


The pharmaceutical industry is often put forward as the best example of the need for patents. Patents are extremely important in reaping the benefits of innovation in the pharmaceutical industry due to two factors: the high cost of discovering, developing, and gaining regulatory approval for a new medicine, and the very low imitation costs relative to the expenditures incurred by the innovator in drug discovery and development. Footnote 1 Many laboratories can replicate drugs because it is relatively easy to imitate a product once it is available on the market and after researchers analyze its composition. Thus, the pharmaceutical sector depends heavily on the patent system to protect innovative designs from imitation. Footnote 2 It is only through enforceable patent protection that drug companies can generate sufficient revenues to undertake the costly and risky research and development (R&D) that makes the introduction of new products possible. In a few sectors, patents are essential to the development and introduction of innovations, and the pharmaceutical sector is one such example. Footnote 3
However, patents can also be employed as a strategy to harm the competitive process by adopting measures to restrict price competition; to extend the breadth and duration of their patent protection; or to delay, discourage, or block the market entry of competing products (e.g., patents thickets, secondary patents, and defensive patents). Footnote 4 According to Henry Steele in a seminal work, firms use patents not only as a way to protect their innovations but also as a strategy to increase their market share and to limit price competition. Footnote 5 For instance, patent or patent application holders may take measures such as cross-licensing agreements, licensing, and litigation settlements to restrict competition. Footnote 6 Specifically, to maximize the value of an innovation, patentees usually include field-of-use restrictions in licensing and cross-licensing agreements. These field-of-use restrictions are provisions under which licensees are prohibited from selling drugs in bulk form; that is, the form in which drugs are manufactured prior to being packaged into finished dosage form. This means that licensees can only sell patented drugs in finished dosage form, packaged form, or specialty form; that is, pharmaceutical drug products in the form in which they are marketed for use such as pills, tablets, capsules, or syrup. In this way, licensees prevent competitors from converting the bulk drug into finished and packaged tablets—tableting, bottling, packaging, and labeling—and from selling them either generically or under their own brand names and at their own prices.
The inclusion of a field-of-use restriction was commonplace in the agreements signed by big pharmaceutical firms in the period discussed in this article. Footnote 7 The patent holder usually decided not to license to small firms, so this type of restriction blocked small competitors from accessing the active ingredient in bulk form and from selling the drugs at significantly lower prices than those of large firms. This is because, on the one hand, smaller firms had a cost advantage over large companies because they did not have to recoup extensive R&D costs and the costs of advertising, promotion, and marketing. On the other hand, these costs were entry barriers for smaller companies into the prescription retail market. If the patent licensing agreement included a field-of-use restriction, small firms could only buy the drug exclusively in finished packaged form at a higher price. In selling drugs not in bulk form but under a brand name, big firms had to spend much more money on costly promotional campaigns. This expense ultimately made the drugs more expensive, because the big firms had to recover their marketing investment from the consumer. The inclusion of field-of-use restrictions in patent licensing agreements theoretically provided a solution that reduced competition because these restrictions constrained price competition from small companies.
This article focuses on the ethical pharmaceutical industry in the United States—particularly the tranquilizer sector—in the 1950s and early 1960s, when psychopharmacology in the treatment of mental illness really took off. Footnote 8 In this period of exponential growth of the pharmaceutical industry, large manufacturers challenged each other vigorously in product competition in order to gain a significant market share through their major investments in R&D and marketing. Moreover, there was rapid growth in the number of new medicines to treat mental illness, sustained efforts by pharmaceutical firms to develop markets for their products, and rising demand for drugs. Consumers were optimistic about the possibilities of pharmaceutical science, and this culture led to increasing demand among patients despite the high prices. Footnote 9
Concerns over the consequences of excessively high consumer pricing arose in the 1930s when inflation and its consequences for consumers became increasingly important issues in U.S. politics. It was in this context that economic New Dealers promoted government price control as a way of preventing under-consumption. In particular, by the late 1950s, Footnote 10 Congressional Democrats, the Federal Trade Commission (FTC), consumer groups, and some physicians became preoccupied with the high costs of prescription drugs and the advertising practices of prescription drug firms.
In response to these concerns, the influential Democrat Senator Estes Kefauver started a Congressional investigation to study the price inflexibility of firms producing ethical drugs (or prescription drugs) in certain product categories that were characterized by their novelty and their importance to medicine. Footnote 11 The high demand for drugs and the role of patents as a strategy to harm price competition came to public attention in late 1959 during Kefauver’s hearings on the pharmaceutical industry. These hearings were an important development in the political history of prescription drugs and the first attempt to regulate their prices. Footnote 12 In particular, the senator focused on the relationship between patent protection and high prescription drug prices in the U.S. pharmaceutical sector. On May 8, 1961, the Subcommittee on Antitrust and Monopoly of the Senate Judiciary Committee submitted its report on the ethical drugs industry Footnote 13 (ethical drugs are discussed below). The report found that monopolistic drug pricing and abuses of patent privilege existed in the U.S. pharmaceutical industry. Following the hearings, Kefauver introduced a bill proposing several reforms, one of which was to restrict the use of pharmaceutical patents in order to increase competition and reduce the price of prescription drugs. His greatest legislative achievement was the passage of the Kefauver–Harris Drug Act of 1962, one of the most significant consumer safety laws of the twentieth century. Consequently, these hearings were fundamental to modern consumer politics and to the history of U.S. medicine after World War II.
This article is related to a growing body of literature focusing on the use of patent licensing as an instrument to limit competition. Footnote 14 Specifically, our purpose is to study whether field-of-use restrictions in patent licensing contracts truly impeded price competition, as the Kefauver Committee concluded. The main source employed for this study was the original Senate Subcommittee Hearings on Antitrust and Monopoly during the period 1959–1961 (the Kefauver Committee). Footnote 15 The article, in turn, links to other strands of the literature such as the joint importance of R&D and marketing in the pharmaceutical industry.
