


INTRODUCTION
Crimean-Congo haemorrhagic fever (CCHF) is a tick-borne viral zoonosis widely distributed in Africa, Asia and eastern Europe within the distribution range of ticks belonging to the genus Hyalomma [Reference Hoogstraal1]. The virus is a member of the Nairovirus genus of the family Bunyaviridae and has a single-stranded, negative-sense RNA genome consisting of three segments L (large) 4·1–4·9×106 Da, M (medium) 1·5–1·9×106 Da and S (small) 0·6–0·7×106 Da, each contained in a separate nucleocapsid within the virion [Reference Clerx, Casals and Bishop2]. The virions contain three major structural proteins: two envelope glycoproteins, GN and GC encoded by the M segment, and a nucleocapsid protein, encoded by the S segment, plus minor quantities of viral transcriptase or L (large) protein. The M segment of CCHF virus has one open reading frame which encodes a precursor polypeptide, with a highly variable amino-terminal domain and fairly conserved carboxyl-terminal region. The precursor polyprotein is cleaved to form the two mature glycoproteins, GN (37 kDa) and GC (75 kDa) [Reference Marriott, Nuttall and Elliott3].
Recent reports on molecular characterization of CCHF virus include the first complete sequence data for all three RNA segments [Reference Marriott and Nuttall4–Reference Meissner8], characterization of the M segment [Reference Papa6], genetic analyses based on nucleotide sequence data from the S and M segments from various geographic regions [Reference Sanchez, Vincent and Nichol5–Reference Deyde14], and evidence for the natural occurrence of reassortment and recombination [Reference Hewson10, Reference Deyde14, Reference Lukashev15]. RNA viruses with segmented genomes have the ability to reassort when dual infection occurs, and this can play a role in pathogenicity and epidemiology of the viruses. This in turn implies that phylogeny based on nucleotide sequence data for the S segment alone [Reference Burt and Swanepoel12] may be inadequate. Hence we extended our earlier observations to the M and L segments of the genome to determine whether reassortment plays a role in viral pathogenicity, or correlates with geographic distribution within southern Africa.
METHODS AND MATERIALS
Virus isolates
Details of the origin of 23 CCHF isolates included in the study are summarized in Table 1; 21 isolates were from southern Africa and two were from Pakistan. Sequence data were determined retrospectively for isolates collected in southern Africa from 1985 to 2001 and stored at −70°C as freeze-dried 10% suckling mouse brain suspensions at the level of mouse brain passage 2–3. The suspensions were inoculated into Vero cell cultures and total RNA extracted from the infected cells using the acid guanidium thiocyanate–phenol–chloroform method. For two isolates viral RNA was extracted directly from serum samples submitted from patients in Pakistan using the QIAamp viral RNA isolation kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol.
Table 1. Origins of CCHF virus isolates included in the present study
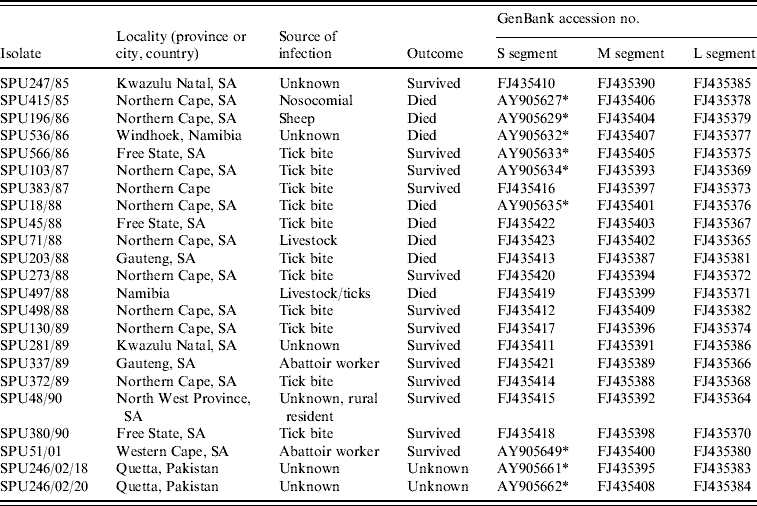
* Sequence data for S segment determined in this study retrieved from GenBank [Reference Burt and Swanepoel12].
