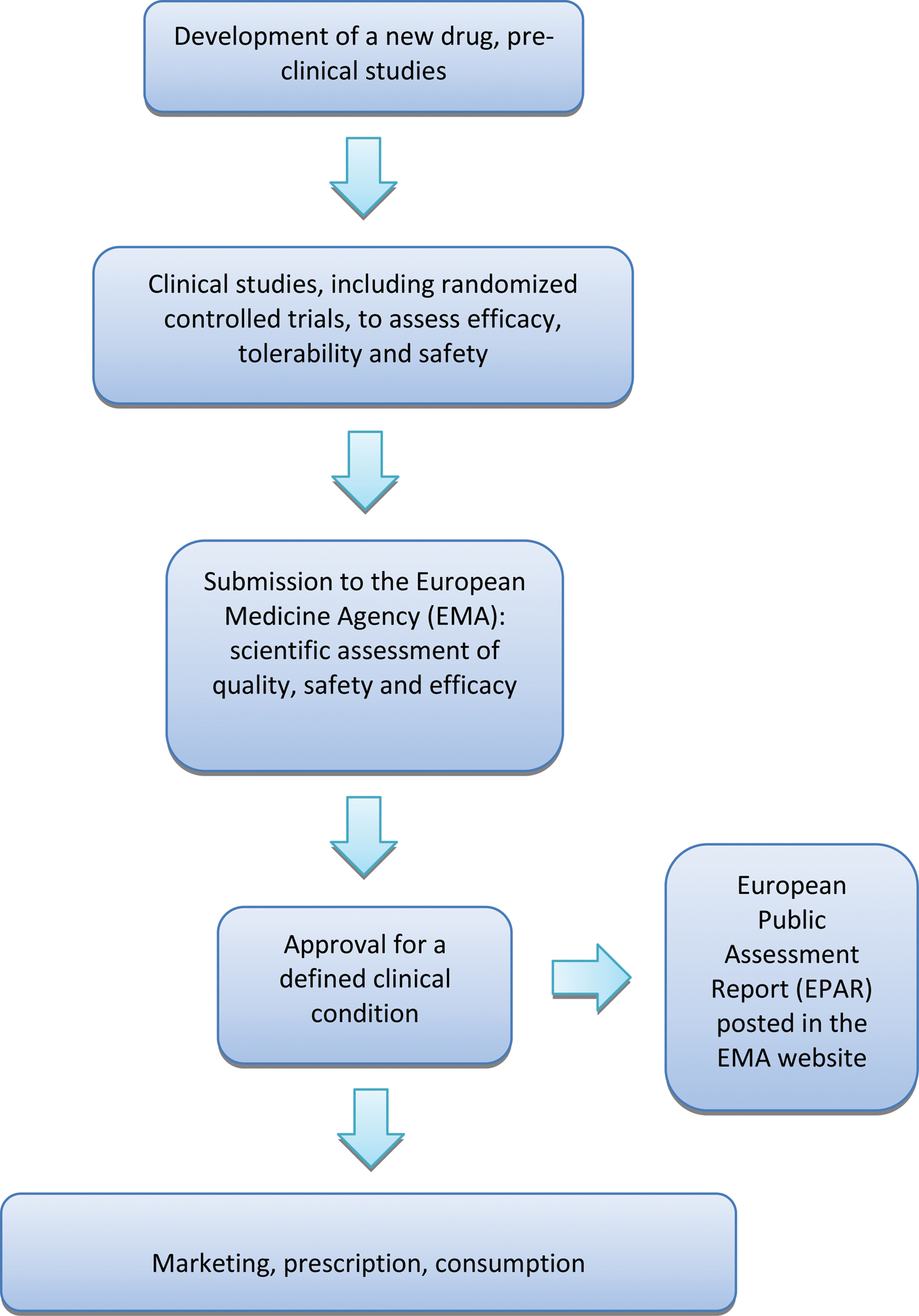
A crucial aspect of the whole process is that the EMA approves or rejects a new medicine on the basis of the results of randomized controlled trials (RCTs), as RCTs are considered the reference research design to assess the beneficial and harmful effects of therapeutic interventions (Cipriani & Geddes, Reference Cipriani and Geddes2009). Once the EMA has given marketing authorization for a medicine, it publishes a European Public Assessment Report (EPAR), a document that summarizes the preclinical and clinical data produced by the manufacturer, including a detailed description of the results of RCTs that led to the decision of granting a marketing authorization. (Fig. 1).