We show how patent licensing restrictions, in fact, facilitated the erosion of price competition for prescription drugs only in the institutional market, in which lower prices were the major tool of competition. In this submarket, the presence of smaller firms as competitors stemmed from the fact that licensing contracts did not include field-of-use restrictions, which forced leading pharmaceutical companies to enter into price competition in order to sell their patented drugs. However, because smaller firms would have had to buy the drug in finished dosage form if field-of-use restrictions were included, they then would also have had to pay a price similar to retail druggists. Therefore, they would be in no position to engage in sales to hospitals or to the government at lower prices. This strategy among leading firms—including field-of-use restrictions in patent licensing contracts to exclude small firms as competitors and thereby to preserve their market position—made no sense in the retail prescription submarket. There was no price competition in this submarket, and product differentiation and advertising were the major competitive weapons for increasing company market share. Smaller firms were not considered threats because the high costs of marketing and promoting a drug made it impossible for small sellers of generic-name products to obtain any significant share of the retail prescription market, even though they were able to sell drugs cheaper than brand-name drugs because they incurred lower costs (that is, no research and marketing costs).
Some previous drug industry studies have concluded that there was an absence of competition in the drug industry as a result of patent privilege. Footnote 16 Other studies found significant rivalry based on factors other than price in the pharmaceutical industry. Footnote 17 The evidence presented in this article indicates that before any conclusions can be drawn, it is crucial to define the concepts of competition and of the market—institutional or retail—in which firms sold their patented drugs. Thus, we demonstrate that there was indeed a lack of price competition in the retail market but not in the institutional market. In the first submarket, this was true even when smaller firms were present as competitors, although product competition was relevant among patented drugs. Finally, our research also provides empirical evidence of the following assertions:
-
• The grant of a patent does not always confer monopoly power to patentees.
-
• Patents are necessary as a means to stimulate investment in R&D.
-
• In the pharmaceutical sector, investment in R&D—and its alignment with marketing—is a major form of competition. Footnote 18
The remainder of the article is organized as follows. The second section begins with the main characteristics of the retail prescription market. The third section contains a short description of the ethical tranquilizer sector in the United States in the 1950s. In the fourth section, we show the likely harm to competition caused by field-of-use restrictions in patent licensing contracts. The main conclusions are set out in the final section.
Selling Drugs in the Prescription Tranquilizer Sector
The golden age of drug discovery, or the therapeutic revolution, began around 1935 with the introduction of vitamins, hormones, sulfonamides, and antibiotics and their derivatives, and it came to an end in the 1970s. Footnote 19 During this period, drug development was a highly profitable activity, firms invested heavily in R&D, and competition increased as new products with incremental advances were introduced. Footnote 20 These years of the therapeutic revolution also witnessed American’s growing enthusiasm for prescription pills, the rise of national health services, the expansion of the market, increasing government control over drug development, and concerns about rising health care costs. Footnote 21 The exponential growth of the pharmaceutical industry from this therapeutic revolution and the expansion of the market are reflected in the fact that, by 1960, around 70 percent of all sales came from products that had not existed before 1951. Footnote 22
Following World War II, the pharmaceutical sector was characterized by intense product differentiation and product competition. Footnote 23 Different manufacturers could market the same chemical entity under a number of separate and distinct brand names or under its generic name. During the patent period, the original manufacturer could license other firms to sell the drug, some with their own brand name and others with the generic name. Footnote 24 Thus, the same drug was often marketed under different names. This approach is known as product differentiation.
Product competition was also intense in the pharmaceutical sector. When one firm was awarded a patent, other firms attempted to modify the molecular structure of the relevant compound in order to discover drugs with limited incremental advances in therapeutic value. Such drugs might be similar to those products already on the market yet sufficiently different to win a patent. Footnote 25 Pharmaceutical companies generally recognized that they were in a race with other firms to develop innovative drugs. Footnote 26 It was the era of molecular modification, meaning that not all drugs on the market were really innovative; rather, they were duplicate drugs under different names. Such products are called false innovations, me-too, or duplicative drugs, and are defined as drugs with chemical properties and therapeutic effects similar to those of drugs already on the market. Footnote 27 They were considered innovative due to their new characteristics; for example, better taste or better metabolism. The importance of molecular modification was reflected in the fact that the bulk of spending on research by pharmaceutical companies was “to exploit and market the foreign advances or to modify the original drugs just enough to get a patentable derivative, but not to change it enough to jeopardize the original effect.” Footnote 28
The vast majority of “new” drugs were variations of older drugs already on the market, although they may have been important for some patients due to their therapeutic properties. Footnote 29 However, the number of new chemical entities (that is, a drug or chemical without precedent among regulated and approved drug products because of its significant therapeutic advance) was comparatively small. More than 4,560 new ethical drugs were introduced in the period 1951–1961, of which only 360 were truly innovative drugs; that is, new chemical entities.
The me-too business was possible because the Food and Drug Administration (FDA), which had been authorized starting in 1938 to approve prescription drugs; it usually approved a drug simply if it was better than a placebo, but it was obligated to certify only its safety, not its efficacy. Footnote 30 Sometimes there was not even the slightest difference between different products; nevertheless, competing companies advertised them as different compounds. The FDA was required to give clearance only to new drugs, so new combinations of familiar drugs did not have to be re-evaluated.
