



Background
Reports have found that newly authorised medicines often have questionable or no added patient-relevant benefits compared to older treatments (Motola et al., Reference Motola, Ponti, Poluzzi, Martini, Rossi, Silvana, Vaccheri and Montanaro2006; van Luijn et al., Reference Van Luijn, Gribnau and Leufkens2010; Prescrire, 2019; Wieseler et al., Reference Wieseler, McGauran and Kaiser2019; Erhel et al., Reference Erhel, Scanff and Naudet2020). In specialities like oncology, this seems to be particularly prevalent (Kim and Prasad, Reference Kim and Prasad2015; Davis et al., Reference Davis, Naci, Gurpinar, Poplavska, Pinto and Aggarwal2017; Gyawali et al., Reference Gyawali, Hey and Kesselheim2019; Naci et al., Reference Naci, Davis, Savovic, Higgins, Sterne, Gyawali, Romo-Sandoval, Handley and Booth2019). In a cohort of drugs approved by the German drug regulatory agency between 2011 and 2016, the Institute for Quality and Efficiency in Health Care (IQWiG) judged that 10% had substantial benefits compared to already available treatments, while 58% had no proof of added benefits (Wieseler et al., Reference Wieseler, McGauran and Kaiser2019). Out of 18 newly approved drugs in the combined field of psychiatry/neurology, only one was judged to have substantial added benefits compared to already available treatments (Wieseler et al., Reference Wieseler, McGauran and Kaiser2019).
Systematic reviews of commonly used psychiatric drugs, particularly of antidepressants for depression (Jakobsen et al., Reference Jakobsen, Katakam, Schou, Hellmuth, Stallknecht, Leth-Møller, Iversen, Banke, Petersen, Klingenberg, Krogh, Ebert, Timm, Lindschou and Gluud2017; Munkholm et al., Reference Munkholm, Paludan-Müller and Boesen2019) and of central stimulants for attention deficit hyperactivity disorder (ADHD) (Storebø et al., Reference Storebø, Ramstad, Krogh, Nilausen, Skoog, Holmskov, Rosendal, Groth, Magnussen, Moreira-Maia, Gillies, Rasmussen, Gauci, Zwi, Kirubakran, Forsbøll, Simonsen and Gluud2015; Punja et al., Reference Punja, Shamseer, Hartling, Urichuk, Vandermeer and Nikles2016; Castells et al., Reference Castells, Blanco-Silvente and Cunill2018; Cândido et al., Reference Cândido, Menezes de Padua, Golder and Junqueira2021), have highlighted potential problems with low generalisability of psychiatric drug trials. Methodological limitations include small sample sizes, short trial durations, restricted trial populations in terms of allowed psychiatric comorbidity, risk of withdrawal effects due to previous exposure to the drug of interest, and use of surrogates and rating scales rather than patient-relevant outcomes.
Researchers have advocated for psychiatric pivotal trials, i.e. clinical trials conducted with the purpose of obtaining a marketing authorisation for a new drug or a new indication, with active comparators (i.e. already approved medications instead of placebo only) and a focus on hard, functional outcomes such as hospitalisation admissions and suicide rather than symptom rating scales (Barbui et al., Reference Barbui, Cipriani, Lintas, Bertelé and Garattini2007; Barbui and Garattini, Reference Barbui and Garattini2007; Barbui and Bighelli, Reference Barbui and Bighelli2013). An assessment of the evidence base for new psychiatric drug approvals in Europe categorised the general evidence as being ‘poor’ (Erhel et al., Reference Erhel, Scanff and Naudet2020), and the European Medicines Agency (EMA)'s guidelines for designing pivotal psychiatric drug trials have been highlighted as important to improve the usefulness of these trials (Barbui and Bighelli, Reference Barbui and Bighelli2013). On a more general note, it has been argued that the threshold for new drug approvals has been lowered to accommodate the pharmaceutical industry's interests (Davis et al., Reference Davis, Lexchin, Jefferson, Gøtzsche and McKee2016).