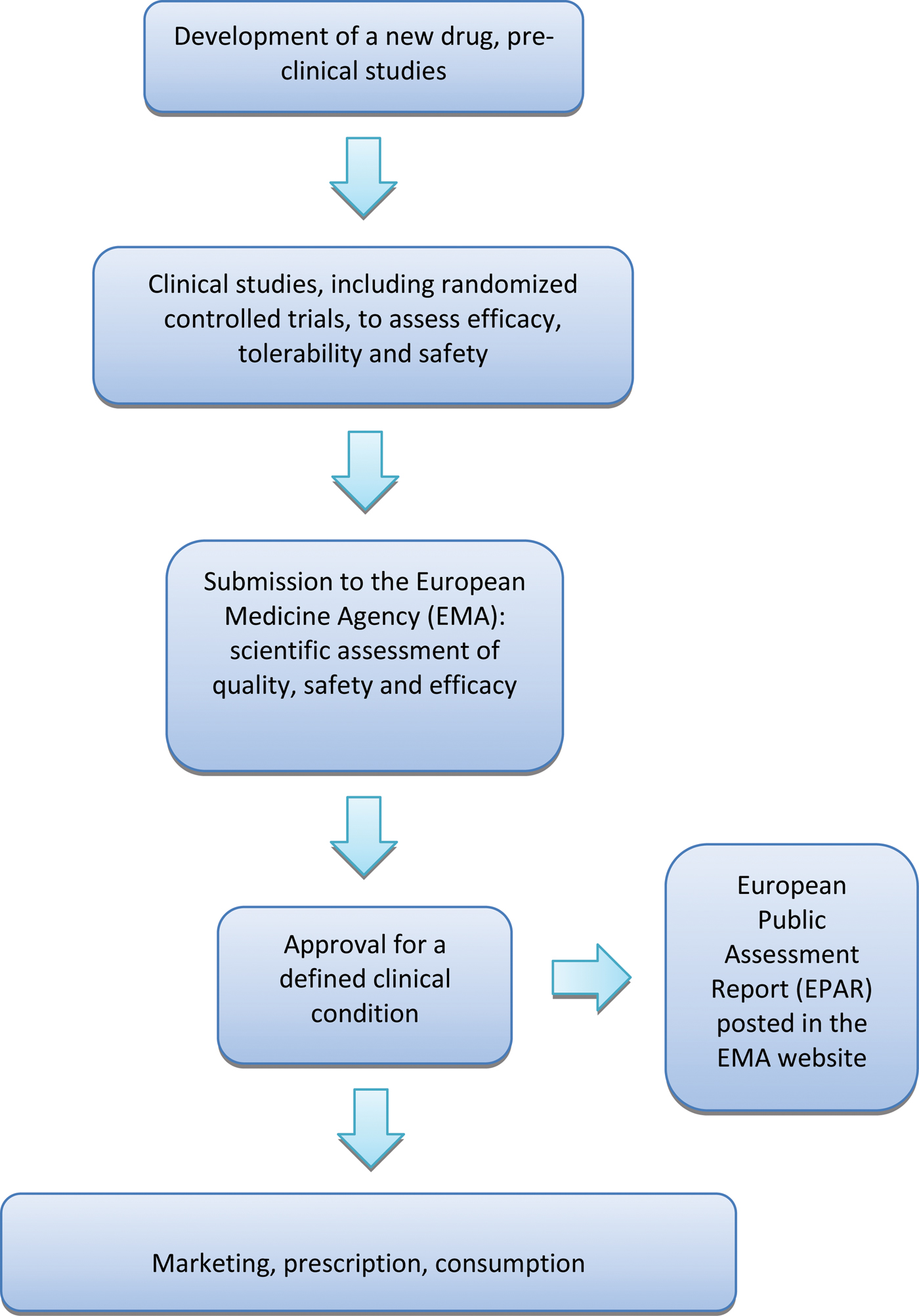
Fig. 1. European process of approval of a new drug. Details are discussed in the text.