Reverse transcriptase polymerase chain reaction (RT–PCR) and nucleotide sequencing of amplicons
Partial nucleotide sequences were determined for each segment of the genome. Pairs of primers were identified from alignment of sequence data retrieved from GenBank for five complete M and L segments of southern African isolates (SPU41/84, SPU128/84, SPU97/85, SPU415/85, SPU103/87) (accession numbers AY900142, AY900141, DQ211633, DQ211635, DQ211634, DQ076417, DQ076414, DQ211620, DQ211622 and DQ211621). To confirm that incongruencies resulted from segment reassortment, two regions of the M segment were sequenced and the topology of the trees constructed from the data were compared.
The M primer sequences (positions relative to SPU415/85) were:
MF3 (4804–4821): 5′-G(C/T)T GCT GG(C/T) TAG AGT CAG-3′
MR3 (5365–5346): 5′-CTC AAA GAT ATA GTG GCT GC-3′
MF2 (2756–2774): 5′-CCC TGG A(C/T)T GTC CAT TTG C-3′
MR2 (3392–3372): 5′-CTT (A/G)TT GCC TCT GTG TTC (C/G)AC-3′.
L segment primer sequences were:
LF1 (2120-2139): 5′-CAG TTG CAT CCA GAG TTC AG-3′
LR1 (2969-2951): 5′-CTT TGC GCA TTG CCT GTT C-3′.
Partial sequence data for the S segment of southern African isolates were obtained from a previous study using published primer pairs F2 and R3 [Reference Burt and Swanepoel12, Reference Schwarz16, Reference Burt17]. In addition, sequence data were retrieved from GenBank for isolates from other geographic regions, to determine their relationships to southern African isolates (Table 2).
Table 2. Data retrieved from GenBank, accession numbers provided
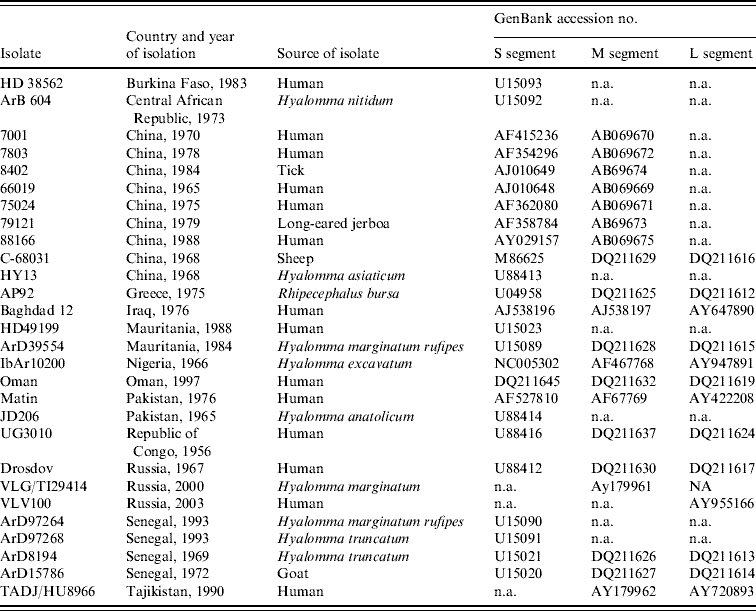
n.a., Not available.
For all amplifications RT–PCR was performed using the Titan One Tube RT–PCR system (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer's instructions and cycled using standard cycling conditions and annealing temperatures determined from the T m of the oligonucleotide primers. The nucleotide sequences of the amplicons were determined using Big Dye™ Terminator Sequencing Ready Reaction kits (Applied Biosystems, CA, USA) according to manufacturer's instructions.
