


INTRODUCTION
Avian influenza virus (AIV) has two major surface proteins, the haemagglutinin (H) and neuraminidase (N) [Reference Alexander1], whose coding genes are subject to a rapid rate of change [Reference Chen and Holmes2]. To date, 16 H and nine N subtypes of AIV have been recognized. The pattern of genomic change and the geographical separation of viral lineages make it possible to perform molecular tracing of AIVs [Reference Alexander1–Reference Munster4]. In addition to wild-bird AIVs [Reference Munster4], the molecular epidemiology of outbreaks of avian influenza (AI) in domestic birds has been studied [Reference Perk, Schudel and Lombard5]. In Europe, close antigenic and genetic similarity was observed between the H genes of low-pathogenic avian influenza viruses (LPAIVs) detected in wild mallard and those of highly pathogenic AIVs from all contemporaneous outbreaks of highly pathogenic AI in poultry [Reference Munster4]. It has also been demonstrated that the outbreak of H9N2 LPAI in poultry in Israel during 2000–2004 was the result of three separate viral introductions [Reference Perk, Schudel and Lombard5]. The source of these introductions was considered to have been wild waterfowl [Reference Perk, Schudel and Lombard5].
Although it is possible that LPAIV may have been introduced to Ireland from both North America and Europe by migratory birds [Reference Wernham6, Reference Crowe and Merne7], phylogenetic data has not been available to establish the source of AIVs in this country. The isolation of a number of LPAIVs from wild birds [Reference Raleigh8] provided an opportunity to study their likely origin and to compare viruses from wild birds to those detected in farmed birds during the same period.
The purpose of the work reported here was to determine the most likely origin of LPAIVs detected in wild birds in Ireland, to establish if there was year-to-year viral persistence and to compare AIVs from migratory wild birds with those from sedentary mallard and poultry.
METHODS
Viruses
A total of 35 AIVs, isolated in Ireland between 2003 and 2007, and RNA from one specimen from which no isolation was possible, was used in the study (Table 1) [Reference Raleigh8]. Virus isolation and serological characterization of all subtypes was performed according to the protocol stipulated in Directive EC 92/40 (1992), with subtyping confirmed by reverse transcription–polymerase chain reaction (RT–PCR) [Reference Raleigh8]. Thirty-four AIVs were from wild waterfowl (all dabbling ducks), one was from free-range commercial chickens and the other was from a flock of farmed mallard. All but one of the wild-bird viruses had been obtained from hunted birds at the Wexford Sloblands during 2003–2007 and all had been detected during the winter months [Reference Raleigh8].
Table 1. Avian influenza virus subtypes isolated from samples collected from waterfowl in Ireland during 2003–2007Footnote *
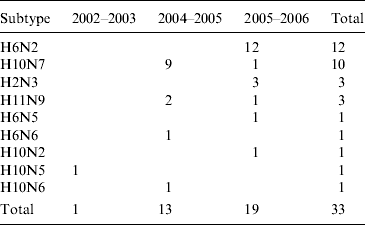
* In addition, the H gene from one H5 virus detected in 2007 (which could not be isolated) was sequenced directly from cDNA (not in table). One H6N2 LPAIV was isolated from commercial chickens (not in table). One H11N7 LPAIV was isolated from farmed mallard (not in table). Samples were not collected during the winter of 2003/2004.
Extraction of RNA
Viral RNA was extracted using the Qiamp RNA viral mini kit (cat. no. 52906 Qiagen Ltd, UK) according to the manufacturer's instructions.
RT–PCR
Amplification of the H gene was achieved by modified RT–PCR protocols based on published methods [Reference Lee9–Reference Tsukamoto12] (Table 2). The main modification consisted of combining primers from different protocols in order to ensure that sufficiently large gene fragments were amplified (Table 2). All H RT–PCR assays were performed in a 50 μl reaction volume, using the Qiagen One-step RT–PCR kit containing Qiagen One-step RT–PCR enzyme mix, 5× Qiagen One-step buffer, dNTP mix, RNase-free water, 5 μl RNA, and a final concentration of 10 pmol of each forward and reverse primer.
