


INTRODUCTION
Since Escherichia coli O157:H7 was first recognized as a cause of illness in 1982 [1, Reference Riley2] it has emerged as an important foodborne pathogen, causing outbreaks worldwide that are often widely dispersed [Reference Karch3]. The first reported case of E. coli O157:H7 infection in New Zealand occurred in 1993 [Reference Baker4]. Since then incidence has increased markedly, with 128 cases of verotoxigenic E. coli infection reported in 2008 (3.0 cases/100 000 population) [5]. Of these, 120 isolates were available for serotyping with 118 (98·3%) O157:H7 and two non-O157:H7 [5]. Phage-type 2 (PT2) has been the most common phage type identified in E. coli O157:H7 isolates referred to New Zealand's reference laboratory, accounting for 48% of isolates in 2006 (C. Nicol, unpublished data).
E. coli O157:H7 has a low infectious dose and patients can develop the life-threatening condition haemolytic uraemic syndrome [1, Reference Riley2]. Early detection of an E. coli O157:H7 outbreak could have a significant impact on the burden of disease. Identifying dispersed outbreaks quickly requires a realtime surveillance system that can distinguish isolates resulting from exposure to a common source from those causing sporadic infections. Within the USA surveillance is carried out using PulseNet USA, who monitor and collate E. coli O157:H7 typing data, amongst other foodborne pathogens [Reference Gerner-Smidt6]. Many foodstuffs are now distributed worldwide, which significantly increases the potential for an international outbreak. Therefore, methods that characterize E. coli O157:H7 must be internationally comparable, easily transportable, and epidemiologically relevant.
Currently, genomic DNA macrorestriction analysis using pulsed-field gel electrophoresis (PFGE) is widely used to characterize E. coli O157:H7. One application for the method is within the PulseNet USA and other PulseNet surveillance networks in the Asia-Pacific region, Canada, Europe, and Latin America [Reference Swaminathan7]. PFGE offers good discrimination between E. coli O157:H7 strains, particularly when two restriction enzymes are used [Reference Gupta8], although it has a number of limitations that have led to alternative methodologies being investigated. Multiple-locus variable-number tandem-repeat analysis (MLVA) is one alternative subtyping method and like PFGE it could also be used within the PulseNet platform [Reference Gerner-Smidt6, Reference Hyytia-Trees9]. MLVA is a polymerase chain reaction (PCR)-based typing method that discriminates between isolates based on differences in the number of variable number tandem repeat (VNTR) sequences at multiple loci [Reference Lindstedt10]. It has a number of distinct advantages over PFGE, as results are available faster, they are easily comparable between laboratories, and the method is amenable to high-throughput analysis [Reference Lindstedt10]. MLVA data for E. coli O157:H7 isolates generally show good correlation with PFGE data although MLVA may be more discriminatory and epidemiologically relevant [Reference Noller11–Reference Murphy16].
Prior to this study no MLVA data were available for E. coli O157:H7 isolated from cases in New Zealand. Case isolates are routinely analysed using single-enzyme (XbaI) PFGE analysis although the profiles for E. coli O157:H7 PT2 isolates are often very similar, making discrimination challenging. In this study we compared data generated by XbaI PFGE analysis and the MLVA methodology described by Hyytia-Trees et al. [Reference Hyytia-Trees9]. These methods were used to characterize E. coli O157:H7 isolates referred to the New Zealand's national Enteric Reference Laboratory (ERL) at the Institute of Environmental Science and Research Ltd. (ESR), to determine if MLVA would be suitable for analysis of New Zealand case isolates.