Data analysis
After editing of the data and allowing for inclusion of data retrieved from GenBank, the phylogenetic analyses were performed using a 413 base pair (bp) region from the conserved 3′-carboxyl terminal of the M segment, a 590 bp region targeting the central region of the M gene, a 436 bp region of the S segment and a 754 bp region of the L segment. The data were analysed using Molecular Evolutionary Genetics Analysis (MEGA) version 3.1 and bootstrap neighbour-joining method with 1000 replicates [Reference Kumar18]. Sequence divergence was determined using MEGA to calculate the average P distances within groups and between groups.
RESULTS
Genetic analysis
The genetic relationship of isolates determined using neighbour-joining analysis is shown in Figure 1 based on partial nucleotide sequence data for (a) S segment (436 bp), (b) M segment, 413 bp 3′-region (the tree topology was identical for a 590 bp region from the middle of the M segment, data not shown), and (c) L segment (754 bp) of CCHF virus. The phylogenetic relationships determined for CCHF isolates using partial segment data were in agreement with findings previously obtained with complete segment sequence data [Reference Deyde14, Reference Morikawa, Saijo and Kurane19], and the groups were designated I–VIII following the nomenclature of Deyde et al. [Reference Deyde14]. Tree topologies based on the S and L segment data showed good correlation, with no incongruencies in the grouping of the southern African isolates. Group switching of non-southern African isolates ArD8194 and ArD15786 from S group I to L group III was previously described by Deyde et al. [Reference Deyde14] who concluded that it was probably the result of L segment reassortment. Comparison of the present tree topologies for each segment showed incongruencies in the M segment grouping for 15 southern African and one Asian isolate. Two isolates, SPU247/85 and SPU281/89 (shown in bold in Fig. 1) from group II in both S and L trees acquired M segments from group III; one isolate (SPU498/88, shown in bold in Fig. 1) from group III in both S and L trees acquired M segment from group VII; one Asian isolate (SPU246/02/18) from group IV in both S and L trees acquired M segment from group III; and 12 isolates (shown in upper case in Fig. 1) from group III in both S and L trees (SPU380/90, SPU497/88, SPU51/01, SPU130/89, SPU383/87, SPU18/88, SPU71/88 (group I in S), SPU45/88, SPU566/86, SPU196/86, SPU536/86, SPU415/85) acquired M segments from group IV. In the absence of complete nucleotide sequence data for the CCHF strains included in the present study, and to support the hypothesis that the discrepancies in grouping were the result of segment reassortment and not recombination events, an additional section of the M segment was sequenced and the tree topology based on this region was identical to that shown in Figure 1 (data not shown). Group IV in both S and L trees comprised isolates solely from Asia. The apparent acquisition of Asian group IV M segments by southern African isolates suggests movement of CCHF virus between continents.