Intense product rivalry due to the high levels of investment in R&D, as new product introductions and incremental advances were introduced, forced pharmaceutical firms to engage in costly marketing campaigns in order to increase sales of a particular brand. Footnote 31 New products were especially important in the pharmaceutical industry, as treatments of diseases were continually changing and new diseases were conceptualized by pharmaceutical marketing. Footnote 32 Clinical research results and laboratory science were extensively used for (scientific) marketing purposes, particularly after the 1950s, and they were fundamental in shaping the drug market. The rise of scientific marketing, including medical information in promotional material for physicians, was closely related to the rapidly changing market caused by the introduction of new drugs. Marketing was also used to promote and redefine disease categories and to reshape prescription patterns, and thereby create new markets and expand existing markets for establishing drugs. As Jean Paul Gaudillière analyzed, Ciba for example, transformed a tranquilizer into an antidepressant, redefined a new form of depression to be treated with antidepressants, and focused on the idea of the preventive action of antidepressants. Footnote 33 By doing so, this company was able to build a market for antidepressants to treat a great number of patients suffering from mental problems. Footnote 34
This product competition created increased rivalry in the drug industry for patented drugs and made it inevitable that firms would spend huge amounts of money on marketing in order to increase their sales. Footnote 35 The introduction of numerous drugs with very similar therapeutic effects prompted companies to engage in major marketing efforts, as they could not compete on the basis of these effects. Footnote 36 If they wished to survive competition in the market, firms had to advertise their products heavily through different channels, including medical journals, films, radio, television, samples, and pharmaceutical representatives. Footnote 37 This strategy resulted in an increase in advertising by firms in the 1950s from approximately 10 percent to roughly 15 percent of sales to promote their innovations. Footnote 38 The bulk of advertising and promotional efforts went into persuading doctors to prescribe specific brand names; by the 1960s, more than 90 percent of the pharmaceutical companies’ spending on marketing was aimed at doctors (only 10 percent was toward pharmacists and hospitals). Footnote 39
This high level of advertising focused on doctors explains why, after 1938, decisions regarding patients’ use of prescription drugs were mainly in the hands of physicians, as they were the people prescribing the drugs. Footnote 40 Moreover, druggists were limited to issuing the trademark written on the prescription. During the 1950s, forty-four U.S. states enacted antisubstitution laws to prohibit brand substitution as a result of an alliance among physicians, pharmacists, and the brand-name pharmaceutical industry. These laws constrained the use of drugs sold under their generic names. In this situation, pharmacists were legally bound by their state laws to fill prescriptions as written by doctors. Footnote 41 However, purchase decisions of prescription drugs did not lie solely in the hands of physicians. Footnote 42 In the 1940s, wide prescription drug abuse started “adding up to an astonishing 15 million Americans by the late 1960s. The FDA estimated that half of all amphetamine pills in the 1960s were dispensed without the benefit of prescriptions.” Footnote 43 Moreover, David Herzberg indicates that consumers with problems of addiction pressured their doctors to change their types of drugs, increasing the number of refills or increasing the quantity written on the prescriptions. Footnote 44
These antisubstitution laws also encouraged firms to focus on marketing and promotion. The proliferation of brands meant that advertising became a significantly important strategy to increase sales. Footnote 45 Patent-holders used their patent period to promote their product’s brand name to physicians and tried to persuade them to use it for reasons of quality, rather than price. Footnote 46 Leading pharmaceutical companies justified their enormous expenditures on promotion and advertising by arguing that a trademark gave a guarantee of purity and strength, as well as the quality of a well-known firm with a recognized reputation. The industry argued that drugs with the same generic name but different proprietary names could differ in their therapeutic effects. These are the reasons the industry cited to oppose Senator Kefauver’s proposal in S. 1552 to encourage physicians to prescribe drugs by their generic name. Generic prescribing would be unsafe because of the existence of substandard drugs sold under generic names, with substitution being defined as a health hazard. It was argued that brand-name products inspired confidence among physicians, and this then explained the high percentage of drugs prescribed by brand name. Footnote 47 In addition, a risk-averse physician would not prescribe a drug by its generic name until its efficacy was proved. The private doctor would be much more inclined to trust the trademark on the assumption that the relevant company would be safe. They, therefore, placed their trust in firms’ sizes and reputations, since they had no way of knowing the quality of all the manufacturers. Footnote 48 Moreover, doctors were not sensitive to price differences, and even tended to be unaware of prices. Habit, therefore, played a major role in physicians’ prescription practices. Footnote 49 The result was that doctors prescribed only a few of the many available brand-name drugs—that is, the most heavily advertised drugs, which were those of the big companies—and this meant that smaller firms never developed the capacity to finance selling campaigns.
For a small company, it was very difficult to finance a selling campaign. Footnote 50 Promotional expenditures were far greater than those for R&D. For example, the amount spent on R&D in 1958 by the twenty largest companies with the highest annual sales of drugs represented 6.4 percent of their total pharmaceutical sales ($2.3 billion). Footnote 51 This proportion was not particularly high, given that the companies’ selling expenses were 24 percent (one-third of the total cost of production Footnote 52 ) and net profits were 13.1 percent. Footnote 53 These data (see table 1) show that selling costs were almost four times that of research expenses.
Table 1. Breakdown of sales dollar, 20 major drug companies, 1958 (in percent of sales*)

Source: “Administered Prices,” 86th Cong., 1960, 2nd session, part 16, at 9,176–9,177.
Note: * = includes royalties and other income. ** = includes labor costs, material costs, bottling, finishing into tablets or capsules, and depreciation on plant. *** 3 percent of the 13.9 is for the patents to the Swiss company, with U.S. research being about 10 percent.
In sum, big pharmaceutical companies integrated marketing techniques with R&D activities to increase their market shares and to withstand competition from drugs that were chemically different but offered the same therapies. This explains the high rate of product obsolescence and rapidly growing markets, which stimulated competition and provided an impetus to innovate. Footnote 54 In turn, the profitability of investments in drug R&D and marketing led to more innovation and more drugs. Footnote 55
In the tranquilizer sector, there were many medications but not many significant improvements, although a small reduction in the incidence of side effects could be demonstrated with the new drugs. For example, although there were several types of tranquilizers, medical opinion inclined to the view that the later modifications of the original phenothiazines were therapeutically similar to the first ones that were released. The advances made following the discovery of chlorpromazine, the first of the antipsychotics, were small and incremental. There were drugs with similar properties patented and marketed as competing products that were as effective but had fewer or different side effects. Footnote 56
The importance of promotional expenses for a particular drug is well illustrated in this sector. Tranquilizer advertising was the major component of medical advertising, as indicated by the executive director of the National Committee Against Mental Illness. Miltown and Equanil, for example, were heavily advertised as a condition of Wyeth’s license with Carter: Wyeth would spend at least 20 percent of total meprobamate sales revenue on its promotion. Despite its high price, Miltown was the most widely used. It was the first psychotropic blockbuster and the fastest-selling drug in U.S. history. Footnote 57
The cost of Miltown per tablet for a 1,000-tablet production run is shown in table 2. Advertising, promotion, and clinical samples cost one cent per tablet—Carter’s total advertising and promotion cost for Miltown was $9.20 per year per doctor in 1959—as compared to seven-tenths of one cent, which was the manufacturing cost per tablet, or about one-and-a-half times more. Research and royalties were four-tenths of a cent per tablet, which was less than half the expenditure for advertising and promotion. The net profit for the manufacture and sale of Miltown was 23 percent per tablet.
Table 2. Miltown costs and profit per tablet by Carter, March 31, 1959 (cents per tablet)

Source: “Administered Prices,” 86th Cong., 1960, 2nd session, part 16, at 9,161.