METHODS
Bacterial strains
All E. coli O157:H7 isolates used in this study originated from clinical cases of gastrointestinal disease and were referred to ERL for confirmation and toxin testing. The identification of E. coli O157 was confirmed biochemically and a multiplex PCR performed to detect the presence of the virulence genes stx1, stx2, eaeA, and hlyA [Reference Wang, Clark and Rodgers17]. The somatic O157 and flagellar H7 antigens were determined by latex agglutination (Prolex E. coli O157 Latex test reagent kit, ProLab Diagnostics, Canada) and confirmed using Mast Assure E. coli mono factor O157 antisera (Mast, UK). A PCR specific for the gene encoding the H7 antigen [Reference Fields18] was used if the agglutination tests were positive for O157 but negative for H7. Phage typing [Reference Ahmed19, Reference Khakhria, Duck and Lior20] was undertaken using a standard set of typing phages (International Centre for Enteric Phage Typing, UK).
Isolates selected for use in this study consisted of nine E. coli O157:H7 isolates from a panel of E. coli PFGE reference strains (New Zealand Reference Culture Collection, ESR, New Zealand), all 80 E. coli O157:H7 isolates referred to the ERL in 2006, and 29 E. coli O157:H7 PT2 isolates from 2005.
PFGE
Macrorestriction analysis using PFGE was performed according to the PulseNet standardized protocol [Reference Ribot21]. Chromosomal DNA from all isolates was restricted using XbaI (Roche, Germany) and the resulting band profiles were analysed using the BioNumerics Software programme version 5.1 (Applied Maths, Belgium). Dendrograms were generated using the Dice similarity coefficient and the unweighted pair-group method with arithmetic mean (UPGMA) clustering. Clusters were defined at 0·5% optimization, 1·5% band tolerance, and >95% similarity.
Preparation of DNA template
E. coli O157:H7 isolates were cultured on tryptic soy agar plates incubated at 37°C for 18 h. Four to five colonies were suspended in 500 μl sterile distilled water. The suspension was heated to 95°C for 10 min and the debris pelleted by centrifugation (5 min at 13 768 g). The supernatant was used as the source of DNA template for PCR amplification.
PCR
MLVA was performed as described by Hyytia-Trees et al. [Reference Hyytia-Trees9] except, following correspondence with the authors of that paper, the VNTR-10 locus was not assessed. The remaining eight loci were amplified using three multiplex reactions (reactions 1a, 1b, 2) instead of two (Table 1) as, in our laboratory, the VNTR34 amplicon often failed to amplify in the presence of VNTR3 and/or VNTR9 primers. The labelled forward primers were synthesized by Applied Biosystems (USA). All unlabelled primers were synthesized by Invitrogen (USA). All amplifications were performed in 10 μl reactions containing 1×PCR buffer without magnesium (Invitrogen), 2·0 mm MgCl2, 1·0 mm PCR nucleotide mix (Invitrogen), 1·0 U Platinum Taq polymerase (Invitrogen), primers at concentrations described in Table 1, and 0·8 μl DNA template. Reactions were cycled in a GeneAmp® 9700 thermocycler (Applied Biosystems). The amplification conditions used were: denaturation at 94°C for 5 min, followed by 35 cycles of 94°C for 20 s, 65°C for 20 s, and 72°C 20 s, and a final extension at 72°C for 5 min.
Table 1. Primer concentrations and multiplex reactions used to amplify the eight loci assessed in the E. coli O157:H7 MLVA method. Primer sequences are described by Hyytia-Trees et al. [Reference Hyytia-Trees9]
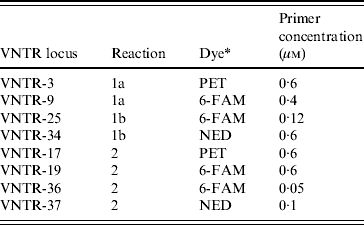
* Dye used to label the forward primer.
DNA sequence analysis
DNA sequencing was performed according to the manufacturer's instructions using the BigDye® Terminator v. 3.1 Cycle Sequencing kits (Applied Biosystems) and an ABI 3130xl Genetic Analyzer (Applied Biosystems). Primers used for sequencing were the (non-labelled) forward and reverse MLVA amplification primers (Table 1). Sequencing reactions that initially failed were repeated in the presence of 1 m betaine (Sigma-Aldrich, USA), using an annealing temperature of 56°C [Reference Zhao, Haqqi and Yadav22]. Data analysis was performed using the ChromasPro sequence analysis program (Technelysium Pty Ltd, Australia).