Given the high value of RCTs in the overall assessment process, the Agency provides scientific guidance on methodological issues related to the conduct of RCTs in general, and specifically intended to guide the design of efficacy studies in different disorders and patient populations. The main rules governing the registration of new compounds can be summarized as follows. According to the EMA RCTs may be designed to show superiority or equivalence or non-inferiority. Sample size calculation and hypothesis testing may differ according to this trial classification. In equivalence and non-inferiority trials, a conclusion of equivalence or non-inferiority depends on the maximum difference between competitive treatments acceptable as clinically irrelevant. The EMA states that most active comparator trials are not designed to demonstrate superiority or equivalence, but to show non-inferiority of the new drug in comparison with the standard one; these studies often include a placebo arm for comparison to establish efficacy in absolute terms. This implies that, for example in clinical trials for patients suffering from schizophrenia, the EMA states that although the use of a placebo arm for comparison raises ethical problems, placebo-controlled trials are required to show the efficacy of new antipsychotic compounds (EMA Committee for Medicinal Products for Human Use, 2011). In addition, comparison with a standard product is generally needed, with the aim of showing a similar balance between the new drug and the active comparator. Similarly, in major depression, since in around one-third of RCTs no difference is found between the effect of placebo and that of the active comparator, placebo-controlled trials are necessary and a three-arm approach is the recommended design (EMA Committee for Medicinal Products for Human Use, 2010).
Over the years, the European medicine approval process has been criticized along several lines of arguments. A first criticism refers to the use of placebo in disorders where active treatments are available, as for example in schizophrenia and depression (Barbui & Garattini, Reference Barbui and Garattini2007; Sorenson et al. Reference Sorenson, Naci, Cylus and Mossialos2011). RCTs may be designed to compare new drugs with placebo to make them eligible for registration. As a consequence, new drugs may be evaluated and approved with no comparison with active alternative treatments. If comparisons are made with active-controls, studies may rely on demonstrating therapeutic non-inferiority, as this is in agreement with current EMA requirements. This means that new drugs can be proved effective and safe on their own, even though they might in fact be potentially less effective or less safe than other drugs currently in use.
A second compelling issue is that the EMA takes regulatory decisions on the basis of the results of individual studies with no role for a systematic review of all the evidence available at the time of submission for approval. In addition, there is no role for aggregating efficacy data using meta-analytic techniques (Cipriani et al. Reference Cipriani, Furukawa and Barbui2011). Clearly, this leaves the possibility to submit only a selection of clinical trials, and lack of statistical re-analyses does not allow producing overall estimates of treatment effect, which would be extremely useful to make evidence-based judgments on the value of new drugs.
Another major issue refers to the quality of the information that is currently available in the EPARs, especially the quality of reporting of RCTs. A recent analysis of the EPARs for psychiatric disorders highlighted that RCTs are described in a way that is of little value (Barbui et al. Reference Barbui, Baschirotto and Cipriani2011). It is not possible to ascertain the degree of difference between active compounds and placebo, and consequently, it is difficult to make an informed judgment on the evidence that makes a new drug eligible for approval.
It has been argued that these limitations can be overcome. The following suggestions have been made: (i) the concept of added value should be introduced into the legislation to induce investigators to design and conduct clinical trials aimed at demonstrating superiority between active compounds, with large-scale two-arm trials, without placebo, in which a new drug must be compared with a reference one (Garattini, Reference Garattini2005; Barbui & Garattini, Reference Barbui and Garattini2007; Garattini & Bertelè, Reference Garattini and Bertele'2007; Sorenson et al. Reference Sorenson, Naci, Cylus and Mossialos2011); (ii) the EMA should base its decisions on a careful analysis of all the randomized evidence available at the time of submission, and meta-analytical techniques should be introduced to assist the assessment process; (iii) the EMA should require at least some trials to be conducted by independent scientific organizations, as the monopoly that drug companies have in evaluating their own products can lead to biased evidence (Garattini and Chalmers, Reference Garattini and Chalmers2009); (iv) the reporting of RCTs in the EPARs should be improved (Barbui et al. Reference Barbui, Baschirotto and Cipriani2011), and a more transparent information policy should be implemented (Lancet, 2010; Gotzsche & Jorgensen, Reference Gotzsche and Jorgensen2011), with an expected beneficial effect for the whole medicine approval process.