Table 2. Details of primers used to amplify segments of the H and N2 genes of AIVs detected in Ireland during 2003–2007
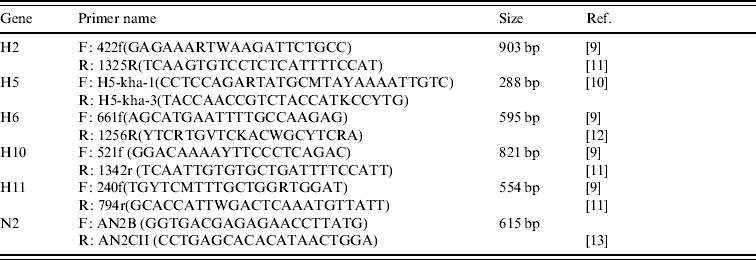
Amplification was performed using an MJ Research Engine thermocycler (MJ Research Inc., USA) with the following cycling conditions: a reverse transcription cycle of 15 min at 48°C followed by 30 min at 50°C, followed by an activation cycle (of the Taq polymerase) of 95°C for 15 min, followed by seven cycles of denaturation at 94°C for 20 s, annealing at 60°C for 20 s (decreasing by 1°C per cycle) and extension at 72°C for 1 min. This was followed by 38 cycles of denaturation at 94°C for 20 s, annealing at 54°C for 20 s and extension at 72°C for 1 min followed by a final extension at 72°C for 10 min.
Amplification of the N2 gene was performed as described by Takao et al. [Reference Takao13].
Identification of H and N subtypes by gel electrophoresis
PCR products were visualized in a 2% agarose gel using SYBR® Safe DNA gel stain (Invitrogen Corp., UK) and a permanent photographic record was made using the Fluorochem Gel Documentation System (Flourochem SP, USA). A 100-bp DNA ladder (Invitrogen Corp.) was included in each electrophoresis run.
Sequencing
PCR products were purified using the GenElute PCR Clean-Up kit (Sigma-Aldrich, Ireland) according to the manufacturer's instructions.
Sequencing was performed on each purified DNA fragment using the forward and reverse primers employed in the RT–PCR assay. Sequencing was based on DYEnamic ET Terminator protocols (GE Healthcare UK Ltd, UK). The reaction mix in each tube contained 4 μl DNA Template (PCR product), 8 μl Dye Terminator reaction premix and 5 pmol primer. The reaction mix was made up to a total volume of 20 μl with ultra-pure H2O. The tubes were mixed thoroughly and centrifuged at 750 g. The cycle sequence was performed in an MJ Research thermocycler (MJ Research Inc.). Sequencing reactions with DYEnamic ET Terminators were performed using the following programme: 95°C for 20 s; 55°C for 15 s; 60°C for 1 min for 25 sequencing cycles.
Excess Dye Terminators were removed from the sequencing reaction mixtures using SigmaSpin Post-Reaction Purification Columns (Sigma-Aldrich) according to the manufacturer's instructions. The final sequencing reaction mixtures were homogenized by vortexing and transferred to a 96-well plate (AbgeneUK).
Sequencing was performed using a MegaBACE 1000 DNA sequencer (GE Healthcare UK Ltd). Samples were injected into the MegaBACE at 3 kV for 75 s, and separation was carried out at 6 kV for 200 min.
Phylogenetic analysis
A segment of the H gene of each subtype and the N gene of N2 subtypes was sequenced twice using forward and reverse primers, followed by the generation of consensus sequences using the Seqman Pro program of the Lasergene 8 Analysis software suite (DNASTAR Inc., USA).
Homology searches of the Influenza Virus Resource of the National Centre for Biotechnology Information (NCBI) [Reference Bao14] Entrez Nucleotide database were performed with the Basic Local Alignment Search Tool (BLAST) of the Megalign program (DNASTAR Inc.). Importation of Irish and NCBI sequences was performed using the Entrez sequence retrieval system of Megalign. Multiple alignment of the top matching sequences was performed using the ClustalW (Slow, accurate, IUB) alignment algorithm [Reference Thompson, Higgins and Gibson15] in the Megalign program (DNASTAR Inc.). Sequences were then imported into Mega 4 [Reference Tamura16], realigned by the ClustalW method and trimmed before phylogenetic analysis.
The bootstrap consensus tree, inferred from 1000 replicates [Reference Felsenstein17], was constructed using the neighbour-joining method [Reference Saitou and Nei18]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches [Reference Felsenstein17]. The trees were drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method [Reference Tamura, Nei and Kumar19] and are in the units of the number of base substitutions per site. Codon positions included were 1st+2nd+3rd+non-coding. All positions containing gaps and missing data were eliminated from the dataset (complete deletion option).
RESULTS
PCR products
PCR products were obtained for an H gene segment of each subtype detected between 2003 and 2007. Sequences of the N gene were also obtained for the N2 subtypes of these viruses.