Pivotal trials are often designed in a bilateral agreement between the pharmaceutical company and the drug regulator (Fig. 1). The EMA and the US Food and Drug Administration (FDA) publish guidelines on how to design and conduct these pivotal trials for new drug approvals and these guidelines offer a roadmap in supporting the design of pivotal trials. EMA publishes ‘Clinical Efficacy and Safety Guidelines’ (EMA, 2020a), which the agency ‘strongly encourages’ their applicants to follow. These guidelines ‘reflect a harmonised approach of the EU Member States and the Agency on how to interpret and apply the requirements for the demonstration of quality, safety and efficacy […]’ (EMA, 2020b). The FDA publishes ‘Guidance Documents’ (FDA, 2021). These documents are ‘intended to assist the pharmaceutical industry’ in designing and conducting pivotal trials, but they are not ‘legally binding or enforceable’ (FDA, 2005). The documents ‘represent current Agency thinking, sponsor submissions that conform to current guidance should be considered acceptable’ (FDA, 2005). Pharmaceutical companies and regulatory agencies may communicate at any stage of medicines development, e.g. EMA provides ‘scientific advice’ (EMA, 2021a).
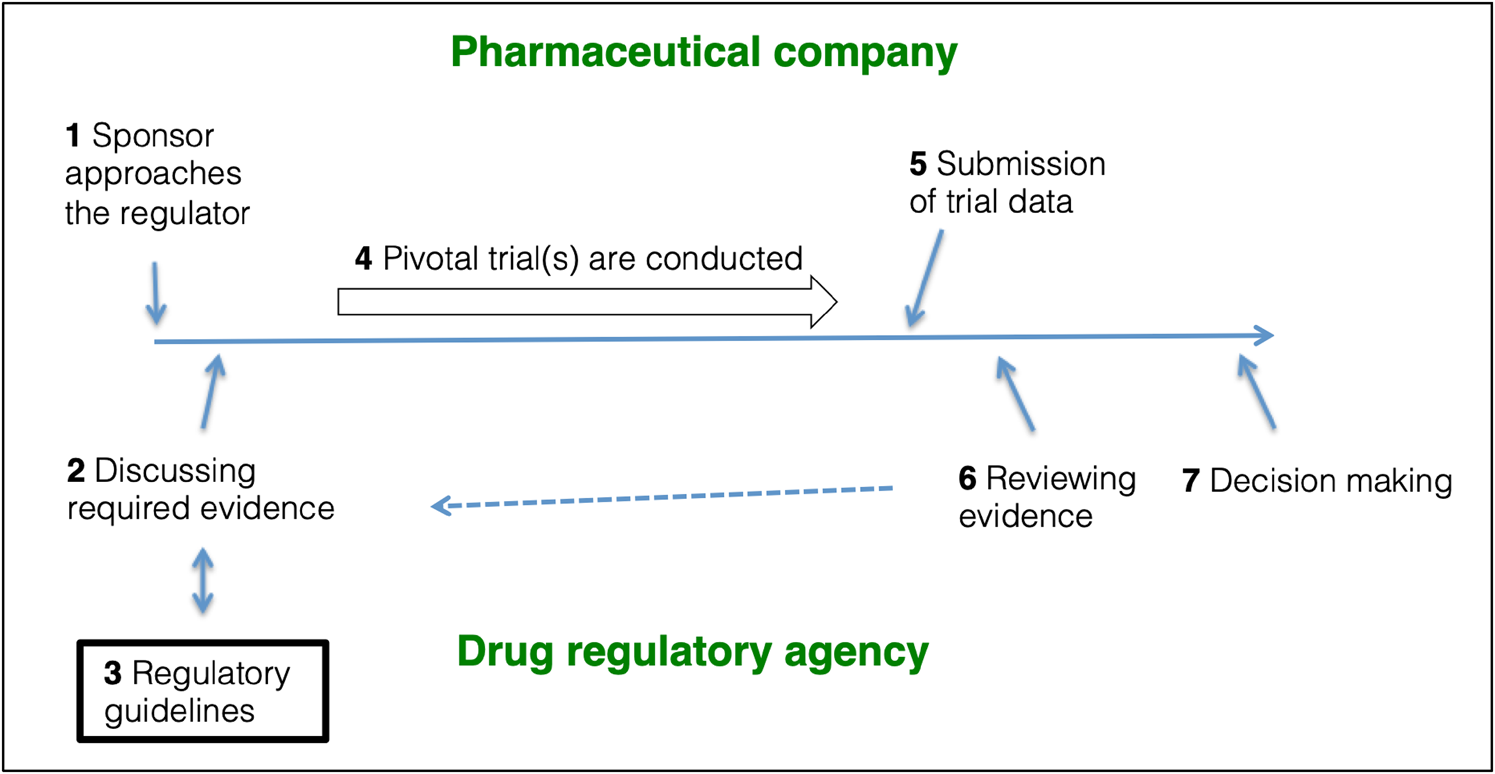
Fig. 1. The role of regulatory guidelines in pivotal trial designs. (1) The sponsor approaches the relevant drug regulator with the purpose of getting a new drug authorised. (2) Sponsor and regulator discuss (and often agree) on a drug development program. (3) The design and conduct of the pivotal trials will often follow design recommendations outlined in regulatory guidelines. (4) The sponsor conducts the pivotal trials. 5) The acquired data are submitted to the regulators as a Marketing Authorisation Application (EMA) or a New Drug Application (FDA) using the Common Technical Format. (6) The regulator reviews the data, and, if necessary, steps 2, 3 and 4 are repeated to obtain additional information. (7) The regulator decides whether to authorise or reject the application. Figure based on FDA (2019d); EMA (2021b).
