


Rabies is caused by a virus within the genus Lyssavirus of the family Rhabdoviridae. According to the official ICTV Master Species List 2009 – version 6, the genus Lyssavirus is composed of 11 species: Aravan virus (ARAV), Australian bat lyssavirus (ABLV), Duvenhage virus (DUUV), European bat lyssavirus 1 (EBLV-1), European bat lyssavirus 2 (EBLV-2), Irkut virus (IRKV), Khujand virus (KHUV), Lagos bat virus (LBV), Mokola virus (MOKV), rabies virus (RABV) and West Caucasian bat virus (WCBV) [1]. RABV is the only lyssavirus present in terrestrial mammals throughout the world and associated with bats only in the Americas.
The RABV genome consists of a single-stranded, non-segmented, negative-sense RNA of ~12 kb in size and encodes for five structural proteins: the nucleoprotein (N protein), phosphoprotein (P protein), matrix protein (M protein), glycoprotein (G protein), and the RNA-dependent RNA-polymerase (L protein). The N, P, and L proteins form the internal helically packaged ribonucleocapsid complex (RNP) while the M and G proteins form the inner and outer lipid bilayer envelopes surrounding the RNP core, respectively [Reference Wunner2]. Many of these genes have been targeted for molecular studies. The N genes are highly conserved and have been extensively employed for rabies diagnosis using reverse transcription–polymerase chain reaction (RT–PCR) and other genetic analyses [Reference Denduangboripant3–Reference Bourhy6]. These studies have provided clearer understanding of the distribution of RABV throughout the world.
Rabies is a fatal zoonotic disease. It is endemic in Asia, where it causes about 31 500 human deaths each year despite the availability of effective vaccines [Reference Knobel7]. Of these cases, an estimated 20 000 human deaths occur every year in India alone. Lack of comprehensive rabies control programmes and an inability to pay for post-exposure treatments after dog bites are responsible for high incidences of rabies in Asian countries [Reference Knobel7, Reference Wilde8]. Over the past decade many molecular epidemiological studies have been conducted in Asian countries, and have provided a better understanding of the RABV variants circulating in Asia and the transmission dynamics of the disease [Reference Denduangboripant3, Reference Nanayakkara, Smith and Rupprecht9–Reference Hyun13]. This information is needed for implementation of effective rabies control programmes in the region.
Bhutan is located between China and India and canine rabies is prevalent in southern Bhutan along the border with India [Reference Tenzin14]. As in other Asian countries, domestic dogs in Bhutan play an important role in the maintenance and transmission of the disease to other domestic animals and occasionally to humans. In recent years, rabies outbreaks have increasingly occurred in endemic southern Bhutan as well as in some other areas where rabies had not been reported previously [Reference Tenzin14]. Despite frequent outbreaks, molecular epidemiological studies of rabies in Bhutan have not been undertaken and the RABV variants circulating in the country are unknown.
In the present study, we performed genetic characterization of RABV based on the partial nucleoprotein (N) gene. A phylogenetic analysis of these N gene sequences was performed in order to investigate their genetic relationship with other RABV variants circulating in the world, especially in Asia. The information generated from this research could help in planning a more effective rabies control programme in Bhutan.