Phylogenetic analysis of H genes
A group of three almost identical H2N3 viruses was isolated from wild birds at the Wexford Sloblands in the winter of 2005/2006. A 98·8% identity with A/mallard/Sweden/45/2005(H2N3) and 98·4% with A/wigeon/Norway/2006(H2N6) (Table 3) were the closest matches with European sequences while the most closely aligned North American wild-bird sequence (89·8%) was A/herring gull/Delaware/670/1988(H2N9) (Table 4).
Table 3. Comparison of sequence identity of the H genes of Irish LPAIVs with Eurasian viruses
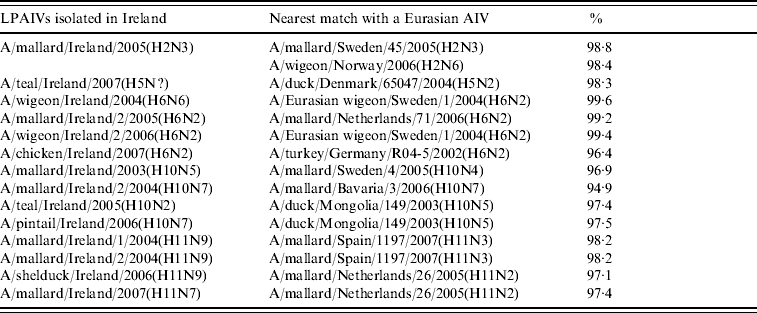
Table 4. Comparison of sequence identity of the H genes of Irish LPAIVs with North American viruses

An H5 gene was sequenced directly from RNA extracted from a cloacal swab collected from a wild teal during 2007. This sequence aligned very closely to those of European lineage, 98·3% identity with A/duck/Denmark/65047/2004(H5N2) (Table 3) being the nearest match.
H6 subtype AIVs isolated from wild birds comprised H6N6 (n=1) in 2004/2005, H6N2 (n=12) and H6N5 (n=1) in 2005/2006 (Table 1). All of these wild-bird viruses were isolated at the Wexford Sloblands [Reference Raleigh8]. In 2005/2006, H6N2 viruses formed two distinct clades (Fig. 1), consisting of viruses (n=9) with ⩾99·8% identity with each other isolated in November–early December 2005 and AIVs with ⩾99·4% identity with each other (n=3) but ⩽97·3% identity with the 2005 group isolated at the end of January 2006. The H gene of the H6N5 isolate from November 2005 was similar to (⩾99·8%) that of the H6N2 viruses of that period. The gene of the 2006 H6N2 AIVs was similar (99·6%) to that of the H6N6 viruses from November 2004. H6 sequences were less divergent from North American strains than other subtypes. The closest match between an Irish and Eurasian wild-bird AIV was A/wigeon/Sweden/1/2004(H6N2) (99·6%) (Table 3) while A/mallard/Minnesota/Sg-00220/2007(H6N1) (96·8%) was the most closely related North American sequence (Table 4). One H6N2 LPAIV was isolated from chickens in 2007. The H gene of this chicken-derived H6N2 AIV had ⩽94·0% identity with wild-bird subtypes in Ireland and 96·4% similarity to LPAIVs from poultry in Germany (Fig. 1).
