



Introduction
Schizophrenia is a still all-too-often chronic and debilitating psychiatric disorder that is characterized by a combination of positive and negative symptoms, cognitive dysfunction, affective and motor disturbances that often result in functional impairment and poor quality of life.Reference Kahn, Sommer and Murray1 , Reference DiBonaventura, Gabriel and Dupclay2
Current treatment guidelines suggest long-term treatment with antipsychotic medications in conjunction with psychological interventions, for people with first episode psychosis, an acute exacerbation or recurrence of psychosis.3 Despite pharmacologic advances in recent years, an effective treatment of schizophrenia remains an issue, as knowledge about the pathophysiology of this complex disorder is lacking.Reference Kahn, Sommer and Murray1 , Reference Kane and Correll4 The most challenging components of treatment effectiveness are adherence and efficacy for negative and cognitive as well as residual positive symptoms and adverse effects.Reference Kahn, Sommer and Murray1
Dopamine modulating antipsychotics remain the primary and only currently approved treatment for schizophrenia, and over the past years many different antipsychotics have been developed and tested for their efficacy and safety in reducing acute symptoms of schizophrenia and maintaining stability.Reference Leucht, Leucht and Huhn5 –Reference Kishimoto, Hagi and Nitta9 However, currently available drugs are mostly effective against positive symptoms (i.e., hallucinations, delusions, disorganized thought or speech, and bizarre behaviors). This situation leaves most patients with diverse residual symptoms relatively untreated, especially negative symptoms (lack of motivation, drive, enjoyment, social interactions) and cognitive dysfunction (deficits in attention, memory, executive functioning, grasp of social situations/interactions), which lead to poor quality of life outcomes and lifelong impairments,Reference Lieberman10 –Reference Mosolov, Potapov and Ushakov18 as cognitive and negative symptoms persist for most of the patients’ lives.Reference Correll15 , Reference Millan, Fone and Steckler19 To date, there are no FDA-approved medications for negative and cognitive symptoms, and augmentation strategies have been largely disappointing so far.Reference Carbon and Correll20 , Reference Correll, Rubio and Inczedy-Farkas21
Furthermore, as at least positive symptoms in schizophrenia have primarily been associated with dopamine dysfunction, all currently available effective antipsychotics have dopamine D2 receptor occupancy as a key feature, which is also associated with a varying degree of adverse effects, including motor impairments and prolactin abnormalities, with additional dopamine or off-target receptor activity-related effects on sedation, weight gain, metabolic disturbances and cardiovascular risk factors, morbidity and mortality.Reference Solmi, Murru and Pacchiarotti22 –Reference Vancampfort, Stubbs and Mitchell29 In addition, too much dopamine blockade has been associated with secondary negative and cognitive symptoms.Reference Carbon and Correll20
However, drugs with dopamine D2 receptor occupancy have remained the only approved and effective pharmacologic treatment for schizophrenia, since its discovery in the 1950s.Reference Insel16 Currently, only clozapine, which has considerable adverse effects,Reference Nielsen, Correll and Manu30 has been found superior to other antipsychotics in treatment-resistant schizophrenia,Reference Siskind, Siskind and Kisely31 defined as a failure to at least two non-clozapine antipsychotics.Reference Howes, McCutcheon and Agid32 Thus, less side effect–prone agents in monotherapy or as augmentation are needed for patients with suboptimal positive symptom response or treatment-resistance.
Recent findings suggest that the core pathophysiology of schizophrenia may also involve dysfunction of glutamatergic, serotonergic, cholinergic, and gamma-aminobutyric acid (GABA) signaling, with an imbalance within any of these influencing the entire system.Reference Yang and Tsai33 Therefore, novel treatment approaches have also been focusing on molecular targets beyond the dopamine hypothesis, including glutamate, serotonin, acetylcholine, GABA, and inflammatory cytokines.Reference Garay, Citrome and Samalin34 , Reference Köster, Carbon and Correll35
Due to the goal of maintenance treatmentReference Correll, Rubio and Kane8 and long-standing problems with adherence in schizophrenia,Reference Kane, Kishimoto and Correll36 long-acting injectable antipsychotics (LAIs) have been developed and largely shown benefits for prevention of relapse, hospitalization, and mortality in patients with schizophrenia.Reference Kishimoto, Hagi and Nitta37 –Reference Kishimoto, Robenzadeh and Leucht42 Novel LAI formulations have emerged, aiming to improve acceptability and avoid the need for oral cotreatment or loading strategies.
