


INTRODUCTION
Influenza viruses are common causes of respiratory infections in humans and seasonal outbreaks occur annually with about 600 million cases, 3 million cases of severe illness and 250 000–500 000 deaths/year [1]. Hospitalization rates from severe illness can be as high as 3/1000 for children aged 6–23 months and as high as 9/1000 for children aged <6 months [Reference Grijalva2]. In the USA, the virus causes more than 200 000 hospitalizations and 30 000 deaths every year [Reference Thompson3, Reference Thompson4]. Influenza viruses are enveloped RNA viruses belonging to the Orthomyxoviridae family and exist as three distint types, A, B, and C, according to the antigenicity of their structural proteins, nucleoprotein (NP) and matrix protein (M1). Types A and B cause epidemic human disease. Type A virus causes the most severe disease and is divided into subtypes on the basis of two surface antigens: haemagglutinin (HA) and neuraminidase (NA). Sixteen HA and nine NA subtypes have been identified, the most prevalent influenza A virus subtypes that infect humans are H1N1 and H3N2 [Reference Palese and Knipe5, Reference Ren6].
Genetic reassortment and accumulation of mutations in gene segments may result in ‘minor’ (antigenic drift) or ‘major’ (antigenic shift) changes that produce new virus strains. Antigenic drift strains escape from the host defence mechanism of acquired immunity and cause annual epidemics. The antigenic shift can lead to a pandemic if the novel subtype of influenza virus contains an HA protein to which humans have no pre-existing immunity and if it can readily spread from human to human by worldwide distribution, with serious consequences for human health [Reference Girard7].
Influenza vaccination is a key public health strategy for influenza prevention and control [Reference Cox and Subbarao8]. The vaccine includes two influenza A virus subtypes and one influenza B virus strain. Major targets are the highly conserved external domain of HA proteins that induce an HA-specific antibody immune response. The HA protein consists of two subunits, HA1 and HA2. HA1 contains at least five antigenic domains (A–E) and undergoes constant antigenic mutations that produce new variants that allow the virus to evade the humoral immune response of the host [Reference Hampson and Mackenzie9]. Continuous genetic mutations of HA1 cause an antigenic drift that modifies the antigenic properties over time [Reference Blackburne, Hay and Goldstein10]. Evolutionary analyses have provided significant information about the antigenicity, pathogenicity, receptor-binding specificity, glycosylation and drug susceptibility of the influenza virus [Reference Furuse and Oshitani11].
In Italy, the Italian Network for Surveillance of Influenza (INFLUNET), active since 2000, implements weekly updated clinical epidemiological surveillance [12, Reference Zizza13]. The World Health Organization (WHO) and the Italian National Influenza Center (NIC) guide virological surveillance activity. The National Institute of Health and the Inter-university Centre for Research on Influenza and other Viral Infections (CIRI-IV) in collaboration with the regional health authorities, contribute to the Italian National Influenza Surveillance system [12, 14].
In this study, virological analysis of influenza virus co-infections in humans and changes in the genetic variability of A(H3N2) virus strains in Apulia from 1999 to 2009 were examined. We performed sequence analysis of isolated influenza viruses to study the genetic differences and compared them with the vaccine strain.
METHODS
Human samples
From 1999 to 2009, 1269 oropharyngeal swabs were collected from patients with influenza-like illness (ILI), and demographic and clinical data were collected. Virological surveillance of influenza started on week 46 of each year and continued until week 17 of the following year. Sample collection was carried out during the influenza seasons in the province of Lecce (Apulia, Italy) by the epidemiological and virological sentinel network which is made up of 29 general practitioners (GPs) and nine paediatricians who survey about 1·3% of the total population.
An ILI case was defined as a person with sudden onset of fever >38°C accompanied by one or more respiratory symptoms (non-productive cough, sore throat, rhinitis) and one or more general symptoms (myalgia, headache, malaise, sweating, asthenia, shivers) [12, 14]. The ILI case definition used was always the same during the 10 years of surveillance.