Some of EMA's trial design recommendations (choice of comparator, outcomes, and patient population) for psychiatric drug trials have previously been described (Barbui and Bighelli, Reference Barbui and Bighelli2013). However, the development and content of EMA and FDA guidelines for psychiatric drug trials have never been systematically assessed. We wanted to assess (1) the guideline committee members and their declared conflicts of interest; (2) the organisation of commenting phases on draft and concept papers; (3) the stakeholders who comment on guideline draft and concept papers; and (4) the EMA and FDA design recommendations for five trial characteristics in pivotal psychiatric drug trials.
Methods
Inclusion of drug regulatory agency guidelines
We applied the following inclusion criteria: (1) the guideline (draft or final version) should be published by the EMA or FDA on how to design pivotal trials for drug approval; (2) the guideline should cover all aspects of pivotal trial design (i.e. what the FDA calls ‘umbrella guidance’) and not address specific trial aspects only, e.g. inclusion criteria or specific outcome measures; and (3) the guideline should cover psychiatric diagnoses as defined in current or previous versions of the Diagnostics and Statistics Manual of Mental Disorders (DSM) or the International Classification of Diseases (ICD). We included the newest guideline (draft or final version) if more than one guideline was published on the same subject. See eMethods for details.
Guideline committee members' conflicts of interest
The guideline committee members and/or EMA and FDA employers that are responsible for drafting and revising the guidelines are not publicly disclosed. Similarly, the committee members' declared conflicts of interest are not publicly available. We therefore filed Freedom of Information Act requests to the EMA and FDA to access the committee member lists and their declared conflicts of interest. We wanted to report the proportion of committee members with declared conflicts of interest for each guideline.
Guideline development
We assessed the development of draft guidelines in regards to, (1) public availability of draft versions for commenting; (2) active recruitment of specific stakeholders to comment on drafts; (3) applied restrictions on who may comment on the documents; (4) announcement of drafts and final guidelines, e.g. on social media, in newsletters or in relevant journals.
We searched the EMA and FDA websites for information. It was not necessary to contact the regulators for additional information. See eMethods for details.
Stakeholder comment documents
EMA publishes dedicated documents with stakeholder comments submitted on draft guidelines along the final guidelines. The stakeholder comments and EMA's corresponding replies are collated in a single document and made available for each guideline (EMA, 2020a).
FDA manages and operates public correspondence on federal regulations and other rule-making documents through the Docket's Management (US Government, 2020). All official US Federal documents, such as drafts or final Guidance Documents, have their own designated entry, called a Docket Folder Summary, where the public can submit and also view submitted comments on the Guidance Documents.
For each included stakeholder document related to a specific guideline, we counted the total number of stakeholders, total number of comments, and total number of comments by each stakeholder. We categorised the stakeholders according to their conflicts of interest as ‘industry’ (e.g. pharmaceutical companies or other industry), ‘not-industry but with industry-related conflicts’ (e.g. organisations, associations or individuals reporting financial conflicts of interest related to pharmaceutical companies), ‘independent’ (organisations, associations and individuals with declared no conflicts of interest related to the pharmaceutical industry) or ‘unclear financial relationship’ (there was insufficient information to identify and/or categorise the stakeholder). We searched for information on Google, PubMed, OpenPaymentsData, and stakeholder websites regarding funding, annual budgets, and disclosed conflicts of interest. For organisations and associations, we focused on board and executive members. We did not define a lower monetary limit for conflicts of interest. See eMethods for details.