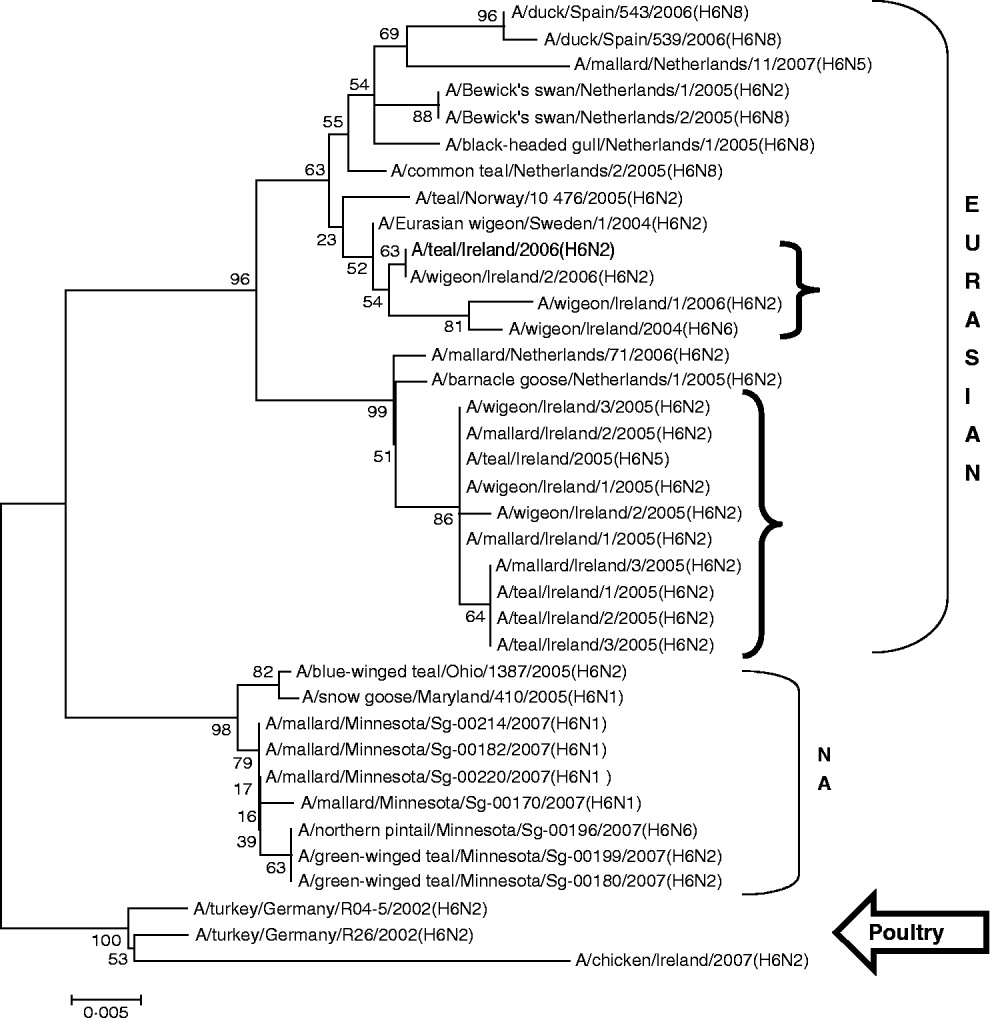
Fig. 1. Phylogenetic tree of H6 AIVs showing the evolutionary relationships of sequences of Irish isolates (![]() ) obtained between 2003 and 2007 with sequences of Eurasian and North American (NA) AIVs. The taxa were chosen from the closest matches to the Irish H6 sequence by homology searching.
) obtained between 2003 and 2007 with sequences of Eurasian and North American (NA) AIVs. The taxa were chosen from the closest matches to the Irish H6 sequence by homology searching.
H10 AIVs comprised 13 isolates from wild birds; H10N5 (n=1) in 2003/2004, H10N7 (n=9) and H10N6 (n=1) in 2004/2005, H10N2 (n=1) and H10N7 (n=1) in 2005/2006 (Table 1). The closest similarity of the 2003 isolate with a 2004/2005 isolate was 94·6%, while all the 2004/2005 viruses shared >99·7% identity with each other. The two 2005/2006 isolates shared 99·7% identity with each other but only 96·4% identity with the 2004/2005 isolates. The gene sequences were most closely aligned with Eurasian lineages of AIV (Fig. 2). The closest agreement (97·5%) was with an H10N5 sequence from Mongolia (Table 3). The most similar North American virus was a teal-derived H10N7 LPAIV from Louisiana, with which there was 81·8% agreement (Table 4). The H gene of the H10N2 virus from 2005 was almost identical to that of the H10N7 AIV isolated during the same over-wintering period.