Notes: * = these expense items are average for Carter’s ethical drug business.
Although there was little overuse of potent tranquilizers—patients did not like to take them owing to their unpleasant side effects—the less potent ones were overused to a tremendous degree. The minor tranquilizers were everywhere in the popular media, thanks to intense marketing and the widespread view that anxiety was not so much a sign of mental illness as a symbol of success. Footnote 58 The consumerist culture also explains why these drugs were initially quite popular. Footnote 59 The abuse of minor tranquilizers—especially of meprobamate—was remarkable, although there was little evidence that the drug was qualitatively different in its clinical effects from a number of other hypnotics, including barbiturates. Footnote 60 The danger with this practice was the problem of addiction; for example, large doses of meprobamate (Miltown, Equanil, and various other trade names) over a long period of time could cause addiction.
The development, introduction, and marketing of potent and dangerous drugs increased concerns regarding the regulation of the industry as a means to protect consumers. At this time, there was major government and public concerns regarding drug safety regulation, the abundance of me-too drugs, and industry marketing practices. Kefauver’s hearings were initially predicated on concerns about monopolistic pricing, but as the hearings continued attention focused increasingly on pharmaceutical marketing, consumer safety, and drug efficacy. Specifically, Kefauver’s greatest achievement was the Kefauver–Harris Drug Act of 1962 (also called the drug amendments) because it directly related to the drug industry and health issues. This was the first major change of the preceding law: the Federal Food, Drug, and Cosmetic Act of 1938. This act required manufacturers to show that a drug was safe, and drugs had to receive the FDA’s approval before they could be marketed. The Drug Act of 1962 was one of the most significant consumer safety laws of the twentieth century: to receive approval by the FDA, a new drug’s efficacy and safety had to be demonstrated prior to marketing. Footnote 61 All drugs approved between 1938 and 1962 were reviewed under the new law, and those which were found to be ineffective were removed from the market. Thus, the drug hearings triggered major progress for consumer safety legislation and expanded the FDA’s authority over the testing, manufacturing, and marketing of new drugs. Footnote 62 Furthermore, Kefauver’s investigation encouraged crucial debates on contemporary decisions in U.S. health policy and practice. Footnote 63
As a result of the hearings held by the Subcommittee on Antitrust and Monopoly, Kefauver introduced a bill (S. 1552) to the subcommittee in 1961 designed to increase competition, to enhance the use of generic names, and to reform the patent system. It introduced compulsory licensing and the granting of patents only in relation to those drugs that were true innovations (and not me-too drugs). The bill met with strong resistance from two Democratic senators (Everett Dirksen and Roman Hruska), the American Medical Association, and the Pharmaceutical Manufacturers’ Association (PMA), the industry’s trade association. The PMA included only 140 firms from among more than 1,300 mainly medium- and large-size firms that produced almost 95 percent of all sales of prescription drug in 1960. President John Kennedy and the White House staff were also reluctant to support the bill. Two of the arguments advanced by the PMA to oppose the charges made by Kefauver were: (1) the superiority of the U.S. free enterprise model, and (2) the benefits of the competitive system as the best asset in the struggle against Communism. Footnote 64 Finally, however, Kennedy signed the bill into law in October 1962 (the Kefauver–Harris Drug Act), but the patent provisions were rejected. Footnote 65 The Kefauver–Harris Drug Act was the first legislation to regulate the efficacy and safety of new drug products and reflected changes in the U.S. economy toward a politics of consumption. Regulation of pharmaceuticals became a national problem in the United States following Kefauver’s hearings. Footnote 66
The Rise of the Tranquilizer Sector in the United States
In the 1950s mental illness was a major health issue in the United States, with at least 25 percent of visits to doctors due to mental health problems, Footnote 67 and filling more than half the hospital beds in the country. Around 10 percent of Americans suffered from some form of mental illness, which cost the nation $3 billion a year. Footnote 68 In 1958 sales of tranquilizers at the manufacturers’ level were estimated at nearly $200 million a year (total annual sales of drugs for the United States were roughly $1.8 billion). Footnote 69
The introduction of effective drug therapy and its extensive use in the treatment of the mentally ill began in the 1950s. It was widely used to treat psychosis, depression, anxiety, and mania. Footnote 70 This changed the course of psychiatry, as practitioners began to consider both psychotherapy and psychopharmacology as regular treatments. Footnote 71 Interestingly, the discovery of these drugs was accidental and began with the synthesis of a number of phenothiazine antihistamines in the 1940s, which manufacturers then discovered were effective sedatives. Footnote 72
The arrival of new drugs capable of altering moods and mental illness challenged ideas about psychiatry and mental disorders, which were now attributed to brain chemistry. Anxiety was the common term used to refer to mental problems treated on an outpatient basis. The new psychiatry paradigm led to a conception of anxiety as a general psychic consequence of the demands and pace of conditions of modern life. Because anxiety was common and related to many ills, the introduction of drugs to treat problems of generalized stress and to enhance well-being created a mass market, and doctors became increasingly oriented toward prescribing pharmaceuticals. Footnote 73 This new market included those populations who began to think that mental distress could be treatable by drugs, having been persuaded by public relations and marketing efforts to grab the consumers’ attention. Footnote 74 The popularity of these drugs explains psychiatry’s pharmacological revolution, which began in the 1950s. In many publications from those years, one can read about the value of minor tranquilizers for a great number of life problems, including tension, nerves, irritability, menstrual stress, psychosomatic symptoms, insomnia, menopause, juvenile delinquency, family and marital difficulties, and problems at work.
At the end of the 1950s, there were three main classes of patented tranquilizers sold under a generic name or a brand name (table 3): phenothiazine derivatives (the most common of which were Compazine and Thorazine); alkaloids of Rauwolfia serpentina (the most important was reserpine); and finally, a miscellaneous group, principal among which was meprobamate. All of these drugs, termed ethical drugs, were sold by prescription. The first two groups included potent tranquilizers—or major tranquilizers or antipsychotics—that were largely used for the treatment of those hospitalized and other seriously ill mental patients. These proved to be extremely effective in combating major mental diseases. The other classes of tranquilizers, which received the most publicity, were usually simple sedatives, and their effects were mainly of a symptomatic nature (that is, mild tranquilizers). They were advertised for states of anxiety or tension and were suitable for treating the nonpsychotic patient. Footnote 75
Table 3. Major marketers of mental drugs, 1958

Source: “Administered Prices,” 86th Cong., 1960, 2nd session, part 16, at 8,887.