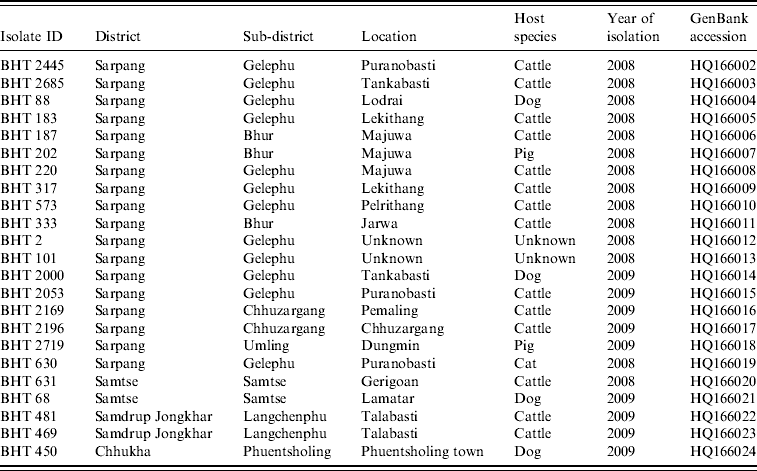
Twenty-three fresh brain tissue samples were obtained from cattle (16), dogs (4), cat (1), and pigs (2) that died of clinically confirmed rabies from four southern districts (Samtse, Chhukha, Sarpang, Samdrup Jongkhar) in Bhutan during 2008 and 2009 (Table 1, Fig. 1). These samples were collected from areas near the border between Bhutan and the Indian states of Assam and West Bengal. Rabies was confirmed in each case by the fluorescent antibody test (FAT). For RT–PCR and nucleotide sequencing, ~10 mg of each FAT-positive brain tissue sample was smeared onto FTA® Gene Guard System (a commercial product consisting of filter paper impregnated with patented chemicals supplied by Whatman, USA), air-dried, and then transferred to the laboratory (WHO Collaborating Centre for Research and Training on Viral Zoonoses, Faculty of Medicine, Chulalongkorn University) in Thailand.
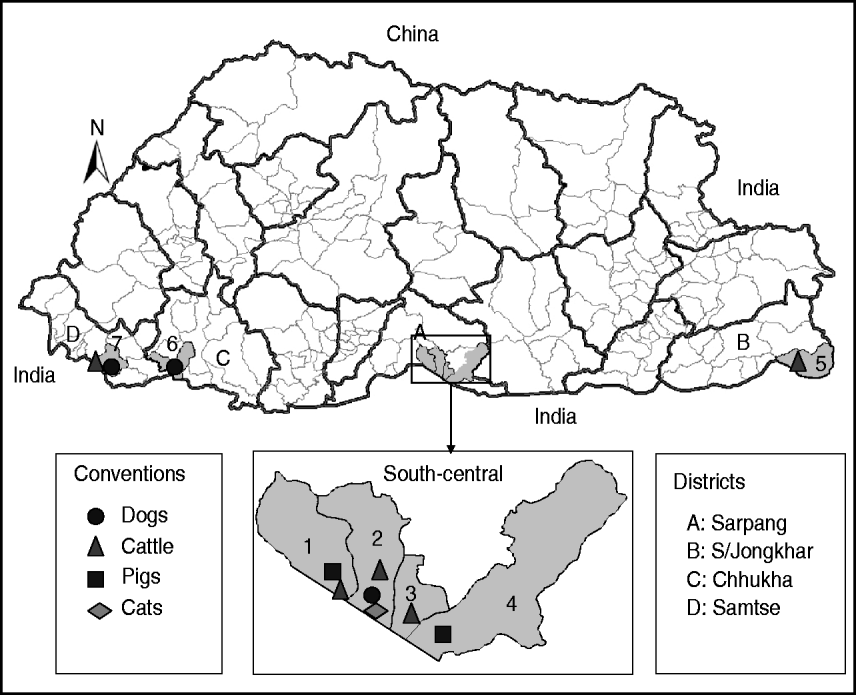
Fig. 1. Map of Bhutan showing the geographical locations where the RABV isolates were obtained from different species of animals. Thick and thin lines on the map indicate the district and sub-district boundaries, respectively. Numerical values on the map indicate the names of sub-districts where RABV isolates were obtained (1, Bhur; 2, Gelephu; 3, Chhuzargang; 4, Umling; 5, Langchenphu; 6, Phuentsholing; 7, Samtse; see Table 1 for details). Symbols represent the species from which the virus was isolated.
Table 1. Rabies virus isolates obtained from Bhutan and used in the present study
The brain tissue sample from the filter paper was eluted by rocking each dried brain spot in 9 ml lysis buffer (NucliSens; bioMérieux, The Netherlands) at room temperature with a rotator mixer (RS-60; Biosan, Latvia) at 60 rpm for a period of 2 h. The filter paper was then removed from the buffer solution.