Trial design recommendations
For all included research guidelines, we extracted recommendations regarding five trial design characteristics: (1) duration of follow-up, also after the randomised phase ended; (2) exclusion criteria related to psychiatric comorbidity; (3) allowing previous exposure to the drug or drug class, which may be called an ‘enriched design’ or ‘enrichment strategy’ (FDA, 2019a); (4) efficacy outcomes (primary and secondary); and (5) choice of comparator (e.g. placebo, active comparator, or both).
We wanted to put the trial design recommendation in a clinical context for each of the psychiatric diagnoses and corresponding guidelines, i.e. what is the natural history of the different conditions in terms of expected symptom duration; what are the most common comorbid psychiatric diagnoses and how common are they; and whether previous exposure to the drug might introduce potential withdrawal effects and also bias the sample of participants in favour of those who tolerate the drug. We extracted this basic clinical epidemiology information from the guidelines, when available. We further searched the Core Outcome Measures in Effectiveness Trials (COMET) database (COMET, 2020) for standardised sets of outcome measures for the individual indications, and we assessed the most recent systematic review (Eiring et al., Reference Eiring, Landmark, Aas, Salkeld, Nylenna and Nytrøen2015) of patient preferences for outcome measures in psychiatric drug trials.
See the Supplementary material for differences between the protocol (Boesen et al., Reference Boesen, Gøtzsche and Ioannidis2020) and the final manuscript.
Results
Overview of results
We searched the EMA and FDA websites in February 2020 and identified 13 eligible EMA Clinical Efficacy and Safety Guidelines and five FDA Guidance Documents (Table 1). The 13 EMA guidelines were published between 2005 and 2017; the panic disorder guideline could not be downloaded. All EMA guidelines were adopted (i.e. final) versions, two (insomnia and schizophrenia) had been revised once, and the depression guideline had been revised twice. Concept papers for upcoming revisions were published for the depression and bipolar disorder guidelines. The five FDA guidelines were published between 2015 and 2019; four were draft versions and one (opioid use disorder) was a final guideline. The 18 guidelines covered 15 unique indications (depression, ADHD, and alcohol dependence were covered by both agencies). The full dataset is available here (https://osf.io/3xcdu/?view_only = 957edc6293894497ba7aaf5bb8fc8205).
Table 1. Overview of included regulatory research guidelines

Listed in order of publication year.
a A concept paper for a revision of the guideline has been published.
b The link did not work so we could not include the guideline. Links to the guidelines can be found in Supplement 1.
Guideline committee members' conflicts of interests
The EMA informed us they were not able to respond within their regular response time caused by EMA's relocation to Amsterdam. After 11 months from submission, EMA had not processed our request of access to committee member lists. See correspondence with EMA (Supplement 2).
The FDA Freedom of Information Team informed us during a telephone meeting that, (1) federal employees are not allowed to have financial conflicts of interest; (2) Guidance Documents are authored internally in the Agency by divisions or offices and not by individual employees; and (3) therefore the FDA is not in possession of conflicts of interest declarations related to Guidance Document development (see summary of the meeting in Supplement 1). We partly confirmed statement (1) from FDA's website (FDA, 2017a), where it is stated that FDA employees are generally prohibited from holding financial interests, e.g. stocks, in any FDA ‘significantly regulated organisation’. However, there are exceptions to the rules. For instance, only designated FDA employees, called ‘confidential filers’, are required to disclose conflicts of interest. Employees that are not required to disclose their financial conflicts of interest may hold financial interests up to US$15,000 under certain conditions (FDA, 2017a). We found some information relevant to statements (2) and (3) in the FDA Manuals of Policies and Procedures (MAPPs) ‘Developing and Issuing Guidance’ (FDA, 2005) and ‘Developing indication-specific Guidances’ (FDA, 2014), specifically that offices and divisions are responsible for writing the Guidances (FDA, 2014). See correspondence with the FDA in Supplement 2.
Guideline development
EMA and FDA produce their research guidelines in a similar fashion, summarised in Table 2.
Table 2. Overview of commenting phases on regulatory guidelines

Exact references and verbatim statements can be found in the full dataset (https://osf.io/3xcdu/?view_only=957edc6293894497ba7aaf5bb8fc8205).