Note: * Sparine is half as potent as Thorazine, which is less potent than Vesprin. Compazine and Trilafon are more potent than Thorazine: the more potent the phenothiazine derivatives, the fewer side effects produced. Rauwolfia derivatives are safe compounds; serious side effects have been rare.
In 1959 the most significant firm selling phenothiazine derivatives in the United States was the American company Smith Kline & French (SKF), which was primarily a producer of ethical drug specialties. Footnote 76 During the late 1940s, SKF was interested in researching a compound that might potentiate other drugs, especially sedatives. In 1952 the French company Rhône-Poulenc discovered a new allergy drug it called chlorpromazine, which slowed down bodily processes. The development of this drug came from an interest in the 1940s in antihistamines, as they were effective sedatives. Footnote 77 In 1952 SKF obtained an exclusive license from Rhône-Poulenc to use, sell, and distribute the compound chlorpromazine in specialty form only. Rhône-Poulenc, which had applied in 1951, obtained the U.S. patent rights on July 14, 1953. Footnote 78 Thorazine was the trade name chosen by SKF for chlorpromazine; the product went on sale in 1954. Footnote 79 The introduction of chlorpromazine was a major milestone in the treatment of psychotic patients, Footnote 80 and Thorazine became the first widely available antipsychotic medication. In 1952 SKF also obtained a license from the French company to sell prochlorperazine under the trademark Compazine—a phenothiazine compound similar to chlorpromazine but much more potent. The patent was issued to Rhône-Poulenc in September 1959. SKF was the only domestic supply source for Thorazine and Compazine, and it had estimated sales of $24 million of Thorazine and $20 million of Compazine, representing around 20 percent of the tranquilizer market in 1958 and around 60 percent of phenothiazine derivatives (see table 3). Footnote 81 SKF signed more patent licensing contracts with Rhône-Poulenc. In 1956 SKF entered into an agreement with the French company to use Thorazine in combination with other products. That new product was named Thoradex.
Rhône-Poulenc also signed patent licensing contracts with other companies, such as with American Home Products Corp. (hereafter AHP), which received an exclusive license in 1957 to sell promazine (a molecular modification of the phenothiazine nucleus). Through its Wyeth Laboratories Division, AHP offered promazine under the brand name Sparine; it was a potent tranquilizer for patients seriously agitated with delirium tremens.
In 1952 reserpine, one of the first antidepressants introduced commercially in the United States, was isolated in Switzerland by Ciba from the dried root of Rauwolfia serpentine. This root had been used medicinally in Europe for more than three hundred years for the treatment of anxiety states, but it was in 1933 that a group of Indian researchers reported its value as a hypotensive agent. Ciba Pharmaceutical Company (a wholly owned subsidiary of Ciba) held the patent starting in 1956, and it sold reserpine under the trade name Serpasil. Several firms also sold reserpine, as we explain in the next section.
Meprobamate was the first minor tranquilizer and the most sold in 1958 (see table 3). Footnote 82 Dr. Frank Berger accidentally discovered mephenesin, a muscular relaxant, in England; eleven years later, he and his colleague, Dr. Bernard Ludwig, discovered a molecular modification of mephenesin—meprobamate—as a tranquilizer. They filed an application in 1950, and the patent rights to meprobamate were assigned in 1955 to Carter Products Inc., which sold the drug in finished form under the trademark Miltown through its ethical drug division, Wallace Laboratories. Carter licensed one other firm, AHP, through its Wyeth Division, to sell finished meprobamate in the United States under the trademark Equanil. Footnote 83
Patents and Price Competition in the Tranquilizer Sector
In the tranquilizer sector, all drugs were patented. In principle, only the owner of a patent—thus, a monopolist—could manufacture and sell the relevant product; this is, the patented drug. However, patent licensing agreements were used in the pharmaceutical industry. Licensing to other firms meant that more than one firm could produce and sell the product. One common practice, as noted earlier, was to license only to big firms, with smaller firms being excluded from licensing contracts. Usually, the patent-holder decided not to license small firms so as to exclude them from competition. This was because of their lower costs: there were no economies of scale in production and small firms did not commit significant resources to R&D and marketing, which were two barriers to entry in the prescription retail market. With lower costs, if smaller firms had access to bulk powders, they could sell licensed drugs cheaper than the licensor. This situation was only possible if the licensor decided to give a license to a small firm, or in those few cases in which the bulk drugs were available to all makers of preparations containing them. This happened when licensing contracts did not include field-of-use restrictions. Footnote 84
However, another frequent practice was for patent licensing contracts to include a restrictive clause, specifically a field-of-use restriction; that is, a provision stipulating that the firm would be granted a license only to make and sell in final packaged form, not in bulk form. Therefore, if the patent licensing contract included this restriction, small firms were not allowed to buy ingredients in bulk and could not manufacture tablets but had to purchase the drugs exclusively in finished packaged form from the licensees and the patent holder, and thus at a higher price. As a result, in most cases, the only producers were those big firms that held patents on the drug ingredients and the licensees.
In this section, we examine whether field-of-use restrictions in patent licensing contracts truly prevented competition in the tranquilizer sector. This restrictive clause was included in all patent licensing contracts regarding the main new tranquilizers discovered in the 1950s, except for reserpine. Thus, for example, there were field-of-use restrictions prohibiting licensees to sell in bulk form in the licensing contracts that Rhône-Poulenc issued to SKF to manufacture and sell the drug chlorpromazine under the trademark Thorazine and the drug prochlorperazine; in the contract that Rhône-Poulenc signed with Wyeth, a subsidiary of AHP, to produce Phenergan and Sparine; and in the contract that Carter Products signed with Wyeth to sell meprobamate under the trademark Equanil.
We studied whether field-of-use restrictions in patent licensing contracts truly prevented competition in two different markets in which ethical tranquilizers could be sold: the retail market (which included chain drug stores, mass merchandizers, independent pharmacies, supermarket pharmacies, and mail order pharmacies) and the institutional market (private nonprofit hospitals, state and local governmental hospitals, clinics, dispensaries, and federal agencies). In the former, as noted earlier, the people who usually decided which drugs to prescribe were doctors, and doctors tended not to care about the prices of the drugs they prescribed.