Therefore, this selective review aimed to (i) summarize the mechanisms of action and, if available, the results of novel, emerging, and currently investigated pharmacologic treatments for total, negative, and cognitive symptoms of schizophrenia, as well as residual and resistant positive symptoms, (ii) summarize the latest clinical data of newly approved formulations of LAIs, and (iii) evaluate the data for the addition of opioid receptor modulation to mitigate olanzapine-associated weight gain.
Methods
We conducted a selective review of (i) different pharmacological drug targets for the treatment of total, negative, and cognitive, as well as residual or resistant positive symptoms of schizophrenia, (ii) new FDA-approved and investigated LAIs, and (iii) samidorphan-olanzapine combination. For this purpose, we conducted in March 2019 a series of targeted literature searches in PubMed and clinicaltrials.gov for “schizophrenia” paired together with the following mechanisms of action or psychopharmacological agents: phosphodiesterase 9 inhibitor, PDE-9 inhibitor, phosphodiesterase 9 (PDE9) inhibitor, phosphodiesterase 10A (PDE10A) inhibitor, d-amino acid oxidase (DOAA) inhibitor, cannabidiol, glycine transporter-1 (GLYT1) inhibitor, dopamine D1 antagonist, dopamine D1 partial agonist, dopamine D2 antagonist, dopamine D2 partial agonist, dopamine D3 antagonist, dopamine D3 partial agonist, trace amine-associated receptor-1 (TAAR1) agonist, 5-HT2A inverse agonist, sigma-2/5-HT2A antagonist, lumateperone (ITI-007), roluperidone (MIN-101), sodium benzoate, sodium nitroprusside, LuAF-11167, LuAF-35700, BI-425809, F17464, MK-8189, RO5263397, SEP-363856, TAK-063, TAK-831, Aripiprazole Lauroxil NanoCrystal® or Aristada Initio, Doria® or risperi-done ISM®, paliperidone palmitate, risperidone extended release, TV-46000, Perseris and RBP-7000. Excluded were agents or trials evaluating patients with other psychotic disorders than schizophrenia, or mixed samples.
Results
Total symptoms of schizophrenia (including positive and negative symptoms)
Although many first- and second-generation antipsychotics are available, all modulate dopamine with or without serotonergic transmission modulation, but additional mechanisms of actions conferring efficacy, either added on separately or combined with dopamine-serotonin antagonism, are needed. Furthermore, improved safety and tolerability also remains a goal. Several agents are at different stages of testing.
Cannabidiol
While tetrahydrocannabinol (THC), the major component of cannabis, is pro-psychotic, the potential antipsychotic and precognitive properties of cannabidiol for schizophrenia and its efficacy for other neuropsychiatric disorders are actively being pursued.Reference Ibeas Bih, Chen and Nunn43 –Reference Leweke, Mueller and Lange45
In an exploratory 6-week, double-blind study, patients with schizophrenia were randomized to cannabidiol (n = 43) or placebo (n = 45) added to preexisting antipsychotics.Reference McGuire, Robson and Cubala46 Although no differences between cannabidiol and placebo emerged regarding the Positive and Negative Syndrome Scale (PANSS) total or negative and general scale scores, cannabidiol had significantly lower PANSS positive scores at week 6. Furthermore, cannabidiol was superior to placebo in being rated as improved (Clinical Global Impressions-Improvement (CGI-I)) and as not severely unwell (CGI-Severity). However, cannabidiol was not superior to placebo on the Global Assessment of Functioning (GAF) Scale or the Brief Assessment of Cognition (BACS) (Table 2). Cannabidiol was well tolerated, with similar adverse event frequencies between cannabidiol and placebo.