EMA guidelines
The development of EMA's Clinical Efficacy and Safety Guidelines is primarily described in the guideline ‘Procedure for European Union guidelines and related documents within the pharmaceutical legislative framework’ (EMA, 2009), to which stakeholder comments have also been published (EMA, 2005). There are three stages where stakeholders may comment on the guidelines during the development: (1) first, a concept paper, which outlines the overall purpose of the guideline, is made available for commenting for 2–3 months. It is unclear if, and when, a concept paper is written with input from outside stakeholders or comments are actively solicited (EMA, 2009; section 4.4); (2) then a draft guideline is made available for comments, usually for 3–6 months. Also for this stage, it is unclear if and when comments are actively solicited (EMA, 2009; section 4.6); finally (3) the adopted guideline is published and may be commented on at any given time. EMA announces all documents for public consultation on a designated website called ‘Open consultations’ (EMA, 2020c).
FDA Guidance Documents
The Guidance development is described in the FDA Manual of Policies and Procedures ‘Developing and Issuing Guidance’ (FDA, 2005) and ‘Developing indication-specific Guidances’ (FDA, 2014), the Code of Federal Regulation, Part 10, Section 10.115 (CFR 2019), and FDA's report on Guidance Documents (FDA, 2011). There are two stages, possibly three if the draft development is included, where stakeholders may comment on Guidance Documents: (1) First, FDA develops a draft Guidance. This can be done in collaboration with individuals or organisations outside FDA, e.g. through input from workshops or Advisory Committees. Stakeholders may also submit draft proposals directly to FDA; (2) a draft Guidance Document is made publicly available for comments usually for 60 days through announcement (called a ‘notice of availability’) in the Federal Register (Federal Register, 2020), the official journal of the United States Government, and a designated website (FDA, 2020a); (3) the final Guidance Document is published and announced in the Federal Register and online and may be commented on at any given time. Every year, FDA also publishes a ‘Guidance Agenda’, a list of guidances they plan to publish or revise during the year (FDA, 2020b).
Stakeholder comments
There were stakeholder comments available for six of the 13 EMA research guidelines and four of the five FDA guidelines. Seventy stakeholders submitted a total of 1014 comments, of which 947 comments could be assigned to a stakeholder. For 67 comments the stakeholder was unknown, as EMA's post-traumatic stress disorder (PTSD) guideline did not assign the individual comments to the stakeholder. There were 52 unique stakeholders of which two, International Society for CNS Clinical Trials and Methodology and Lundbeck, commented on both EMA and FDA documents. See the full dataset for details.
EMA guidelines
Thirty-eight stakeholders made 771 comments on six EMA guidelines. There were 25 unique stakeholders; 19 stakeholders commented on one guideline and six stakeholders commented on two or more guidelines, Lundbeck and the European Federation of Pharmaceutical Industries and Associations (five guidelines), Merck Sharp & Dome (three), and European College of Neuropsychopharmacology, International Federation of Association of Pharmaceutical Physicians, and Hoffman-La Roche (two each). We categorised 25 (66%) stakeholders as ‘industry’, nine (24%) as ‘not-industry but with industry-related conflicts’ and four (10%) as ‘independent’ (Table 2). On the comment level, we categorised 534 (76%) comments as ‘industry’, 148 (21%) as ‘not industry but with industry-related conflicts’, 22 (3%) as ‘independent’ and 67 were not categorised (Table 3).
Table 3. Stakeholders categorised by conflicts of interest

FDA Guidance Documents
Thirty-two stakeholders made 243 comments on four FDA Guidance Documents. There were 29 unique stakeholders; 26 stakeholders commented on one document and three stakeholders, Lundbeck, Anthem Inc., and International Society for CNS Clinical Trials and Methodology, commented on two Guidance Documents each. We categorised 13 (41%) stakeholders as ‘industry’, nine (28%) as ‘not-industry but with industry-related conflicts’, two (6%) as ‘independent’ and eight (25%) as ‘unclear’ (Table 2). On the comment level, we categorised 106 comments (44%) as ‘industry’, 95 (39%) as ‘not-industry but with industry-related conflicts’, 22 (9%) as independent and 20 (8%) as ‘unclear’ (Table 4).
Table 4. Stakeholder comments categorised by conflicts of interest

a The comments were not assigned to the individual stakeholders.
b Calculated as the proportion of assigned comments, not the total number of comments.