However, the institutional market was price-sensitive. In this submarket, there was no longer a captive aspect to the market, and the manufacturer had to compete on a generic basis. Thus, in the case of hospitals, hospital pharmacists were encouraged to prescribe those products listed on the hospital’s formulary, and hospital physicians agreed that prescriptions written under a brand name should be interpreted as if they were generic prescriptions, unless the physician indicated otherwise. Footnote 85 The idea was not only to reduce costs but also to avoid duplication in a pharmacist’s orders and duplication of drug names. A hospital formulary committee and the chief pharmacist would decide on the drugs required, and quantity purchases were made on a best-bid basis. Footnote 86 Hence, in contrast to the retail submarket, in which physicians and pharmacists did not have any incentive to prescribe the cheapest drug, in the institutional submarket there was a motivation to seek price cuts. Footnote 87
Price Competition in the Retail Market
A field-of-use restriction in a patent licensing agreement could theoretically have been a solution to erode price competition and to increase a company’s market share in the pharmaceutical market in which product competition was intense. In order to understand the relationship between restrictive licensing clauses and price competition in the retail market, we distinguished three different possible situations, dependent on the number of firms producing finished drugs from bulk powder active ingredients: those in which there was only one producer; those in which there were a few big firms producing the drug; and those with several manufacturers, both large and small. The distinction between these three scenarios is important in order to understand the different pricing policies of the large drug firms in the tranquilizer sector.
The first scenario is one in which only one company produced the drug; this happened with phenothiazine derivatives. In this case, Rhône-Poulenc, the owner of the patent, was a foreign company that did not operate in the United States, so it granted an exclusive license to SFK to manufacture a drug sold under its own brand name. The licensing contract included a field-of-use restriction that specified that SFK could not sell the drug in bulk form to other competitors; only it had access to the production process in the United States. We find this first situation in the contracts that Rhône-Poulenc signed with SKF to produce chlorpromazine and prochlorperazine under the trade names Thorazine and Compazine. Smith Kline and French established a price—$3.09—for wholesale druggists (50-mg Thorazine tablet in quantities of five hundred tablets), plus 15 percent margin to retail druggists (that is, $3.64, which was the price retail druggists paid for it). This price remained the same from 1954 to 1969. This situation was also found with the products Phenergan and Sparine, sold only by Wyeth in the United States at a price to retail druggists of $3 in 1958 (for fifty tablets). It is obvious that no price competition existed in this first scenario, in which there was only one firm producing and selling the patented drug in the U.S. market.
We found in the second scenario that there were only a few big firms with access to the drug in bulk form. This was the case with the tranquilizer meprobamate. As noted in the previous section, only two firms manufactured this patented drug: Carter, the patent holder, and Wyeth, the licensee. The licensing contract included a restriction indicating that Wyeth would make no sales of bulk powder to any other companies, although it had access to the production process; Wyeth tableted and bottled the bulk powder sold to them by Carter. In this case, a lack of price competition was encountered in the retail prescription market, because the licensor and its licensee pursued the strategy of offering the same price for the same drug. The bulk powder sale of meprobamate was reserved for Carter, which, through its Wallace Division, sold the drug under the trade name Miltown. American Home Products Corp., through the Wyeth Division, sold meprobamate under a license that it entered into with Carter in 1955 to produce the drug under the brand name Equanil. The price of Miltown was established in line with the competition, determined in April 1955, and the product was marketed the following month. Carter sold Miltown to wholesaler druggists at 5.2 cents a tablet; retail druggists paid the wholesaler 6.5 cents; and the final price to consumers was 10.8 cents per tablet. The price of Miltown remained the same from 1955 to 1960. Footnote 88 Wyeth set identical prices for Equanil, even though it had to make royalty payments of 13 cents to Carter, and its manufacturing costs were much higher than Carter’s ($0.007 and $0.015 per tablet, respectively). Footnote 89
The tactic of big companies in the prescription retail market of pricing the same patented drug at the same level is also found in the third case, in which there were several firms, both large and small, involved in pharmaceutical manufacturing with access to drug powder. The patented drug was marketed under both different brand names and generic names. We find this third scenario in the case of reserpine, a drug patented by Ciba and in which the licensing contract did not include a field-of-use restriction. In other words, Ciba decided to allow its licensees to sell the drug in bulk powder form to small firms. Hence, smaller firms could manufacture their own tablets and capsules, converting the bulk drug into finished packaged tablets (tableting, bottling, packaging, and labeling), and selling them either generically or under their own brand names and at their own prices.
With the drug reserpine, for example, Ciba charged a wholesale price of $39.50 per bottle of one thousand (25-milligram) tablets. The final price to the consumer was $65.83, given a druggist’s mark-up of 40 percent on the selling price. In contrast, some larger manufacturers including Lilly, Merck, Parke, Davis, Squibb, and Upjohn, all of which obtained their licenses from Ciba, sold reserpine at the same price, which was much lower than that charged by Ciba. Smaller firms, with no advertising costs, reduced prices to as much as 90 percent below Ciba’s price. The lower costs for smaller firms allowed them to reduce their prices to about $1.91 per fifty tablets; Carter’s fixed sale price to wholesale druggists, for example, was $2.60 per bottle of fifty capsules and $3.25 to retailers. Some small firms, such as Panray Corporation, quoted prices as low as $2.65 per bottle of one thousand pills to druggists, given that wholesalers offered a 20 percent reduction to druggists. Footnote 90 However, despite the large difference in the price of the patented reserpine among brand names and unbranded versions (those of small firms), branded products still retained large market shares. Footnote 91 Ciba’s Serpasil, for example, was more widely prescribed than reserpine and was the leading brand. However, Panray sold only to institutions and hospitals; it did almost no business with druggists even though its pricing was much lower than Ciba’s. Footnote 92
It seems clear that a lower price for a patented drug did not imply higher sales in the retail market, as the case of reserpine illustrates. When a drug group was introduced and received approval from the FDA, some manufacturers fought for a share of the prescription retail market, choosing any method that would establish their trade name in the minds of physicians, as discussed above. Big firms frequently employed this policy to maximize the value of their innovations, thus making it possible to achieve a high degree of market control from the supply side. Footnote 93 However, the high costs of marketing and promoting a drug made it impossible for small sellers of generic-name products to obtain any significant share of the retail prescription market.