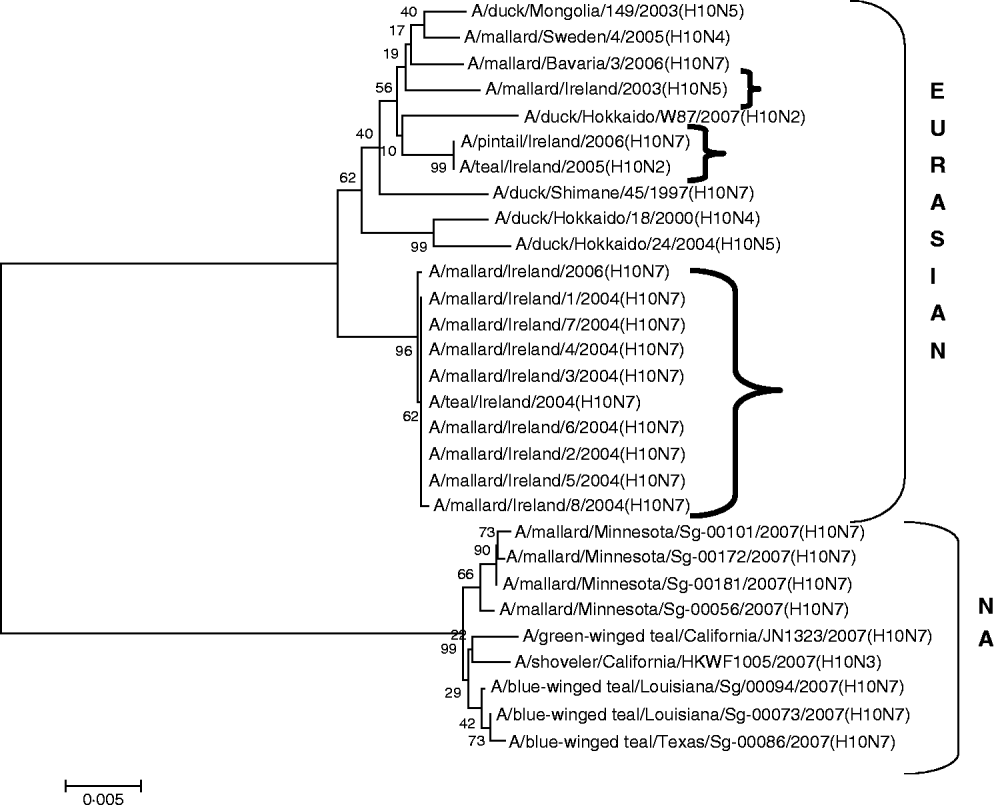
Fig. 2. Phylogenetic tree of H10 AIVs showing the evolutionary relationships of sequences of Irish isolates (![]() ) obtained between 2003 and 2007 with sequences of Eurasian and North American (NA) AIVs. The taxa were chosen from the closest matches to the Irish H10 sequence by homology searching.
) obtained between 2003 and 2007 with sequences of Eurasian and North American (NA) AIVs. The taxa were chosen from the closest matches to the Irish H10 sequence by homology searching.
H11 AIVs comprised four isolates from wild birds, H11N9 (n=2) with 98·2% agreement with each other in 2004/2005, H11N9 (n=1) in 2005/2006 with <97% agreement with the 2004/2005 isolates and H11N7 (n=1) in 2007. The H gene of the H11N7 AIV isolated from farmed mallard in the summer of 2007 shared 99·6% identity with that of the H11N9 virus obtained from a ruddy shelduck in March 2006. The H11 gene sequences were most closely aligned with Eurasian lineages of AIV (Fig. 3). The greatest similarity (98·2%) was with an H11N3 virus from Spain (Table 3) while the closest agreement (81·8%) with a North American virus was with an H11N9 subtype from Ohio (Table 4).
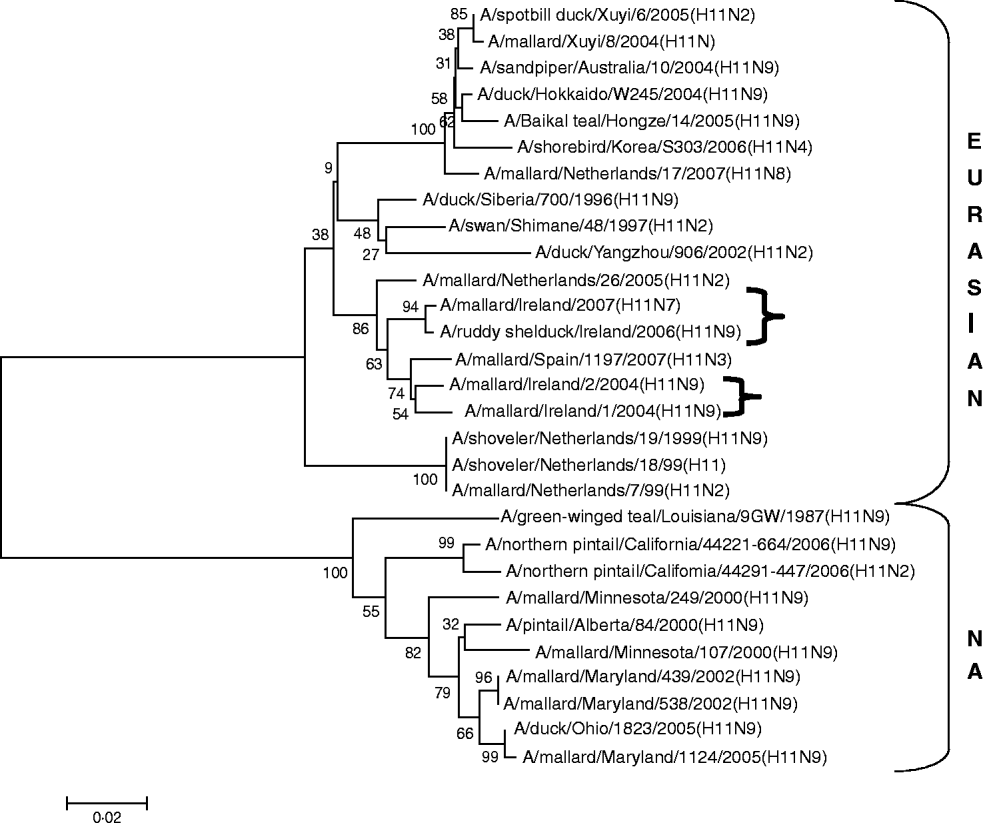
Fig. 3. Phylogenetic tree of H11 AIVs showing the evolutionary relationships of sequences of Irish isolates (![]() ) obtained between 2003 and 2007 with sequences of Eurasian and North American (NA) AIVs. The taxa were chosen from the closest matches to the Irish H11 sequences by homology searching.
