


INTRODUCTION
Salmonellosis is a common foodborne disease in humans worldwide [Reference de Jong and Ekdahl1]. It is caused primarily by consumption of food contaminated with non-typhoidal Salmonella enterica serovars [Reference Hald2]. Illness is often associated with self-limiting gastrointestinal disorders [Reference Zhang3]; however, invasive disease occurs in a proportion of patients [Reference Gordon4] causing severe systemic disease. Salmonella enterica serovars Enteritidis and Typhimurium account for most human infection. S. Dublin (1,9,12[Vi]:g,p:-) has a similar antigenic formula to S. Enteritidis (1,9,12:g,m:-); however, the epidemiology is distinct. S. Dublin is strongly associated with cattle [Reference Kingsley and Baumler5] and it can be transmitted to humans via contact with infected animals or consumption of contaminated products including raw milk and raw milk cheeses [Reference Vaillant6]. S. Dublin has also been detected in other host species including poultry, sheep, dog, horse and mink [Reference Liebana7, Reference Dietz8]. Although human infection with S. Dublin is much less common than infection with S. Enteritidis a higher proportion of S. Dublin infections are invasive. The invasiveness of S. Dublin may be related to expression of the Vi (virulence) antigen which is also commonly found in human-adapted S. Typhi.
Because S. Dublin is a relatively infrequent serotype, specific subtyping methods such as phage typing or multiple-locus variable-number tandem repeat analysis (MLVA) are not widely available although application of the S. Enteritidis MLVA protocol [9] has recently been described as of some value. Pulsed-field gel electrophoresis (PFGE) with XbaI according to the PulseNet protocol [Reference Ribot10] is applicable; however, the indistinguishable PFGE pattern offers limited microbiological support for investigating a suspected S. Dublin outbreak.
Whole genome sequencing (WGS) has the potential to transform the confidence with which laboratories can demonstrate the degree of similarity between isolates of serovars such as S. Dublin which are difficult to type by established methods. In Ireland S. Dublin accounts for between two and seven clinical cases annually. However, approximately one third of human cases are invasive. In October–November 2013 nine isolates of S. Dublin were received at the national reference laboratory. An outbreak investigation was initiated. Here we describe the epidemiological and laboratory aspects of that investigation including the first reported application of WGS to support investigation of an outbreak of S. Dublin.
METHODS
Epidemiological investigations
Outbreak case definition
Outbreak cases were defined as persons with: (i) laboratory-confirmed infection with fully antimicrobial-susceptible S. Dublin exhibiting the MLVA pattern designated as (3-6-1-10-2-3-12); (ii) gastroenteritis symptoms including diarrhoea; (iii) onset of illness between October 2013 and November 2013 and (iv) reported in any region throughout Ireland.
Foodborne illness questionnaire
Cases were contacted and asked to provide their exposure histories using the National Salmonella Trawling Questionnaire. Supplementary information on food purchases was obtained using loyalty card information from a national supermarket chain. Consent was obtained from the loyalty card customer associated with four of the outbreak cases to look for a commonly purchased food item within a 6-week purchasing history prior to disease onset.
Microbiological investigations
The nine human faecal isolates detected during October–November 2013 were characterized as S. enterica serotype Dublin according to the White–Kauffmann–Le Minor scheme [Reference Grimont and Weill11]. They were phenotypically negative for the Vi antigen. Antimicrobial susceptibility testing against a panel of antimicrobials was determined using the broth dilution method [Reference Wikler12]. The interpretation was conducted using the European Committee on Antimicrobial Susceptibility Testing (EUCAST) clinical breakpoints (http://www.eucast.org). PFGE was performed using the PulseNet protocol [Reference Ribot10]. MLVA was also performed. The quoted MLVA pattern is based on the application of the Centers for Disease Control and Prevention (CDC) S. enterica serovar Enteritidis [9].
The nine outbreak-associated S. Dublin isolates isolated during October–November 2013 (Supplementary Table S1) in addition to the apparently epidemiologically unrelated isolate from December 2013 were selected for WGS. The other eight unrelated isolates were chosen in a blinded fashion and were also subjected to WGS for comparison. All isolates are of human origin and isolated from faeces except five isolates were invasive.
Genomic DNA was extracted using the QIAamp® DNA Mini kit (Qiagen, UK). DNA quality and quantity were checked by gel electrophoresis and the Qubit® quantification platform (Invitrogen, USA), respectively. Twenty microlitres of DNA (20–50 ng/μl) from each isolate was submitted for Illumina sequencing using 100-bp paired-end (PE) reads. The depth of coverage was estimated as 100×.