Fragment analysis
Amplification products from reactions 1a and 1b were combined 1:1 and diluted 1:20 with sterile distilled water. Reaction 2 amplification products were diluted 1:40 with sterile distilled water. Samples were analysed on an ABI 3130xl Genetic Analyzer (Applied Biosystems) using the GeneScan™ 600 LIZ® size standard (Applied Biosystems) for comparison. Raw data were analysed using the GeneMapper® Software v. 4.0 (Applied Biosystems) to determine the size and dye label associated with each amplicon and assign an allele number. To confirm the correct allele assignments had been made, sequence data were obtained for a range of alleles at every VNTR locus. The alleles at each locus were entered into BioNumerics as character values and a dendrogram was constructed using a categorical multistate coefficient and UPGMA clustering with a >95% similarity cut-off.
Statistical analysis
The diversity of each VNTR locus was assessed using Simpson's diversity index [Reference Hunter and Gaston23] via the online tool available at the Health Protection Agency website (http://www.hpa-bioinformatics.org.uk/cgi-bin/DICI/DICI.pl). The diversity of phage typing, MLVA, and PFGE was calculated using the Biodiversity Calculator developed by J. Danoff-Burg and C. Xu (http://www.columbia.edu/itc/cerc/danoff-burg/MBD_Links.html). Confidence intervals (CI) were calculated as previously described [Reference Grundmann, Hori and Tanner24].
RESULTS
Initial assessment of the MLVA methodology
The MLVA methodology previously described [Reference Hyytia-Trees9] was initially evaluated using nine E. coli O157:H7 isolates from New Zealand's E. coli PFGE reference panel [Reference Short25]. The isolates had nine unique PFGE profiles and nine unique MLVA profiles, which highlighted the diversity in the isolates assessed. There was little similarity between the clusters identified using PFGE and those identified using MLVA.
Analysis of all E. coli O157:H7 from 2006
In 2006 80 E. coli O157:H7 case isolates were referred to the ERL. Within these isolates eight clusters, involving 24 case isolates, were identified using epidemiological and PFGE data. All eight clusters involved household contacts, with each cluster affecting between two and six cases inclusive. Isolates within each cluster had identical MLVA profiles, even when the PFGE profile differed subtly (Fig. 1). Different PFGE profiles were found for case isolate ERL06-3358 in cluster 7 and the two case isolates in cluster 4 (Fig. 1). Subtly different PFGE profiles may be a consequence of the subjective, bandbased comparison required for analysis of PFGE data or may be truly representative of a different profile. Isolates with an indistinguishable PFGE profile to case isolates associated with a family cluster that did not appear to be epidemiologically related had distinct MLVA profiles (Fig. 1). This was found for ERL06-3357 and ERL06-4287 that had indistinguishable PFGE profiles to those found in family clusters 7 and 2, respectively (Fig. 1).

Fig. 1. MLVA data and the clustering of PFGE data for all 80 E. coli O157 isolates from cases of disease in 2006. The dendrogram for PFGE data was generated using the Dice coefficient and UPGMA clustering. ND, No data; PM, isolates with indistinguishable PFGE and MLVA profiles; P1, P2, P3, P4, isolates with indistinguishable PFGE profiles and distinct MLVA profiles; M1, M2, isolates with indistinguishable MLVA profiles and distinct PFGE profiles.
If PFGE data was relied upon to define clusters within the 80 E. coli O157:H7 isolates referred to the ERL in 2006 a total of 12 clusters would have been identified. This assumes that the definition of a cluster is two or more isolates with indistinguishable XbaI PFGE profiles. Seven of the eight clusters identified using epidemiological data would have been found using only PFGE data (all except family cluster 4, Fig. 1). However in three clusters (2, 5, 7, Fig. 1), PFGE data would have included one isolate that was not part of the cluster (as determined from epidemiological analyses). There were five groups of isolates that had indistinguishable PFGE profiles but had not previously been identified as epidemiologically linked (labelled PM or P1–4, Fig. 1). Of these five groups only one (labelled P1, Fig. 1) involved isolates linked by time and place. The isolates labelled PM had indistinguishable MLVA profiles at eight loci although all other groups had MLVA profiles that differed at three of more loci from other isolates in the same group.