) obtained between 2003 and 2007 with sequences of Eurasian and North American (NA) AIVs. The taxa were chosen from the closest matches to the Irish H11 sequences by homology searching.
Phylogenetic analysis of N2 genes
A total of 13 N2 AIVs was isolated from wild birds, H6N2 (n=12) and H10N2 (n=1) in 2005/2006 (Table 1). In 2005/2006, H6N2 viruses formed two distinct clades (Fig. 1), consisting of viruses (n=9) having ⩾99·4% identity with each other isolated in November–early December 2005 and AIVs with ⩾99·4% identity with each other (n=3) but ⩽92% identity with the 2005 group isolated at the end of January 2006. One H6N2 AIV (n=1) was isolated from commercial chickens in 2007. Gene sequences from wild birds in Ireland aligned most closely with published Eurasian sequences of AIV. The N gene of the H10N2 AIV from the 2005/2006 season was different (92% agreement) from that of the predominant H6N2 subtype of that season and from the H6N2 viruses isolated in January 2006. The N gene of the H6N2 LPAIV isolated from poultry in 2007 shared 93% identity with the 2005 wild bird viruses and 96% identity with those of 2006. The closest agreement (99·8%) between the N2 of a wild bird isolate from Ireland with a published sequence was with an H6N2 virus from Sweden (Table 5) while the closest agreement with a North American virus was 93·2% with an H3N2 AIV from Ohio (Table 6).
Table 5. Comparison of sequence identity of the N2 genes of Irish LPAIVs with Eurasian viruses

Table 6. Comparison of sequence identity of the N2 gene of Irish LPAIVs with North American viruses

DISCUSSION
In this study, analysis of gene sequences of AIVs supported the previously reported existence of separate Eurasian and North American lineages of LPAIVs [Reference Donis20–Reference Olsen22]. The results support the theory that LPAIVs circulating in wild birds in Ireland had been introduced from Eurasia rather than North America [Reference Raleigh8]. However, the findings also strongly suggest that there was viral persistence from year to year and that the introduction of poultry viruses may not have been from wild birds.
All AIVs detected in Ireland during this period aligned more closely with Eurasian than with North American sequences published on the NCBI database. The evidence provided by H gene sequences of H10 and H11 subtypes of AIV, detected during this study, was particularly conclusive (Tables 3, 4; Figs 2, 3). The H gene sequences of Irish H6 isolates were less divergent from North American LPAIVs than those of the H10 and H11 subtypes. However, N2 gene sequences of the H6N2 LPAIVs (12/14 H6 subtypes) isolated during this research aligned very closely with Eurasian rather than North American sequences. The lower degree of divergence of the H2 and H6 subtypes of LPAIV has been documented previously and was attributed to inter-continental transmission of LPAIVs between Eurasia and North America [Reference Makarova23–Reference Koehler26]. Some ducks and shorebirds cross the Bering Straits during migration or may have breeding areas in both the northwestern regions of North America and the far-eastern areas of Russia [Reference Olsen22]. Therefore, the findings of this study supported previous conclusions that the degree of divergence between Eurasian and North American strains of LPAIV varied according to the virus subtype [Reference Koehler26]. Ireland is the only European country that receives a significant number of migratory waterfowl each winter from both North America and Eurasia [Reference Wernham6, Reference Crowe and Merne7]. Mixing of European and North American LPAIVs could occur during the winter season. However, the H genes of all AIVs found in Ireland during this study had a very high level of agreement with the H genes of viruses detected in wild birds in Europe during the same period and were divergent from published sequences of North American isolates. Therefore, the evidence from phylogenetic examination of Irish viruses during this study strongly support the conclusion that LPAIVs had been introduced to Ireland from Europe and not from America. These AIVs were most likely introduced by dabbling ducks migrating along the East Atlantic Flyway rather than by geese migrating from Canada and Greenland. The closer alignment of all Irish subtypes with Eurasian sequences in the NCBI database [Reference Bao14] enabled this conclusion to be made with a high degree of confidence.