The quality of the short-read data was evaluated using the FastQC toolkit (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). Low-quality reads were removed and adapter sequences were trimmed using the ea-utils package (https://code.google.com/p/ea-utils/). Sequence reads of each isolate were mapped against the reference genome of S. Dublin strain CT_02021853 (accession no.: NC_011205) isolated in 2004 from human faeces in Macau, Asia using BWA software [Reference Li and Durbin13]. Single nucleotide polymorphisms (SNPs) were identified using samtools mpileup [Reference Li14]. SNPs over 60 quality threshold (i.e. a 1/1 000 000 chance of a miss-called variant) were only accepted (automated called variants for each isolate are provided in Supplementary Table S2). SNPs were compared to the reference strain and a maximum-likelihood (ML) phylogeny of the isolates was constructed using MEGA6 software [Reference Tamura15] using the GTR+Gamma+Invariant model as the best-fit model of nucleotide substitution. De novo assembly of short PE reads was performed using Velvet [Reference Zerbino and Birney16]. Generated multi-contig draft genomes for each isolate were screened for acquired antimicrobial resistance genes using ResFinder [Reference Zankari17]. Furthermore, housekeeping genes were screened for mutations that are associated with resistance to antibiotics including gyrA and parC genes where certain mutations are linked to resistance to fluoroquinolone antibiotics including nalidixic acid [Reference Saenz18]. Draft genomes were also screened for the Vi antigen genes [Reference Pulickal19] that are responsible for synthesis and transportation of Vi proteins (viPs).
RESULTS
Epidemiological investigations
Outbreak description
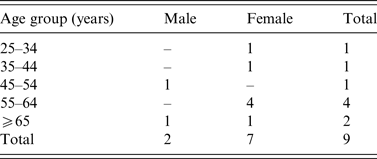
A total nine confirmed outbreak cases were reported with onset dates from October 2013 to November 2013. None was associated with invasive disease. Cases were resident throughout Ireland from North-West to South-East (Fig. 1). Cases were predominantly female (7/9) and ranged in age from 30 to 85 years (Table 1). Two of the nine cases were hospitalized. Where reported, the other seven cases had symptom duration of 3–8 days. All cases recovered.

Fig. 1. Salmonella Dublin outbreak cases by week of onset, Ireland, October–November 2013 (n = 9).
Table 1. Age-sex distribution of Salmonella Dublin outbreak cases (October–November 2013), Ireland
Cases’ exposure
No single food source was identified to be linked to the outbreak based on the completed National Salmonella Trawling Questionnaire and the loyalty card information from a national supermarket chain. Although cheese consumption was considered a possible source of infection, as it was reported by seven of eight cases, no specific cheese product emerged from the epidemiological investigation and Salmonella was not detected from cheese samples obtained from retail and restaurant premises that cases had reported visiting.
Microbiological investigations
The nine outbreak associated isolates were susceptible to all antimicrobial agents tested. They were indistinguishable on PFGE but they were also indistinguishable from many other S. Dublin isolates typed by PFGE. Isolates had the same MLVA pattern (3-6-1-10-2-3-12). A non-complete match to this MLVA pattern (3-6-1-10-2-3-11/12) was observed in a historical isolate from 2011.
WGS analysis
WGS analysis revealed that the nine S. Dublin isolates from October–November 2013 were very closely related with only 1–9 SNP difference. This contrasts with 74–88 SNP difference between the outbreak isolates and an apparently epidemiologically unrelated isolate from December 2013 (O13). Other isolates showed high genetic divergence from the outbreak isolates except a historical isolate from 2011 (J11) that was very closely related to the 2013 cluster with a maximum 15 SNP difference (Fig. 2).

Fig. 2. Maximum-likelihood phylogenetic tree of Salmonella Dublin strains based on single nucleotide polymorphisms (SNP) determined from whole genome sequences. Sequence reads were mapped against the reference genome of S. Dublin (strain CT_02021853). The scale represents the number of nucleotide substitutions per site. Bootstrap support values, given as a percentage of 1000 replicates, are shown on the branches. All S. Dublin isolates had indistinguishable pulsed-field gel electrophoresis profiles. Confirmed outbreak cases (n = 9) in October–November 2013 are highlighted in grey. Other isolates show high genetic divergence to the outbreak cluster including the epidemiologically unrelated isolate (*O13). However, a historical isolatefrom 2011 (J11) is very closely related to the 2013 cluster with a maximum of 15 SNP difference.