Total RNA extraction from eluted specimens was performed by using a silica-guanidine thiocyanate protocol, NucliSens isolation reagent (bioMérieux) according to the manufacturer's instructions. Single-step RT–PCR was performed using the One Step RT–PCR kit (Qiagen GmbH, Germany). Specific sense primer CN8 [3′-GT(TC) GGA TGT TAT ATG GG-5′, nt 1013–1029] and an antisense primer CN4 [3′-GGA TTG AC(AG) AAG ATC TTG CTC AT-5′, nt 1514–1536] were used for the N gene amplification. Binding sites of the primers were referred to the positions of total genomic sequence of the Pasteur Virus (PV) strain of RABV (GenBank accession no. M13215). One cycle of reverse transcription was done at 50°C for 35 min, followed by denaturation at 95°C for 15 min. PCR was followed by 40 amplification cycles of denaturation at 94°C for 1 min, primer annealing at 50°C for 1 min, and extension at 72°C for 1 min. Finally a 10-min extension step at 72°C was done to complete the amplification of the target gene. The final PCR products were run in a 2% agarose gel electrophoresis in TBE 1x buffer stained with ethidium bromide (at 1 μg/μl), and viewed under UV light to observe the specific bands.
Gel slices containing RT–PCR products were excised from the gel and the RT–PCR products were purified using QIAquick PCR Purification kit (Qiagen Ltd, UK) according to the manufacturer's instruction. Direct sequencing of the N gene was performed using primers CN4 and CN8 with ABI PRISM Big Dye Terminator Cycle Sequencing kit (Applied Biosystems, USA) and ABI PRISM 310 DNA sequencer (Applied Biosystems). For each RABV isolate, 395 nt of N gene corresponding to position 1101–1495 of the PV genome (GenBank accession no. M13215) were analysed. All nucleotide sequences generated in this study were submitted to GenBank and their accession numbers were assigned (Table 1).
Multiple sequence alignment was performed using the MUSCLE version 3.8 program [Reference Edgar15]. A neighbour-joining (NJ) analysis employing a Kimura two-parameter model with bootstrap statistic test of the phylogenetic tree (1000 replicates) was performed in the MEGA version 4.0.2 program [Reference Tamura16]. Thirty-seven additional N gene sequences of RABV previously published were retrieved from GenBank and used for comparison. The GenBank accession numbers and other details of the sequences are shown in Figure 2. Bootstrap values of >50% are shown on the tree branches. The Tree Explorer module in MEGA 4.0 was used to obtain the graphic output. Phylogenetic analysis using the Bayesian Markov Chain Monte Carlo (MCMC) method was also implemented in the MrBayes version 3.1 program [Reference Huelsenbeck and Ronquist17] with a GTR+I+gamma evolutionary model. The analysis was run for 2000 000 generations to obtain 200 000 samples from the posterior probability distribution. Posterior probability values >0·95 were considered significant. The estimated tree topology was illustrated using the Treeview program.