Written informed consent was obtained from each enrolled patient or from their parents.
An oropharyngeal swab was taken under direct observation of the posterior throat and tonsil area using a Virocult® sterile swab with plastic shaft [Virocult, green cap, MW950; Medical Wire & Equipment Co. (Bath) Ltd, UK]. After the samples were obtained, swab applicators were cut and placed separately into two tubes containing Hank's balanced salt solution. The contents of the test tubes were frozen within 1 h and kept at −80°C until use.
RNA extraction PCR assays
Viral RNA was extracted from 250 μl of the sample using RNeasy Mini kit (Qiagen GmbH, Germany) following the manufacturer's instructions.
Different molecular methods for the detection of influenza A and B viruses were performed in different years, using primers specific for the M1 and NP genes of influenza A and B.
From 1999 to 2005, cDNA was synthesized by reverse transcription (RT) (RevertAid™ First Strand cDNA Synthesis kit, Fermentas Life Sciences, USA) and a nested RT–PCR was performed to subtype the influenza A viruses using specific primers provided by the Influcheck kit (Euroclone, Arrow Diagnostics S.r.l., Italy).
From 2005 to 2009 this method was replaced by a TaqMan Real-time PCR system in which both the RT and PCR reactions were performed in a single tube. Primers and probes of highly conserved sequences of the influenza A and influenza B matrix gene were used (Fast set InfA/InfB, Arrow Diagnostics S.r.l.). Two separate rooms, one for RNA extraction and one for amplification, were used to avoid cross-contamination of the samples. To subtype type A viruses, specific primers of types 1 and 3 HA gene were used.
Sequencing and phylogenetic analysis
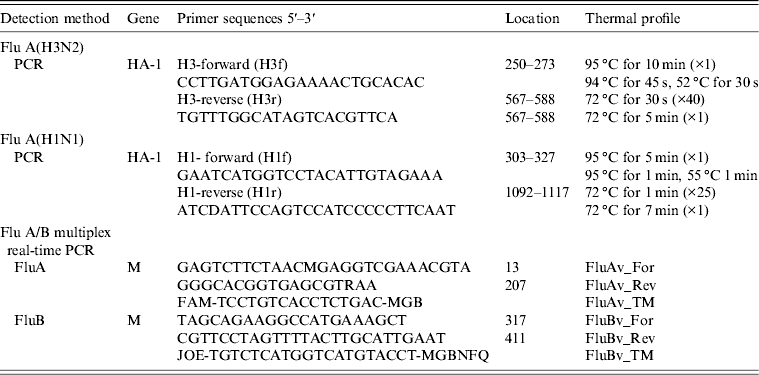
PCR products of the influenza A(H3) HA gene (nt 250–588, corresponding to a 340 bp fragment) was purified with QIAamp nucleic acid purification kit (Qiagen, Germany), according to the manufacturer's protocol. PCR products were sequenced directly or after cloning using the CEQ 8000 (Beckman Coulter) automated sequencer, with the same primers (H3f and H3r; see Table 1) used for the PCR. Phylogenetic affiliation of the obtained sequences was determined by using the BLAST search tool at the NCBI website (www.ncbi.nlm.nih.gov/blast). Multiple sequence alignment was conducted with ClustalW2 version 2.1 (www.ebi.ac.uk/clustalw) and a phylogenetic tree was constructed by the neighbour-joining method and a Kimura two-parameter model using Molecular Evolutionary Genetics Analysis (MEGA) software version 5.0 (www.megasoftware.net). A bootstrap resampling analysis was performed (1000 replicates) to test tree robustness.
Table 1. Flu A(H3) and Flu A(H1) primers and thermal profile used in viral genome amplification
RESULTS
Virological surveillance
During the period 1999–2009, 1269 oropharyngeal specimens were collected from patients with suspected influenza syndrome. However, 100 samples were not processed because they had been stored incorrectly. The typing of viruses isolated from the remaining 1169 samples showed the circulation of different influenza virus strains (Table 2).