Lower prices offered by smaller firms did not jeopardize the licensor’s monopoly in the prescription retail market. In this market, most prescriptions were written under branded trade names, even though small firms might sell drugs under generic names at prices that were a fraction of their larger rivals’ prices. In the 1950s, big companies made the majority of their revenue and profits in this submarket, in which almost 70 percent of total drug sales occurred. It is evident, however, that although there was no price competition in the retail market, the product rivalry from high levels of investment in R&D as new products were created, and as incremental advances were introduced, forced the pharmaceutical firms to engage in costly marketing campaigns to increase sales of a particular brand.
Price Competition in the Institutional Market
Although small companies were unable to gain access to the retail drugstores, because they could neither advertise nor bear the financial costs of detail men—pharmaceutical sales representatives—they were nonetheless very active with regard to sales in the institutional market: the Military Medical Supply Agency (MMSA; the central purchasing agent for all hospitals and dispensaries in the Armed Forces), hospitals, cities, and the Veterans Administration. Small companies were serious competitors in this price-sensitive market because of their lower prices. These institutions were required to purchase drugs under generic names at the lowest possible price from any qualified bidder. To be accepted by the government, each manufacturer’s product had to meet certain specifications for the particular drug involved. Federal and state agencies had certain quality control procedures, such as inspection of possible suppliers’ plants, inspection of manufacturing processes, or random sampling of each lot of drugs for analysis by the FDA. Thus, the government could accept bids by companies selling patented drugs under generic or brand names as long as they were of equal quality.
Institutional buyers usually purchased drugs through a bidding process. The government (federal or state) or a hospital could use two types of solicited bids: a negotiated bid and a competitive or advertised bid. Negotiated bids were usually requested when there was a patented drug product available from only one or very few manufacturers. In this case, there was a chance to negotiate the final price. There was no public opening, and the government contacted each bidder in an attempt to convince that manufacturer that if it reduced the price, it would get a certain amount of business. In a competitive or advertised bid, the government asked for bids on a drug under its generic names. This process was normally used when quality drugs were available from more than one source and there was no room to negotiate the price. With this type of bid, there were specific deadlines by which the bid had to be returned to the purchaser and then there was a public opening, at which time any representative could enter the bidders’ room and listen to the bids as they were offered.
To understand whether the inclusion of field-of-use restrictions of patent licensing contracts eroded price competition in the institutional market—that is, by excluding competition from smaller companies—it is important to distinguish the three different scenarios, as those noted above for the prescription retail market, which were dependent on the number of firms compounding finished patented drugs from bulk-powder active ingredients. First, in which there was only one bidder because the only producer of the drug in the United States was the licensee firm (the owner of the patent was a foreign company that did not operate in the United States), it was found that the bidder offered a discount to the institutional buyer as compared to the price offered in the retail market. This happened, for example, with the drug Sparine, which was produced only by Wyeth in the United States. Wyeth negotiated a bid to the Veterans Administration (VA) at $24.51 per bottle in January 1958 and to the MMSA at $24.42 per bottle in April 1958, whereas retail druggists were paying $32.49. Footnote 94 Similarly, in relation to the product Thorazine, SKF—the only producer of this drug in the United States—offered sales to the federal government at a price lower than that on the retail market. Footnote 95 For example, during the period from 1956 to 1959, the MMSA bought $2,215,113 worth of Thorazine from SKF at negotiated prices. In 1956, they twice purchased the 25-milligram dose in bottles of fifty—at $2.19 and $2.21—whereas a druggist paid $3.03. In 1957, druggists paid $2.27 for the same type and dosage of Thorazine. In 1957, the MMSA made one purchase in the amount of $33.46 for the 100-milligram tablet in bottles of five hundred. However, in that year, the price to retail druggists was $46.32. In 1958 the MMSA made two purchases of the 25-milligram dose in bottles of fifty—at $2.17 and at $2.19. In 1958 the MMSA made one purchase in the amount of $33.37 for 100-milligram tablets in bottles of five hundred. At the same time, the price to retail druggists was $46.32. In 1959 the MMSA made a purchase for $20.80 for 25-milligram tablets in bottles of five hundred, while the price to retail druggists was $28.79. Footnote 96
It seems clear from these data that in the first scenario, when there was only one bidder, the government paid a lower price in comparison to the price paid by retail druggists. This reduction was around 25 percent to 30 percent. Footnote 97 There were two main reasons for giving this price discount: the first was that large institutions bought drugs in great quantities, and the second was that distribution expenses, advertising, and selling costs were not a factor in sales to federal or state governments.
In the second scenario, as noted above, there were only a few bidders and the licensing contract included a field-of-use restriction. This was the case with meprobamate. The federal government advertised this drug by the generic name (meprobamate), but in the United States there were only two bidders: Carter, the patent owner, and Wyeth, the licensee. Carter sold in bulk form to Wyeth, which then tableted, bottled, and labeled it under its own trademark prior to distribution. The MMSA asked for competitive bids, but the outcome was always the same: identical prices but lower than retail prices. Wallace (a division of Carter) and Wyeth bid exactly the same price, so the agency was forced to settle the matter by splitting the awarded contract, drawing lots, or making the decision on the basis of a labor surplus area (see table 4). Henry H. Hoyt, the president of Carter, explained the situation, saying that the firm had a standard price for everyone, from the wholesale druggist in the city, to county and state hospitals, and to military supply depots. The latter two categories received discounts because the orders were placed for large amounts. Footnote 98 The wholesaler who bought meprobamate paid $3.25 for a package of fifty while the government paid $2.50. The price per tablet to druggists was 6.5 cents and to the MMSA was 5 cents, which was a discount of around 23 percent.
Table 4. U.S. Military Medical Supply Agency bids on negotiated contracts, 1958–1959, meprobamate, bottles (five hundred tablets)

Source: “Administered Prices,” 86th Cong., 1960, 2nd session, part 16, at 9,200.
Note: * = awarded the bid because they were in a labor surplus area. ** = in bottles of fifty.
In conclusion, in these first two scenarios—that is, when only one firm or a few firms manufactured a patented drug—either the producer monopolized it or sellers shared the market, respectively. This happened with the patented tranquilizers meprobamate, promazine, and chlorpromazine, purchased by the MMSA at prices that were 25 percent to 35 percent lower than the price to retail druggists.