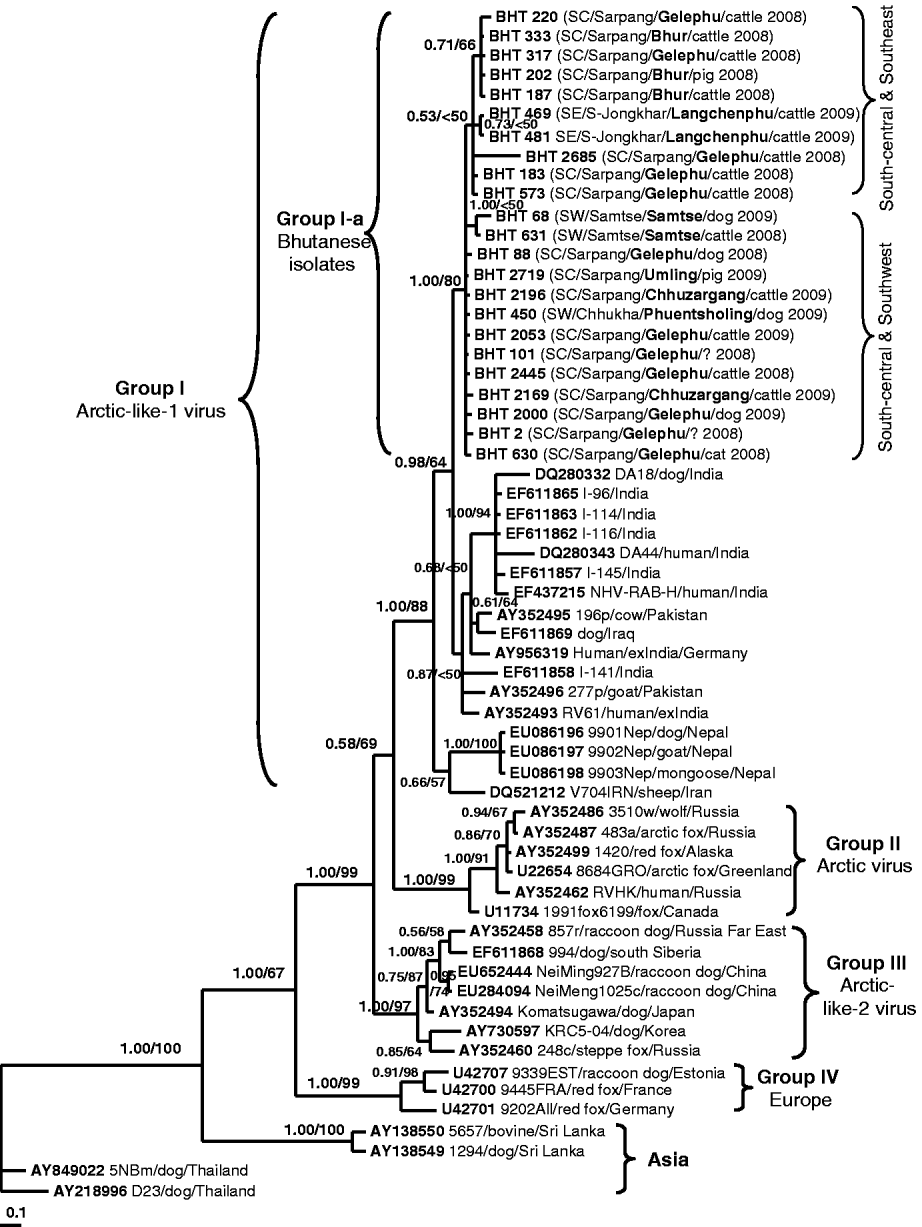
Fig. 2. A phylogenetic tree of 60 RABV isolates analysed in this study. The tree was inferred by the Bayesian MCMC phylogenetic analysis method using a partial nucleoprotein (N) gene (395 bp) of 23 Bhutanese isolates compared to other N rabies sequences obtained from the GenBank database. The posterior probability value of each node is shown along the tree branches. Bootstrap branch-support values >50% (with 1000 replicates) generated from neighbour-joining (NJ) analysis is also indicated. A scale of base substitution number per site is shown at the bottom of the tree. Additional information of each Bhutanese sequence is mapped in parentheses as region/district/sub-district/host species/year of isolation (SW, Southwest; SC, South-central; SE, Southeast). The isolate ID, host species, country of isolation, and GenBank accession numbers of other rabies sequences are also given for examination. Various groupings found in this study are described in the text.
The geographical distribution of the Bhutanese rabies isolates were mapped using Bhutan boundary shape files in ArcGIS 9.3 (ESRI, USA).
Figure 2 shows the Bayesian phylogenetic tree inferred by comparing the partial N gene sequences of 23 Bhutanese isolates with 37 RABV N gene sequences available in GenBank. The topology of the NJ tree (result not shown) was similar to that of the Bayesian phylogeny and only its bootstrap values were mapped on the Bayesian tree. The tree shows four significant genogroups which were supported with posterior probability values >0·95. Group I consisted of RABV isolates from Iraq, Iran, Pakistan, India, and Nepal with an 88% bootstrap supporting value. These isolates were previously described as Arctic-like-1 viruses circulating in the Middle East and South Asia [Reference Kuzmin5, Reference Nadin-Davis18, Reference Kuzmin19]. The Bhutanese isolates formed a monophyletic cluster (labelled as Group I-a) within this Arctic-like-1 virus group. Group II was composed of six Arctic virus isolates originating from Russia, Canada, Alaska, and Greenland (bootstrap value of 99%). Group III consisted of seven viruses originating from Korea, Japan (formerly), southern Siberia, Far East Russia, and northern China (Inner Mongolia) with a 97% bootstrap value. These isolates were previously described as Arctic-like-2 viruses circulating in northeastern Asia [Reference Kuzmin5, Reference Hyun13, Reference Nadin-Davis18, Reference Kuzmin19]. Three isolates from Europe formed Group IV with 99% bootstrap support value, and the other four isolates from Asia (Thailand and Sri Lanka) formed separate clusters according to the country of origin. The phylogenetic analysis clearly revealed that all Bhutanese isolates were closely related to Indian strains and formed a large cluster with the Arctic-like-1 viruses, and were distinct from other Asian RABV isolates from Thailand and Sri Lanka.