The predominance of a different subtype of AIV in each season of surveillance at the Wexford Sloblands led to the conclusion that new AIVs were being introduced to this region by wild migratory birds on an annual basis [Reference Raleigh8]. Analysis of the H genes of the AIVs isolated at the Sloblands detected substantial sequence variation between viruses of the same subtype identified in different years. Season-to-season divergence of the H genes of the H10 isolates was particularly high, strongly supporting the conclusion that different influenza A viruses were being introduced to the region every year by migratory birds. The annual release of a large number of juvenile mallard into the relatively confined space of the Wexford Sloblands resulted in a highly susceptible population of birds in which AIV infections could occur and spread due to the decline or absence of immune protection [Reference Perk, Schudel and Lombard5, Reference Makarova23, Reference Hanson27]. The close similarity between the H genes of viruses isolated from wild birds at the Wexford Sloblands in the same season was probably due to recent active infection of a naive population with a virus newly introduced by migratory waterfowl [Reference Perk, Schudel and Lombard5].
The phenomenon of subtype rotation observed at the Sloblands, with the isolation of a large number of closely matched H10 LPAIVs in the 2004/2005 season and two clusters of closely related H6 subtypes of LPAIV in the 2005/2006 season, was similar to the pattern of rotation of H5 and H7 subtypes detected in Italy in the 2004/2005 and 2005/2006 seasons [Reference Terregino28]. Cyclical patterns of influenza A virus detection in wild birds, characterized by the dominance of one subtype of LPAIV for a period of 1–2 years and then its replacement by another dominant subtype, have also been reported in Canada [Reference Olsen22]. The change in the population of LPAIVs in Canada occurred despite the ability of these viruses to survive from year to year in frozen lake water [Reference Ito29, Reference Kida30]. The phenomenon of abmigration (changing of breeding sites) observed in dabbling ducks may be an important mechanism in facilitating subtype rotation in waterfowl populations [Reference Olsen22, Reference Guillemain, Sadoul and Simon31].
The current study also found some evidence that a year-to-year reservoir of LPAIVs may exist in Ireland. During the summer of 2007 an H11 AIV was detected in farmed mallard in Ireland. The H gene of this virus shared 99·6% identity with an H11 virus isolated during the spring of 2006 from a ruddy shelduck in a captive population of wild birds at Fota Wildlife Park. Young birds hatching in the spring and summer breeding programme are likely to have extended the period of viral circulation in this captive population [Reference Alexander1, Reference Koch and Elbers32]. This finding suggests that this 2006 LPAIV might have been maintained in the resident bird population in Ireland. The finding also indicates that amplification and maintenance of LPAIVs by game bird and zoological collections might have a significant effect on the perpetuation of LPAIV in countries with temperate climates. The close similarity between the H gene of an H6N6 subtype of LPAIV isolated in 2004/2005 with those of the 2006 H6N2 subtypes of AIV may also be the result of seasonal carryover of LPAIV. Breban et al. [Reference Breban33] reported that environmental transmission is an essential element of the recurrence of widespread infections in waterfowl, caused by the same subtypes of LPAIV, with a 2- to 4-year periodicity. This evidence of year-round viral circulation supports the hypothesis that LPAIV may be able to persist in ducks from season to season, even in temperate climates [Reference Olsen22]. The evidence of year-round circulation of LPAIV has major significance for the ecology of LPAIV in Ireland, indicating that the threat to commercial poultry from AI is not confined to the time when migratory waterfowl are present.