Trial design recommendations
The EMA guidelines provided recommendations for all five trial characteristics, i.e. trial duration, psychiatric comorbidity, ‘enriched design’, efficacy outcomes, and choice of comparator in all guidelines, except ‘enriched design’ (alcohol dependence) and psychiatric comorbidity (bipolar disorder). None of the five FDA Guidance Documents contained recommendations for all five trial characteristics; for example, previous exposure to relevant medications was described in one guideline only (opioid use disorder). The trial recommendations for each guideline are summarised in Supplement 1, eTable 1–15.
Overlapping guidelines
EMA and FDA published overlapping guidelines on ADHD (eTable 3), depression (eTable 5), and alcohol dependence (eTable 10). The three FDA guidelines were draft versions and did not contain recommendations on ‘enriched design’ or on psychiatric comorbidity in the ADHD guideline. The overlapping guidelines concurred on trial durations for depression and alcohol dependence, e.g. EMA recommended 4–8 weeks and FDA 6–8 weeks in depression trials, and both agencies recommended a 6-month randomised withdrawal design to assess long-term outcomes. The EMA guidelines seemed more restrictive regarding psychiatric comorbidity, whereas the FDA depression and alcohol dependence guidelines emphasised the inclusion of relevant comorbidity. The guidelines agreed on the use of rating scales in ADHD and depression trials as efficacy outcomes and on the use of full abstinence or no heavy drinking for alcohol dependence.
Trial duration
EMA recommended short- and long-term trial durations in all 12 available guidelines. Short-term durations ranged from 2 to 14 weeks, with the exception of alcohol dependence (3–6 months) and premenstrual dysphoric syndrome (6 months). Long-term durations ranged from 3 to 15 months, of which seven guidelines recommended the randomised withdrawal design, three guidelines (ADHD, insomnia, alcohol dependence) recommended a placebo-controlled trial or the randomised withdrawal design, and two guidelines did not recommend any particular design (bipolar disorder and premenstrual dysphoric disorder).
The FDA recommendations were more varied; 6–8 weeks for depression, 24 weeks for ‘low sexual interest and/or arousal in women’, 6 months for alcohol dependence, and for ADHD they only gave a recommendation for long-term safety trials of 12 months to assess the effect on growth in children.
Psychiatric comorbidity
EMA listed extensive exclusion criteria related to psychiatric comorbidity in ten of the 12 available guidelines. Two guidelines (bipolar disorder and depression) did not specify exclusion criteria.
The three FDA Guidance Documents (depression, alcohol dependence, ‘low sexual interest and/or arousal in women’) that contained recommendations on psychiatric comorbidity were less restrictive than EMA's guidelines and stated that unnecessary restrictions should be avoided.
Enriched design
All EMA guidelines, except alcohol dependence, specified that participants could have been exposed to relevant medication prior to enrolment and there should be a ‘washout’ of such drugs.
Only one FDA Guidance Document (opioid use disorder) referred to previous exposure by stating, ‘patients should be either new entrants to treatment or stable on other treatments’.
Efficacy outcomes
Ten EMA guidelines recommended symptom-rating scales as efficacy measures. The alcohol dependence guideline recommended full abstinence or no heavy drinking, and the insomnia guideline recommended self-rated sleep, polysomnography, and quality of life. Several guidelines stipulated the use of ‘responder analyses’, i.e. dichotomising the participants as ‘responders’ or ‘non-responders’ based on their treatment response on symptom rating scales using arbitrary thresholds.
The FDA Guidance Documents recommended rating scales for two indications (depression and ADHD), no heavy drinking or full abstinence (alcohol dependence), urine toxicology (opioid use disorder), and number of satisfying sexual events or self-rating of sexual interests, arousal, and distress (low sexual interest and/or arousal in women).
Choice of comparator
Ten EMA guidelines recommended three-arm trial designs with both placebo and an active comparator. Two guidelines (alcohol dependence and premenstrual dysphoric syndrome) recommended a two-arm placebo design, but also mentioned three-arm designs as a viable option.
Four FDA Guidance Documents recommended a two-arm design with placebo (ADHD, depression, low sexual interest and/or arousal in women, and alcohol abuse), and one guideline recommended either placebo or an active comparator (opioid use disorder). The alcohol abuse guideline also mentioned the possibility of a three-arm trial design.