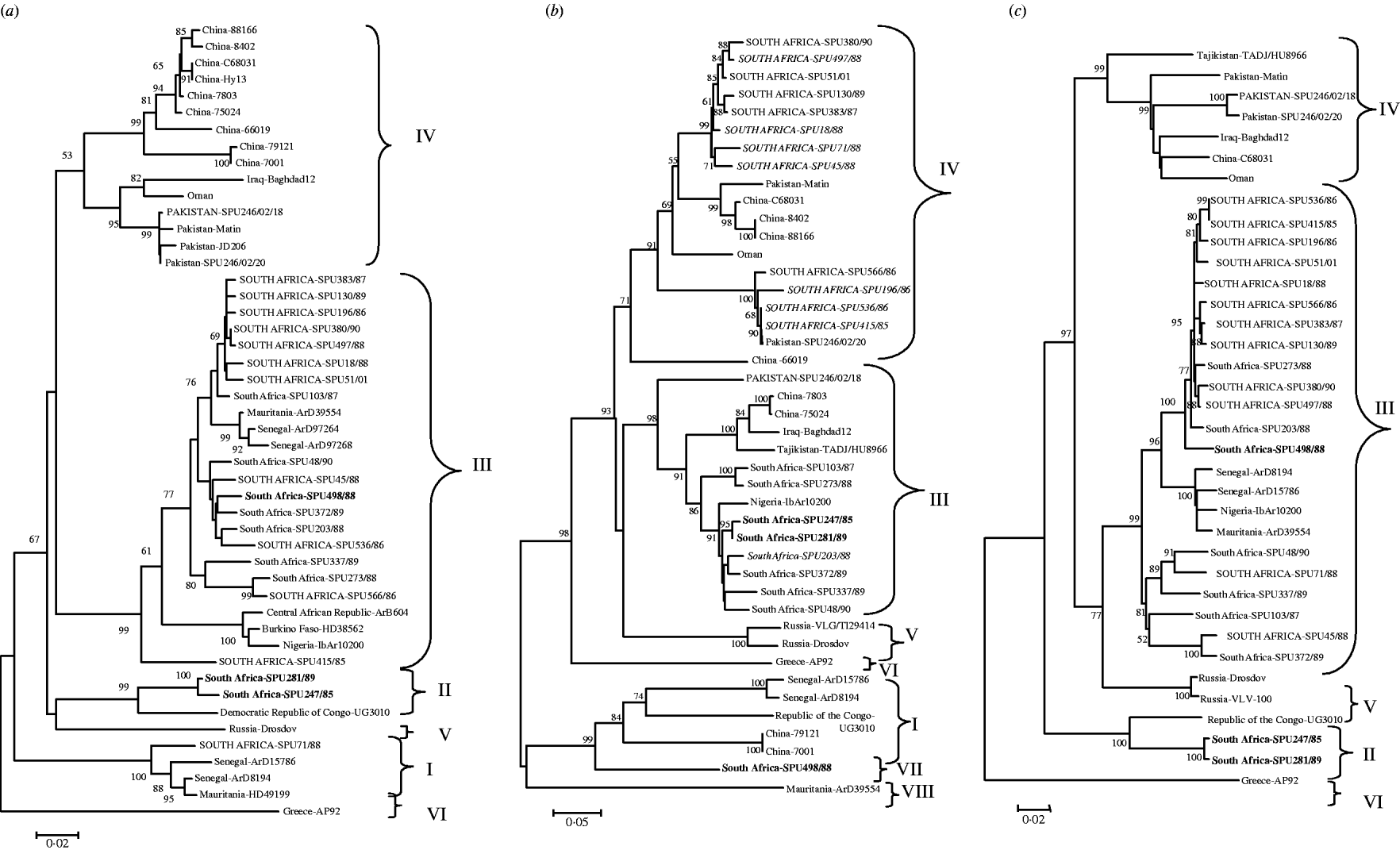
Fig. 1. Phylogenetic analysis of worldwide strains of CCHF virus based on partial nucleotide sequence data from (a) S segment (436 bp), (b) M segment, 413 bp 3′-region (590 bp from the middle region of the M segment, tree topology identical, not shown) and (c) L segment (754 bp) of CCHF virus. Data analysed using a bootstrap neighbour-joining method with 1000 replicates using Molecular Evolutionary Genetics Analysis (MEGA) version 3.1. Patients with a fatal outcome illustrated in italics.
A high degree of nucleotide diversity was evident when sequence divergence was determined using MEGA to calculate the average P distances within groups and between groups for the southern African isolates. The S segment was the most diverse, with nucleotide distances between groups ranging from 29·6% to 43·1%, whereas the amino-acid distances ranged from 2·8% to 4·9%, suggesting that the majority of changes were synonymous. Within each S segment group nucleotide distances were <10·1% and predicted amino-acid distances 1·7%. The L segment was more conserved, with nucleotide distances between groups ranging from 4·1% to 6·6% and amino-acid distances ranging from 7·9% to 11·6%, suggesting that more non-synonymous changes had occurred. Within L segment groups the nucleotide distances were <1·1% and the predicted amino-acid distances 2·1%. The M segment showed the largest number of non-synonymous changes between groups with nucleotide distances ranging from 10·0% to 33·9%, translating to predicted amino-acid differences of 15·3% to 37·3%, while within groups nucleotide distances were as low as 0·6% to 4·3% and predicted amino-acid differences ranged from 1·3% to 5·4%.