WGS data analysis of the outbreak isolates revealed no antibiotic resistance genes or mutations within housekeeping genes gyrA or parC consistent with the susceptible phenotype. However, the mutation detected within the gyrA gene in the epidemiologically unrelated isolate (O13) from December 2013 had changed one amino acid within the gyrase protein and this change is associated with resistance to nalidixic acid. No genetic determinants of antimicrobial resistance were detected within the other isolates consistent with the susceptible phenotype.
Generated multi-contig draft genomes of S. Dublin isolates were screened for the Vi antigen genes. Vi antigen-encoding genes including tviA, tviB, tviC, tviD, tviE (coding for polysaccharide capsule) and vexA, vexB, vexC, vexD, vexE (coding for the polysaccharide transportation proteins) were entirely absent from the outbreak isolates and other unrelated isolates including invasive isolates consistent with the phenotype since all isolated tested negative for the Vi antigen by slide agglutination using Salmonella Vi antisera.
DISCUSSION
A diffuse outbreak of S. Dublin gastroenteritis was detected in Ireland over a period of 6 weeks in October–November 2013. Unusually for S. Dublin all cases were non-invasive. No source of infection was identified.
The wide geographical distribution of the outbreak throughout Ireland suggests that the outbreak might be linked to a widely distributed food item (batch of dairy product), with the shape of the epidemic curve being consistent with a continuous source outbreak. Moreover, the source and/or transmission routes of the S. Dublin gastroenteritis outbreak might differ from the typical sources and/or transmission routes for previously identified sporadic S. Dublin cases within Ireland since the percentage of female cases was higher than normal and all cases suffered gastrointestinal disease while invasive infection was very common in sporadic S. Dublin cases in Ireland. The ability of S. Dublin to cause invasive infection may be related to expression of the Vi antigen which is also commonly found in human-adapted S. Typhi. However, in this study we found that Vi antigen is absent from all S. Dublin isolates including invasive isolates.
Conventional typing methods were of limited value for investigation of the S. Dublin outbreak. There is limited confidence in the laboratory evidence for similarity between S. Dublin isolates because of the limited discriminatory power of currently widely used phenotypic (antimicrobial susceptibility testing) and genotypic (PFGE) typing methods.
Since the outbreak occurred a specific MLVA scheme for S. Dublin has been developed [Reference Kjeldsen20]. MLVA is rapid, simple and suitable for processing large numbers of isolates and it can distinguish between clonal isolates that are indistinguishable by PFGE. However, it has certain limitations [Reference Fabre21] and although MLVA was of value in discriminating an epidemiologically unrelated S. Dublin isolate from December 2013 from the outbreak strain it was not able to provide a conclusive link between the outbreak strain and a historical isolate.
Although PFGE and MLVA allow clustering by similarity/difference they do not provide reliable representation of phylogenetic relationships and isolates that are distantly related can have similar PFGE or MLVA as a result of chance thus limiting their effectiveness in linking historical isolates to identifying transmission routes; therefore, a powerful rapid real-time method is required.
Several retrospective WGS analyses compared to the routine typing methods during outbreaks were carried out. Studies showed that WGS is superior to conventional typing methods including PFGE and MLVA and can provide more detailed information on the genetic characteristic of the strains of foodborne and non-foodborne outbreaks [Reference Koser22–Reference Dallman24]. Furthermore, WGS provides the potential to define the genetic basis for virulence and antimicrobial resistance. It has the potential to replace all currently used phenotypic and molecular typing methods.
We therefore applied WGS technology to S. Dublin outbreak isolates and a number of unrelated isolates to define the genetic relatedness in isolates more precisely. WGS analysis not only confirmed the very close relationship in the outbreak isolates but it also succeeded in identifying a historical isolate from 2011 that was very similar to the outbreak strain. The MLVA pattern of the outbreak isolates designated as (3-6-1-10-2-3-12) was not a complete match for the 2011 isolate (3-6-1-10-2-3-11/12), thus WGS gave greater assurance of similarity. There is no known epidemiological link between the outbreak strain and the historical isolate.
As the cost of the WGS declines and the speed and accuracy of sequencing improves, it is expected that WGS will become a standard tool and replace conventional typing in the detection and investigation of the outbreaks in real time. This ability to show definitive evidence of near identity between outbreak strains and historical isolates as illustrated here will grow in public health importance as databases of WGS of human and food-related isolates increase.
SUPPLEMENTARY MATERIAL
For supplementary material accompanying this paper visit http://dx.doi.org/10.1017/S0950268815001636.
ACKNOWLEDGEMENTS
The authors thank all members of the outbreak control team in Ireland. This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
DECLARATION OF INTEREST
None.