Discussion
Principal findings
To the best of our knowledge, this is the first study to systematically assess the development and content of regulatory research guidelines for psychiatric drugs. Some EMA trial design recommendations have previously been assessed, mainly focusing on the choice of comparator (Barbui and Bighelli, Reference Barbui and Bighelli2013). These regulatory guidelines are intended for industry, but the dearth of non-industry stakeholders was surprising considering the guideline process being apparently open to public comments. The FDA launched in 2010 a ‘Transparency Initiative’ (FDA, 2011) and one of the objects was to improve stakeholder involvement. FDA's stakeholder composition was indeed more varied than EMA's, although still dominated by stakeholders with conflicts of interest. EMA stated in their procedure guideline (EMA, 2009) that they would develop procedures to ensure proactive consultation of patients and patient organisations. For this cohort of guidelines, there was an absence of such stakeholders.
The guidelines recommend the design of clinical trials with limited generalisability, which may contribute to research waste (Chalmers and Glasziou, Reference Chalmer and Glasziou2009). Most recommended trial durations were short and both agencies recommended the randomised withdrawal design for long-term trials. In such a trial, participants who benefit from a drug either in a placebo-controlled trial or in an open-label phase are randomised to continued active drug or placebo (FDA, 2019a; p. 18). Abrupt stopping of psychotropic drugs can lead to withdrawal symptoms, e.g. after use of antidepressants (RCP, 2019) or central stimulants, which carries an FDA black box label warning about the risk of drug dependence (FDA, 2017b). If these drugs are not tapered off slowly, the withdrawal symptoms may be misinterpreted as symptom deterioration and superiority of the drug over placebo. In FDA's opioid use disorder guideline it was stated, ‘Sponsors can conduct trials in patients already stable on other treatments’. This indicates that participants who are adequately treated before the trial may be randomised to placebo and thereby risk experiencing withdrawal symptoms. In EMA's schizophrenia guideline it was stated, ‘typically a few days will be appropriate’ for washing out prior antipsychotic medication during a placebo lead-in, which seems to be insufficient to avoid withdrawal symptoms.
All but two EMA guidelines allowed participants with previous exposure to the drugs to be included in the pivotal trials. Before their inclusion, such participants should ‘wash out’ the drug, i.e. stop the treatment for a certain amount of time. The sample would be ‘enriched’ because participants with a previous negative response would not be allowed to participate, either due to explicit exclusion criteria and/or because participants with a negative response would not want to participate. This trial design detail may seem trivial but it can likely lead to substantial overestimation of the benefits and underestimations of the harms.
Most EMA guidelines recommended extensive exclusion criteria regarding psychiatric comorbidity. The obsessive-compulsive disorder (OCD) guideline described that the population should be ‘pure’ OCD but the guideline also highlighted, ‘lifetime co-morbidity rates of other psychiatric diseases in patients with OCD range from 75 to 84%’. The ADHD guideline mentioned, ‘another Axis I disorder’, ‘severe comorbid symptoms such as depression and anxiety’ or ‘primary axis II disorder’ as exclusion criteria. Such exclusion criteria may reduce the trials' generalisability, e.g. most patients diagnosed with ADHD (Surman et al., Reference Surman, Monuteaux, Petty, Faraone, Spencer, Chu and Biederman2010) or depression (Zimmerman et al., Reference Zimmerman, Balling, Chelminski and Dalrymple2019) in a clinical setting are not eligible for inclusion in pivotal clinical trials. The FDA Guidance inclusion criteria were less restrictive and emphasised the inclusion of participants with comorbidities. In 2019, FDA published a draft guideline entitled ‘Enhancing the diversity of clinical trial populations’ (FDA, 2019b) and the Agency seems cognisant of the problem of restricted trial populations, ‘Sponsors should adopt practices for determining eligibility criteria that will allow the clinical trial population to reflect the diversity of the patients who will be using the drug if the drug is approved’ (FDA, 2019b).
We found that EMA and FDA mostly recommended surrogate outcomes. A recent analysis (Hey et al., Reference Hey, Kesselheim, Patel, Mehrotra and Powers2020) similarly reported that 21 of 27 FDA anti-infective Guidance Documents recommended surrogate outcomes rather than patient-relevant outcomes such as mortality.