The third scenario was in which both large and small firms manufactured the patented drug and were able to make bids to institutional buyers. In other words, patent licensing contracts did not include a field-of-use restriction and small firms compounded finished drugs, converting the bulk drug into finished packaged tablets and selling them at their own prices. This is the case with reserpine, a drug developed by Ciba Pharmaceutical Co. but which was widely licensed, allowing the licensees to sell the drug in bulk powder form. Small firms could set lower prices due to their lower manufacturing costs, given that there were no economies of scale in production and they did not have to undertake R&D or marketing costs in the manufacture of the drug. There were several suppliers that made bids at one time or another. Institutional buyers asked for these bids generically. Footnote 99 The result was a reduction from 1956 to 1959 in the price for reserpine sold to the MMSA from $1.39 to 60 cents, a reduction of more than 60 percent (see table 5). On several occasions, Ciba underbid smaller firms that sold only under the generic name, with the lowest price being offered by Ciba: 60 cents a bottle in February 1959, which was only 1.5 percent of Ciba’s price to retail druggists of $39.90. Footnote 100
Table 5. Military Medical Supply Agency orders of reserpine, 25 mg., bottles of 1,000
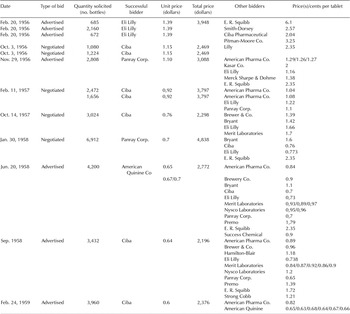
Source: “Administered Prices,” 86th Cong., 1960, 2nd session, part 16, at 9,429.
Reserpine is the only example in which there was a wide variation in prices in the institutional market, even though it was a patented drug. The reason for this unusual situation is that Ciba, which won the patent, licensed any company that wanted to be licensed. Ciba also allowed the licensees to sell the drug in bulk powder form to small firms that manufactured their own capsules—under their own brand name or more frequently under the generic name—because the licensing contracts did not contain a field-of-use restriction. Thus, small firms broke into the manufacturing process and sold reserpine at lower prices than the large firms. The introduction of more bidders led to substantial price reductions in the institutional market. Footnote 101
In is clear from these data that the lowest bids came from major drug companies supplying brand-name drugs in only two cases: in the first, brand-names were purchased when the proprietary drug was one from the few producers available (a negotiated bid), and the second was when the supplier of the proprietary drug was the lowest bidder.
Conclusions
By the late 1950s, Congressional Democrats, the FTC, consumer groups, and some physicians had become preoccupied with the high costs of drugs and the advertising practices of prescription drug firms. In response to these concerns, Senator Kefauver started a Congressional investigation to study the price inflexibility of those firms producing ethical drugs. On May 8, 1961, the Subcommittee on Antitrust and Monopoly of the Senate Judiciary Committee submitted its report on the ethical drugs industry. The report found that monopolistic drug pricing and abuses of patent privilege existed in the U.S. pharmaceutical industry. Kefauver unsuccessfully tried to restrict the use of pharmaceutical patents so as to increase competition and thereby reduce the price of prescription drugs.
In this article, we examined whether patents were employed as a strategy to restrict price competition. We studied the tranquilizer sector in the period when psychopharmacology in the treatment of mental illness really took off. At that time, pharmaceutical firms holding a patent usually decided to include a restrictive clause—a field-of-use restriction—in patent licensing contracts, which allowed licensees to sell only in finished form. In this way, the patent owner prevented small firms from buying bulk sales of the finished powder in order to tablet the powder and sell it, either generically or under their own brand names at lower prices. Smaller firms were able to sell drugs at lower prices because they did not have to recoup extensive R&D and marketing costs and, therefore, they incurred fewer costs than the big firms.
We found that field-of-use restrictions were used as solutions to exclude price competition from small companies only in the institutional market, in which price competition was extremely important to win a bid. On the one hand, licensing contracts that did not contain field-of-use restrictions allowed the presence of smaller bidders, and so the big companies were forced to enter into price competition and to reduce their prices if they wanted to win a bid. Footnote 102 On the other hand, small sellers could not obtain any significant share of the institutional market if the relevant patent licensing contract included a field-of-use restriction.
By contrast, in the retail segment, those companies that sold patented brand-name tranquilizers were able to maintain their market price against a lower-priced drug sold under its generic name. In this submarket, there was no price competition, even when smaller firms were present as competitors. As a consequence, the strategy of including field-of-use restrictions in licensing contracts made no sense in the retail prescription submarket, because the presence of smaller firms selling the same drug at lower prices did not threaten the licensor’s monopoly. High advertising and promotional costs prevented price competition from smaller firms in relation to ethical drugs, regardless of the inclusion of field-of-use restrictions in licensing contracts.
Although there was no price competition from small firms selling patented drugs under their generic names in the retail market, competition among patented drugs was a significant issue, because these drugs were often highly substitutable and could compete with one another. In this situation, the big pharmaceutical firms attempted to sell their products in the retail market, not by offering lower prices but by investing large sums in advertising, promotion, and marketing. The high volume of advertising arose from fierce competition among new products due to high rates of product obsolescence and rapidly growing markets. These conditions promoted competition and provided an impetus to innovate. The final aim of promotional activities was to differentiate products from those of competitors and to attract the attention of physicians. This product competition provided a societal benefit: it increased the rate at which firms developed new drugs and accelerated the introduction of improved (though not innovative) drugs, thus providing patients with a greater range of choices. Footnote 103
Finally, we argued that it is not possible to show that monopolies arose from patent privilege, because product competition undoubtedly existed. High prices for patented drugs allowed the financing of sales campaigns due to product rivalry, although with little or no price difference involved. Patented products were often highly substitutable and could thus compete with one another; additionally, patents did not ensure a monopoly position without a significant selling effort. Therefore, advertising was complementary to innovation: competition between identical chemical entities substantially increased the effectiveness of sales efforts in this industry. Thus, we found a positive and increasing feedback between innovation and advertising that was essential to this industry and was particularly significant in the case of tranquilizers. Pharmaceutical firms used patents and complementary heavy investment in promotion and marketing as a strategy for increasing their market share and to combat competition from ethical drugs that were chemically different yet offered comparable therapies.