Table 2. Results of virological surveillance in Apulia, Italy
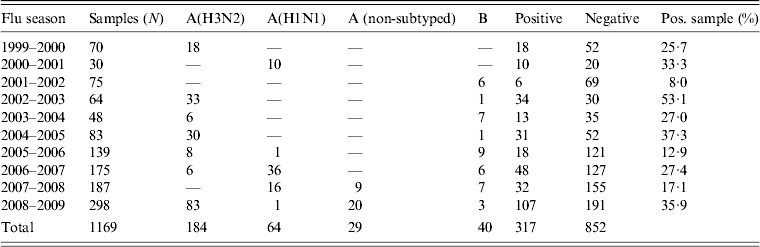
In particular, during the influenza seasons 1999–2000, 2002–2003, 2004–2005 and 2008–2009, the A(H3N2) subtype was the prevalent strain isolated. The A(H1N1) subtype virus was the prevalent strain during 2000–2001, 2006–2007 and 2007–2008 seasons and type B was prevalent in other seasons (2001–2002, 2003–2004, 2005–2006).
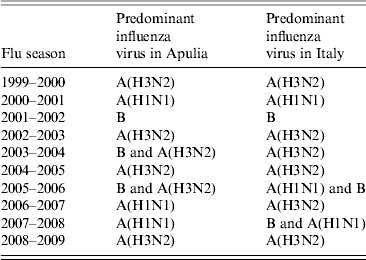
These data were compared with data regarding influenza circulation in Italy (Table 3). Virological surveillance in Apulia from 1999 to 2009 identified the same viruses circulating in Italy in many of the seasons with different strains only in the 2005–2006 and 2006–2007 seasons. In Italy, during the 2005–2006 season, A(H1N1) and B co-circulated, while in Apulia B and A(H3N2) were predominant; however, in the 2006–07 season, A(H3N2) in Italy and A(H1N1) circulated in Apulia [12].
Table 3. Predominant influenza viruses in Italy and Apulia from 1999 to 2009
Phylogenetic analysis of influenza A(H3N2) virus in southern Italy, between 2002 and 2009
The relative prevalence of influenza viruses varies from season to season. In southern Italy, influenza A(H3N2) was the prevalent strain for three seasons in the period between 1999 and 2005 and during the 2008–2009 season, while the A(H1N1) strain dominated during the 2000–2001, 2006–2007 and 2007–2008 seasons. In this study, the partial sequences of the HA gene (nt 250–588) from the A(H3N2) viruses circulating in Apulia in 2002–2009 and the HA reference sequences of vaccine strains from the GenBank database were used to perform a phylogenetic analysis and construct a phylogenetic tree (Fig. 1).

Fig. 1. Phylogenetic tree of the HA gene of the H3N2 influenza A viruses sampled in Apulia from 2002 to 2009.
Overall, HA sequences of the analysed H3N2 were segregated into distinct seasonal clusters. HA sequence (A/Lecce/620/03 and A/Lecce/954/03) isolated in Apulia during the 2002–2003 season appeared to be closely related to the sequence of the vaccine strain A/NewYork/55/01 virus. The 2002–2003 season was characterized by the co-circulation of two viral lineages: A/NewYork/55/01(H3N2)-like virus from the previous season and A/Fujian/411/02(H3N2)-like virus, a new H3 variant. The introduction of the A/Fujian/411/02(H3N2)-like virus caused a ‘jump’ in the evolution of the H3N2 viruses. In fact, in the next season 2003–2004, the HA sequence (A/Lecce/0304/04) isolated in Apulia formed a subclade in the A/Fujian/411/02(H3N2)-like lineage from the 2002–2003 season with the sequence of the vaccine strain A/Wyoming/03/03. In this season, the A/Wyoming/03/03(H3N2)-like virus was the predominant lineage co-circulating with the A/Wellington/1/04(H3N2)-like virus introduced from the southern hemisphere.