If MLVA was relied upon to define clusters within the 80 E. coli O157:H7 isolates a total of 11 clusters would have been identified, assuming that a cluster was defined as two or more isolates with indistinguishable MLVA alleles at all eight loci. MLVA correctly identified all eight clusters found using epidemiological information as well as three groups of isolates that had not previously identified. Two of the three groups not identified previously involved isolates that were not linked by time (ERL06-0828, ERL06-4083, labelled M1, Fig. 1; ERL06-0839, ERL06-1255, labelled PM, Fig. 1), whereas the third involved two cases linked by both time and place with very similar PFGE profiles (ERL06-3139, ERL06-3667, labelled M2, Fig. 1). If a cluster was defined as two or more isolates with indistinguishable MLVA alleles at seven loci a further two groups of isolates would be identified, one with two isolates and the other with three.
Analysis of E. coli O157:H7 PT2 isolates
Phage typing provides a good approximation of relatedness although it is likely to be insufficient for epidemiological investigations. The high percentage of PT2 isolates causing disease in New Zealand means that it is important to be able to discriminate within these isolates. The PFGE (Fig. 1) and MLVA data from 2006 isolates showed that those with the same phage type, including PT2, clustered together. This prompted us to investigate the relationship between phage type, PFGE pattern and MLVA type by performing MLVA on 29 E. coli O157:H7 PT2 isolates from 2005 and comparing it to the data from 2006 isolates.
PFGE analysis indicated that the PT2 isolates from 2005 and 2006 were more closely related to each other than to other phage types. There was no clustering of MLVA or PFGE profiles according to year of isolation and none of the 2005 PT2 isolates were associated epidemiologically with an outbreak. Within the 65 isolates assessed there were 42 unique PFGE profiles and 55 unique MLVA profiles. There were 11 groups of isolates with indistinguishable PFGE profiles, involving 34/65 isolates. There were seven groups of isolates with indistinguishable MLVA profiles, involving 17/65 isolates. Simpson's diversity index was used to provide a statistical measure of the diversity captured by PFGE and MLVA within PT2 E. coli O157:H7 isolates. Simpson's diversity index for PFGE was 0·968 (95% CI 0·944–0·992) and for MLVA it was 0·993 (95% CI 0·987–1·000).
Locus characteristics
In total 118 E. coli O157:H7 isolates from New Zealand were assessed in the current study. The most variable loci were VNTR9 and VNTR3, with 17 different alleles each. These loci had the highest values for Simpson's diversity index: 0·906 (95% CI 0·898–0·914) and 0·892 (95% CI 0·883–0·901), respectively. The locus with the lowest value for Simpson's diversity index was VNTR34 (0·350, 95% CI 0·300–0·401), as 94 isolates (80·0%) had the same allele.
Within the 118 New Zealand isolates assessed there were 80 unique PFGE profiles and 95 unique MLVA profiles. Simpson's diversity index for the 109 isolates for which phage typing results were available was 0·600 (95% CI 0·503–0·695). Simpson's diversity index for all 118 isolates analysed using PFGE was 0·985 (95% CI 0·972–0·997) and for all 118 isolates analysed using MLVA was 0·994 (95% CI 0·990–0·998).
DISCUSSION
This paper describes the first application of MLVA to characterize E. coli O157:H7 isolates from New Zealand. The main advantages of MLVA over PFGE are that MLVA is a molecular-based method that is amenable to standardization, automation, and highthroughput analysis. MLVA subtyping data can be available 5–20 h after sample receipt, depending on the time of day the sample is received and the number of samples. By contrast PFGE typing takes at least 24 h. In our laboratory if double-enzyme analysis is required it takes at least 2 days before PFGE results are available. This is because single-enzyme analysis is undertaken initially, with a second enzyme being used only if more discrimination is required. MLVA is also advantageous compared to PFGE as less staff time is required for processing. Within our laboratory the MLVA protocol, and in particular the primer concentrations, required some optimization although once set up it was found to be easy to use and generated results in a time-frame that would permit subtyping information to play a role in an outbreak investigation.