An interesting finding is the possible association between M segment reassortment and increased pathogenicity. Of 21 randomly selected CCHF isolates from southern Africa, 7/8 isolates from patients with fatal outcome had evidence suggesting the acquisition of M segment RNA from group IV viruses, compared to 5/13 isolates from non-fatal infections, but the difference is not highly significant (P=0·027).
DISCUSSION
Reassortment and recombination events have been described for CCHF virus and other bunyaviruses. Although confirmation of genomic RNA segment exchange and recombination would theoretically require full-length sequencing, previous publications have consistently shown close relationships between segment topologies based on partial and complete sequence data [Reference Deyde14]. Hence we focused on sequencing selected regions. The L segment is largely conserved with three variable regions, the N terminal, C terminal and a variable region from bases 2346–2488 which we selected for sequence determination [Reference Meissner8]. S segment sequence data were available from a previous study [Reference Burt and Swanepoel12]. The M segment has a highly variable region at the N terminal and a more conserved region coding for the glycoprotein precursor. Based on the assumption that if phylogenies generated from different regions of the same segment have different topologies then the incongruencies in grouping are more likely to be attributable to recombination than reassortment, we selected to confirm the consistency of the topology generated for the M segment by determining partial sequence data for two distinct regions.
In a previous study based solely on partial S segment data of southern African isolates it was concluded that despite the potential which exists for dispersal of the virus between Africa and Eurasia, circulation is largely compartmentalized within the two land masses, with the inference that the geographic distribution of phylogenetic groups is largely related to the distribution of tick vectors of the virus [Reference Burt and Swanepoel12]. Evidence that multiple strains were clustered geographically, and that in some instances similar subtypes existed in distant geographic locations, was difficult to explain. It was postulated that genetic diversity within regions resulted from the introduction of multiple lineages of virus over millennia through carriage of infected immature ticks on migrating birds, or through the movement of livestock, as well as from evolutionary pressure within ecological niches. Evidence for the occurrence of reassortment and recombination indicates the existence of additional mechanisms for the generation of genetic diversity, and implies that phylogeny based on nucleotide sequence data from the S segment alone is not adequately informative. The present findings indicate that M segment reassortment is not uncommon and tends to confirm the occurrence of spillover of virus circulation between southern Africa and Asia. It has been suggested, that co-infection and reassortment is more likely to occur over time within vectors than within mammalian hosts, partly because ticks remain infected for longer and are potentially exposed to successive infected hosts [Reference Hewson10, Reference Deyde14, Reference Morikawa, Saijo and Kurane19].
Reassortment has previously been associated with changes in pathogenicity of bunyaviruses. Ngari virus, for example, was found to be a reassortant orthobunyavirus in which the L and S segments are derived from Bunyamwera virus and the M segment from Batai virus. While Bunyamwera and Batai viruses cause benign disease, Ngari was found to be associated with cases of haemorrhagic fever in East Africa [Reference Sall20–Reference Briese22]. The determinants of fatal outcome in CCHF infection are largely unknown, but it is thought that factors such as age and underlying health play a role. Clearly, the possibility that segment reassortment may be associated with increased severity of CCHF infection merits further investigation. While no differences in antigenicity between CCHF isolates has been reported, genetic variation must be taken into consideration when developing new-generation diagnostic assays or vaccines.
ACKNOWLEDGEMENTS
The project was funded by the Polio Research Foundation, South Africa.
DECLARATION OF INTEREST
None.