Finally, we noted that EMA mostly recommended three-arm trial designs with both placebo and active comparator. In contrast, the FDA mostly recommended two-arm placebo-controlled designs, particularly notable for ADHD and depression trials, where many approved medications are available. EMA has previously been encouraged to require comparative evidence prior to regulatory approval (Sorenson et al., Reference Sorenson, Naci, Cylus and Mossialos2011; Barbui and Bighelli, Reference Barbui and Bighelli2013; Wieseler et al., Reference Wieseler, McGauran and Kaiser2019), and such efforts seem warranted for FDA as well.
Implication for practice and research
We identified several cases where the agencies seemed to prioritise the interests of the applicants rather than the public. For instance, FDA wrote in their opioid use disorder guideline, ‘There is great public health interest in assessing additional, clinically meaningful endpoints such as reduction in hospitalizations, emergency department visits, overdose, and death, as well as improvements in the ability to resume work, school, or other productive activity. Though understanding these outcomes would be highly valuable, the Agency recognizes that evaluating these outcomes could require larger trials than those usually conducted for marketing approval’ (FDA, 2019c). If the agencies wish to serve the public, the required pivotal trials should reflect the interests of the public and thus address real-world patient populations and patient-centred outcomes.
FDA informed us that their employees cannot have conflicts of interest. The FDA policy allows non-designated employees to have assets up to US$15 000 (FDA, 2017a). Eleven months after we submitted our request regarding access to the committee members' conflicts of interest, EMA had not processed it. It would be useful for regulatory agencies to make such information publicly available to increase transparency.
Study limitations
The sample of guidelines was small and there was an uneven distribution of stakeholders and comments. There were challenges in counting the number of FDA comments due to the non-uniform format, and we were unable to access a few guidelines and stakeholder documents. We recognise the large variations in the stakeholder comments and that different criteria for quantifying the comments would yield different counts. However, count differences would likely be minor and unlikely to change the overall results. We were also unable to identify several of the individual commenters who submitted comments on the FDA Guidance Documents, since their affiliations were not disclosed. We also did not compare draft and final guidelines to assess if, and how, the submitted stakeholder comments might have served specific industry interests. We believed that such analyses would be too subjective.
It was a challenge to separate the stakeholders according to their conflicts of interest, especially patient groups and organisations. Some evidence points to a dose–response relationship between conflicts of interest and drug prescription patterns (Perlis and Perlis, Reference Perlis and Perlis2016), and a recent systematic review reported that industry funding of patient organisations is common (Fabbri et al., Reference Fabbri, Parker, Colombo, Mosconi, Barbara, Frattaruolo, Lau, Kroeger, Lunny, Salzwedel and Mintzes2020). We did therefore not define a lower monetary threshold, but categorised stakeholders as ‘not-industry but with industry-related conflicts’ if we identified any conflicts of interest.
Conclusion
The EMA and FDA clinical research guidelines for psychiatric pivotal trials recommend designs that tend to have limited generalisability. Independent and non-conflicted stakeholders are underrepresented in the development phases and current guidelines emphasise trials with limited scope that may not offer much clinical value. EMA and FDA should reconsider their guideline development and find ways to promote greater involvement of the public and independent stakeholders. This may require greater advertisement of the guideline process to the public and active invitation of large numbers of scientists and organisations that are known to have no conflicts of interest to comment on the guidelines.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S2045796021000147.
Data
The project protocol was made available on 27 January 2020 on MedRxiv (https://www.medrxiv.org/content/10.1101/2020.01.22.20018499v1). The full dataset is available from the Open Science Framework (https://osf.io/3xcdu/?view_only = 957edc6293894497ba7aaf5bb8fc8205). Questions regarding the dataset should be referred to the corresponding author.
Acknowledgements
We thank the Meta-Research Innovation Center at Stanford (METRICS) lab meeting participants (Nov 2019) for comments on the draft protocol.
Author contributions
KB conceived the idea, collected and analysed data, corresponded with the drug regulatory agencies, and wrote the first draft. JPAI and PCG contributed to the design of the study, commented and supervised the final manuscript. All authors critically revised the final manuscript. All authors approved the final version. KB is the guarantor.
Financial support
METRICS is supported by a grant from the Laura and John Arnold Foundation and METRIC-B is supported by a visiting Einstein fellowship in the Berlin Institute of Health to JPAI from the Einstein Foundation and the Stiftung Charité.
Conflict of interest
None.
Ethical standards
Not applicable.








 Open access
Open access