In the following season (2004–2005) the emergence of the A/California/7/04(H3N2) lineage caused a revision of the vaccine composition from A/Fujian/411/02(H3N2). Accordingly, the HA sequences of the viruses isolated in southern Italy (A/Lecce/0434/05, A/Lecce/0105/05, A/Lecce/321/05) during this season, were closely related to the A/Waikato/3/05 HA sequence that in the phylogenetic tree represents the A/California/7/04(H3N2) lineage. In season 2005–2006 the A/California/7/04(H3N2)-like virus continued to circulate together with the new A/Wisconsin/67/05(H3N2) lineage that became the distinctive lineage in season 2006–2007. As a result, the H3N2 vaccine component for the northern hemisphere in the 2006–2007 season was changed by A/Wisconsin/67/05(H3N2). In this respect, the HA sequences isolated in southern Italy in the 2005–2006 (A/Lecce/0106/06) and 2006–2007 (A/Lecce/462/07) seasons, appeared to be closely related to the A/Wisconsin/67/05(H3N2) lineage. Finally, in the 2007–2008 season the H3 nucleotide sequences of the viruses isolated in southern Italy (A/Lecce/423/08, A/Lecce/536/08) clustered with the sequence of the A/Brisbane/10/2007 virus (northern hemisphere vaccine strain for seasons 2008–2009 and 2009–2010) while, the H3 nucleotide sequences of viruses isolated in the 2008–2009 season (A/Lecce/04133/2009, A/Lecce/03102/2009) were closely related to the A/Perth/16/2009 lineage (southern hemisphere vaccine strain for season 2010–2011).
DISCUSSION
Influenza viruses are rapidly evolving viruses due to being genomically unstable, involving the surface glycoproteins in particular, especially HA. Human influenza A viruses evolve more rapidly than influenza B viruses [Reference Nobusawa and Sato15], while influenza A virus subtype H3N2 shows more amino-acid changes in antigenic sites than subtype H1N1 [Reference Ferguson, Galvani and Bush16]. The mutation rate of the non-structural (NS) genes of A and B viruses was estimated to be about 2·0 × 10−6 and 0·6 × 10−6 mutations/site per infectious cycle, respectively [Reference Nobusawa and Sato15]. However, the substitution rate of human influenza A(H3N2) and A(H1N1) viral sequences was estimated to be about 5·7 × 10−3 nucleotides per year for domain 1 of viral haemagglutinin (HA1) [Reference Fitch17] and on average 3·6 amino acids per year [Reference Smith18], and from 2·34 × 10−3 to 3·67 × 10−3 substitutions/site per year, respectively [Reference Smith19, Reference Smith20].
The rapid evolution of influenza A viruses reduces the effectiveness of vaccine prophylaxis, chemoprophylaxis or chemotherapy. Vaccine composition is based on the antigenic and genetic characteristics of circulating viruses; the occurrence of antigenic changes is of considerable importance in formulating vaccine composition. Immunity to surface antigens, mainly HA, reduces the possibility of infection. Antibody against one influenza virus type or subtype confers limited or no protection against another type or subtype of influenza virus. Furthermore, antibodies produced against one antigenic type or one subtype might not protect against infection due to a new antigenic variant of the same type or subtype [Reference Cox and Subbarao8]. Frequent emergence of antigenic variants through antigenic drift is the virological basis for seasonal epidemics and is the reason for annually reassessing the need to change one or more of the recommended strains in the influenza vaccines [Reference De Donno21].
The effective use of antiviral drugs can help reduce transmission rates of influenza and prevent acquisition. However, continuous genomic reassortment also favours the development of drug-resistant viral variants of H3N2, such as in single 3S1N amino-acid replacement in the M2 internal membrane protein, which functions as an ion channel [Reference Simonsen22, Reference Nelson23].
Evolutionary analysis can provide helpful information on the antigenicity, pathogenicity, and drug susceptibility of the influenza virus.