Analysis of the H gene suggested that all the H10 isolates detected in 2004/2005 and the H2 isolates of the 2005/2006 season resulted from single introductions in the respective years. However, the larger degree of divergence between the H genes of the two H11N9 LPAIV isolates obtained in the 2004/2005 season and the two groups of H6N2 LPAIVs from the 2005/2006 season suggested that these viruses might have been introduced on a number of occasions. The divergence (⩾8%) of the N genes of the two groups of H6N2 viruses isolated in 2005/2006 also strongly support the possibility of multiple introductions.
In the 2005/2006 season, the occurrence of two H6 (H6N5, H6N2) and H10 (H10N2, H10N7) subtypes of LPAIV with almost identical H genes but different N subtypes suggested reassortment between different LPAIVs. Co-infection of ducks with more than one LPAIV has been reported frequently and is an important source of variability due to reassortment between different viruses [Reference Sharp34, Reference Ellstrom35]. As the N2 of the H10N2 isolate was significantly different (8%) from the N2 of the predominant H6N2 subtypes of that season this reassortment is unlikely to have occurred in Ireland.
Wild birds are considered to be the primary source of LPAIVs associated with infection of domestic poultry [Reference Koch and Elbers32, Reference Halvorson36, Reference Campitelli37]. However, there was a significant difference (6·1%) between the H gene of the H6 LPAIV isolated from a commercial flock of chickens compared to those of H6 LPAIVs isolated from waterfowl in Ireland during the same period. There was also a significant difference (about 4–10%) between the N2 gene of the LPAIV isolated from chickens compared to wild-bird isolates. In fact, the H gene of the Irish chicken-derived subtype of LPAIV was more closely related to poultry isolates obtained in Germany in 2002 than to any other previously published sequences of LPAIV, although the N gene sequence of this virus matched most closely that of a virus detected in mallard in The Netherlands in 2006. These findings suggested that this virus might have infected poultry on mainland Europe as a result of contact with wild birds and was introduced subsequently to Ireland by poultry or poultry products, which are considered a significant means of spread of AIVs in domestic birds. These results support similar findings with regard to tracing of H5N1 AIV infection in commercial poultry [Reference Kilpatrick38].
In conclusion, the phylogenetic findings in the present study suggest that migratory birds introduced LPAIVs from Europe to Ireland during each year of the study. However, the findings also showed that LPAIV might in some cases survive from year to year in the native waterfowl population through a combination of the cycling of infection from older to younger birds, environmental transmission and reassortment. Although wild birds were likely to have been the original source [Reference Alexander and Brown39], the introduction of LPAIV to Irish poultry during the study period does not appear to have been as a result of contact with wild birds and measures for prevention and control of AIV must include strict control and monitoring of the movement of poultry and poultry products. The isolation of LPAIV from captive mallard suggested that pre-movement testing and monitoring of this category of bird for the presence of LPAIV, particularly subtypes H5 and H7, should be performed to reduce the risk of viral spread. The finding that all viruses identified in Ireland were of Eurasian origin has practical significance, in that wild geese, which are protected species, can be excluded from future surveillance studies for LPAIV in wild birds in Ireland. This work supports the previous conclusion [Reference Raleigh8] that surveillance should be largely concentrated in dabbling ducks. It is also clear that phylogenetic analysis of AIVs, detected by active surveillance during outbreaks of AI in other European countries, may provide early advance warning of viral introduction to Ireland.
Many of the molecular assays, including most of the real-time (rt)RT–PCR assays, used for identification and characterization of H sequences, have been developed in the USA [Reference Das and Suarez40] and may not be suitable for identification of AIVs circulating in Ireland. The rtRT–PCR assay for characterization of subtype H11 [Reference Das and Suarez40] was unable to identify the H11 AIVs isolated in this country (data not included), probably because of the divergence between the H gene of North American H11 LPAIVs from that of Eurasian viruses of this subtype. The viruses analysed in the current study should provide the sequence data required to modify the primers of the current rtRT–PCR assay [Reference Das and Suarez40] to increase its sensitivity for the detection of Eurasian H11 LPAIVs.
ACKNOWLEDGEMENTS
The support and encouragement of Mr Pat Lenihan, Dr John Ferris and Dr P. J O'Reilly at the Central Veterinary Research Laboratory (CVRL) is gratefully acknowledged. Ms Jean Mooney of the CVRL and Dr Margaret Duffy of the National Virus Reference Laboratory generously provided advice on the presentation of phylogenetic trees.
We gratefully acknowledge the assistance provided during the collection of specimens by members of the National Association of Regional Game Councils and of the National Parks and Wildlife Service.
DECLARATION OF INTEREST
None.