If MLVA is to be accepted as a standard typing method it must provide a high level of discrimination, results must be reproducible, and results must be comparable between both national and international laboratories. We found that the discriminatory ability of the MLVA methodology described by Hyytia-Trees et al. [Reference Hyytia-Trees9] was not significantly different to that of XbaI PFGE analysis, although this may be attributable to the reasonably small number of isolates assessed. It also needs to be recognized that the single-enzyme PFGE method we used is not the most discriminatory PFGE method available [Reference Gupta8]. Our laboratory does not routinely run plugs digested with two enzymes simultaneously and the additional time required to generate double-enzyme PFGE data delays the availability of results. The ability of MLVA to generate rapid results means that it can contribute more timely information for outbreak investigations and would be our preferred method over double-enzyme PFGE. MLVA provided a good level of discrimination within E. coli O157:H7 case isolates from New Zealand, including the predominant PT2 isolates.
One of the roles of subtyping in outbreak investigations is to increase the power of epidemiological studies. This is achieved by guiding epidemiologists on which cases to include in their analyses. Ideally subtyping would permit identification of all clusters resulting from exposure to a common source and exclude all sporadic cases. In reality analysis of subtyping data does not provide this clear-cut result and there needs to be a balance between reporting too many clusters and missing an outbreak. Both MLVA and PFGE facilitated detection of all clusters that were identified using epidemiological data, when clusters were defined as groups of two or more isolates with indistinguishable MLVA or PFGE profiles, respectively. However, MLVA excluded more cases that epidemiological analyses suggested were unrelated. This suggests that, compared to PFGE, MLVA would increase the power of the epidemiological studies by guiding epidemiologists to include the cases in their analysis that are more likely to be associated with a common source.
A recognized artifact of determining the size of a DNA fragment using automated DNA sequencers is that the reported size of an amplicon is different to its actual size [26, Reference Pasqualotto, Denning and Anderson27]. In our study the reproducibility of reported fragment sizes, determined using a genetic analyser, was very high. However, the reported sizes differed from the actual size, which was determined using sequence data, by up to 4 bp. Factors that affect the mobility of a fragment include the length and sequence of the fragment, the genetic analyser used to run the sample, the dye used to label the primer, and the temperature when the sample is run [26, Reference Pasqualotto, Denning and Anderson27]. The observed variability between the reported and actual sizes of an amplicon is problematic for MLVA analysis because it could result in different allele assignments by different laboratories, particularly for small (e.g. 6 bp) repeat sequences. If MLVA is to be truly portable between laboratories then factors that affect the reported size following capillary electrophoresis need to be understood and controlled or accounted for prior to the allele assignment being made. It is unlikely that all the variables affecting the reported size can be controlled between laboratories so it is important to understand and allow for them in the algorithm used to assign alleles. Sequencing PCR amplicons was used successfully in this study to account for variables affecting the reported size of amplicons although analysis of strains with a known number of repeat sequences could also be used.
Our results suggested that MLVA can provide an alternative, rapid method of characterizing E. coli O157:H7 and that the method would be suitable for routine analysis of case isolates in New Zealand. The study enabled a database to be established, which will serve as a reference for comparison of MLVA data in the future. Future use of this methodology in New Zealand will largely depend on the acceptance of the methodology internationally.
ACKNOWLEDGEMENTS
The authors thank the ERL staff for the PFGE and phage-typing data, and the diagnostic laboratories around New Zealand for referring isolates used in this study for confirmatory testing. This project was funded by Capability Development funding (Ministry of Research, Science and Technology) and the Ministry of Health. E. Turbitt is currently affiliated with Murdoch Children's Research Institute, Melbourne, Australia.
DECLARATION OF INTEREST
None.