In this study, we have reported the genetic characterization of A(H3N2) influenza virus in southern Italy from 1999 to 2009. In Italy, the influenza surveillance system conducted on a regional basis allows collection of important epidemiological (i.e. incidence rates, immunization coverage rates) and virological (circulating strains) information.
In our study, virological surveillance showed that the ten epidemic seasons were characterized by a different antigenic and molecular pattern of circulating viruses. In all seasons, at least two viruses co-circulated. Usually, the presence of type B influenza virus and one of the subtype A viruses (H3N2 or H1N1) was observed. A simultaneous presence of three viruses was recorded in only three seasons (2005/2006, 2006/2007, 2008/2009). In many of the seasons studied, the circulating virus belonged to subtype H3N2 (40·0% of the strains identified), which showed a high variability. Genotyping of the isolated strains confirmed this result.
Similar data have been obtained in a study conducted in northern Italy which showed that 49·7% of circulating influenza strains were H3N2 [Reference Lai24]. Virological surveillance in Apulia from 1999 to 2009 identified the same viruses circulating in Italy in many of the seasons, with different strains only seen in the 2005–2006 and 2006–2007 seasons [12, Reference Lai24].
Genetic and phylogenetic analysis of influenza A viruses circulating in our region between 1999 and 2009 confirmed that the genetic structure of influenza A viruses may change from year to year. Usually, the morbidity level varies during the seasons and it is possible to observe some years with low morbidity levels and other years with high morbidity levels. The circulation of new antigenic variants cause annual epidemics characterized by high morbidity, increased hospitalization and mortality [Reference Gabutti25]. Our data, published in a previous paper, showed that in the Apulia region in the 1999/2000 season (Sydney/05/97-like strains), 2002/2003 season (sustained by the new H3N2 variant, A/Fujian/411/02), 2004/2005 season (when a new variant of the H3N2 strain emerged; A/California/07/04) and 2008/2009 season (caused by a new strain of H3N2 virus identified in Australia; A/Brisbane/59/2007), in which subtype H3N2 was prevalent, the morbidity rate was higher [Reference Campa26].
In all analysed seasons, the isolated H3N2 strains were compared with the vaccine strain of each corresponding influenza season. Molecular analysis showed that the H3N2 virus strains, sequenced in this study, did not have any significant mutations and were genetically close to the vaccine strains.
A limitation of the study could be the low number of samples and the short sequence of sequenced nucleotides that may not underline the presence of mutations in important regions for the link between HA and human receptors. Previously, Russell et al. [Reference Russell27] observed H3N2 antigenic variation during an epidemic in different regions and from year to year in the same region.
Despite such spatial and temporal heterogeneities, antigenic evolution has been markedly homogeneous on a global scale. This homogeneity could be due to viruses that circulate globally rather than persist and evolve locally.
Therefore, in our local study, we have not shown significant changes in the gene sequences analysed. One explanation for this could be related to the worldwide dissemination of the same strain or linked to a period of relative stasis (from 3 to 8 years), observed in the antigenic evolution of H3N2 virus [Reference Smith18].
Despite the advances in understanding the antigenic evolution of influenza viruses, more precise knowledge, through careful surveillance to assess the dynamic of the antigenic and genetic changes of circulating strains, may help predict the following season's strains and optimize vaccine strain selection.
ACKNOWLEDGEMENTS
The authors thank the Influenza Collaborative Group: V. Accogli, M. Alba, A. Andrani, M. Antonaci, M. Calsolaro, M. Caricato, C. Casile, R. Costantino, A. Cosi, A. De Benedictis, D. De Giorgi, L. De Giovanni, D. Fiume, D. Frisenna, F. Gerbino, F. Ghionda, M. Greco, C. Inguscio, G. Leuzzi, C. Lillo, C. Macchia, E. Malorgio, C. Mariano, A. Metrucci, S. Minerva, E. Mola, L. Nicoletti, V. Riso, R. Tedesco, and F. Trono for their availability in sample and data collection.
DECLARATION OF INTEREST
None.