This is the first report characterizing the molecular epidemiology of RABV isolates in Bhutan. The result confirmed that all isolates from Bhutan belong to the RABV and were not related to other rabies-related viruses (e.g. from bats) of the Lyssavirus genus. Our analysis showed that the Bhutanese isolates were highly similar and did not form any distinct subgroups although the isolates originated from different geographical areas. This suggests that the RABV variants circulating in southern Bhutan originated from a single common ancestor. Even though the Bhutanese isolates did not form clear subgroups due to low bootstrap values along most branches, they tended to separate into two subgroups based on the geographical locations of the isolates: the Southeast/Central and Southwest/Central subgroups (Fig. 2, see Table 1 for details). However, more sequences from both the southeast and southwest areas of Bhutan would be needed to provide greater phylogenetic support and confirm our hypothesis.
Phylogenetic analyses revealed that Bhutanese RABV isolates were more closely related to the RABV strains from India and Arctic-like-1 viruses circulating in South Asia [Reference Nadin-Davis18, Reference Kuzmin19], and could be grouped together as a large cluster of the South Asian Arctic-like-1 viruses (Group I in Fig. 2). Geographically, Bhutan lies in the same Himalayan region of the Indian sub-continent where the emergence and extensive circulation of Arctic-like-1 rabies viruses have been confirmed in India [Reference Nadin-Davis18]. However, it should be noted that the Indian rabies sequences used for comparison in this study originated mostly from the southern part of India (including a foreigner who died of confirmed rabies after being bitten by a dog in southern India). Even though southern India is far away from Bhutan, it is interesting to observe that Bhutanese and Indian RABV isolates share close genetic relationships, which suggests that they have originated from a common ancestor, probably the Indian strain. Translocation of infected dogs via human activities may be responsible for the spread of RABV between the two countries. Previous studies [Reference Denduangboripant3, Reference Susetya11, Reference Gong12], have hypothesized the translocation of dogs together with their owners migrating for work and trade purposes as a probable mechanism for the spread of RABV from country-to-country or region-to-region in some South East Asian countries (e.g. Thailand, Vietnam, Indonesia).
In the present study, a direct comparison between rabies isolates from Bhutan and those from northeast India (which share a porous border) was not possible because there are no published sequence data available from northeast India. It may be that a similar RABV variant is circulating in dogs in northeast India. This hypothesis needs to be confirmed by surveillance and the performance of molecular studies and genetic analyses.
The limited distribution of RABV within the southern areas of Bhutan provides important information for planning and implementing a rabies control programme in the country. It implies that successful control and even elimination of rabies might be achievable in Bhutan if a sustained vaccination campaign of domestic dogs (>70% coverage) and dog population management is carried out efficiently. Control of rabies within the domestic dog cycle would break the chain of transmission (spillover) into other animals, and would eventually eliminate the disease from the country.
In conclusion, we recommend that more sampling of virus from potential reservoirs from different rabies outbreak areas and time periods is needed to provide detailed information about RABV transmission dynamics in Bhutan.
ACKNOWLEDGEMENTS
We thank The Thailand Research Fund (grant no. DBG5180026) for partial funding of this research work. We also thank Jambay Dorjee, Kesang Jigme and Lungten for providing brain tissue specimens from southeast and southwest Bhutan.
DECLARATION OF INTEREST
None.