TABLE 1. Selected novel and emerging non-pharmacological and pharmacological treatments for schizophrenia
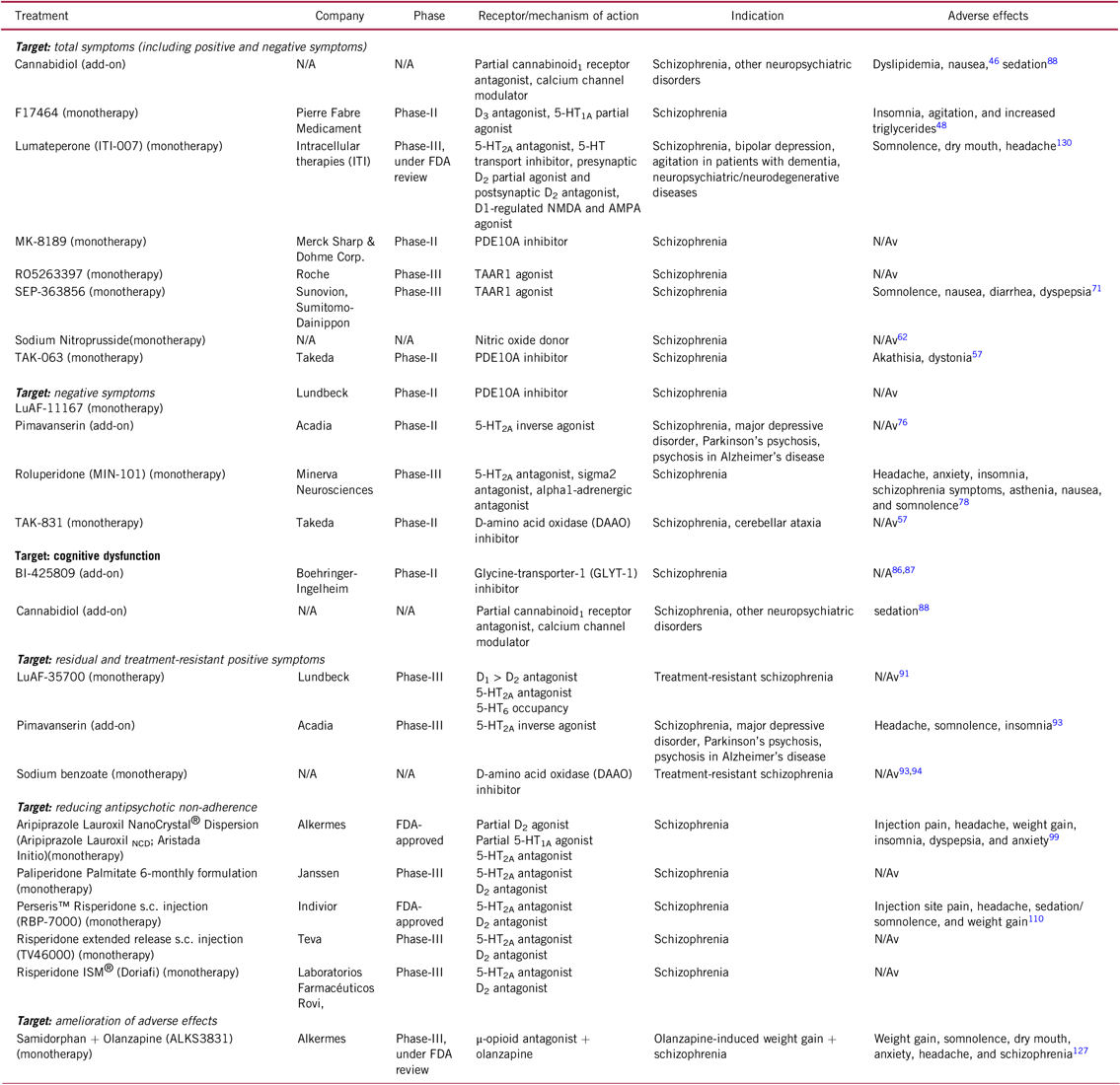
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, N/A, not applicable; N/Av, not available, NMDA, N-methyl-D-aspartate, PDE, phosphodiesterase, s.c. , subcutaneous, TAAR, trace amine-associated.
TABLE 2. Selected novel and emerging pharmacological treatments for schizophrenia targeting total (including positive and negative) symptoms
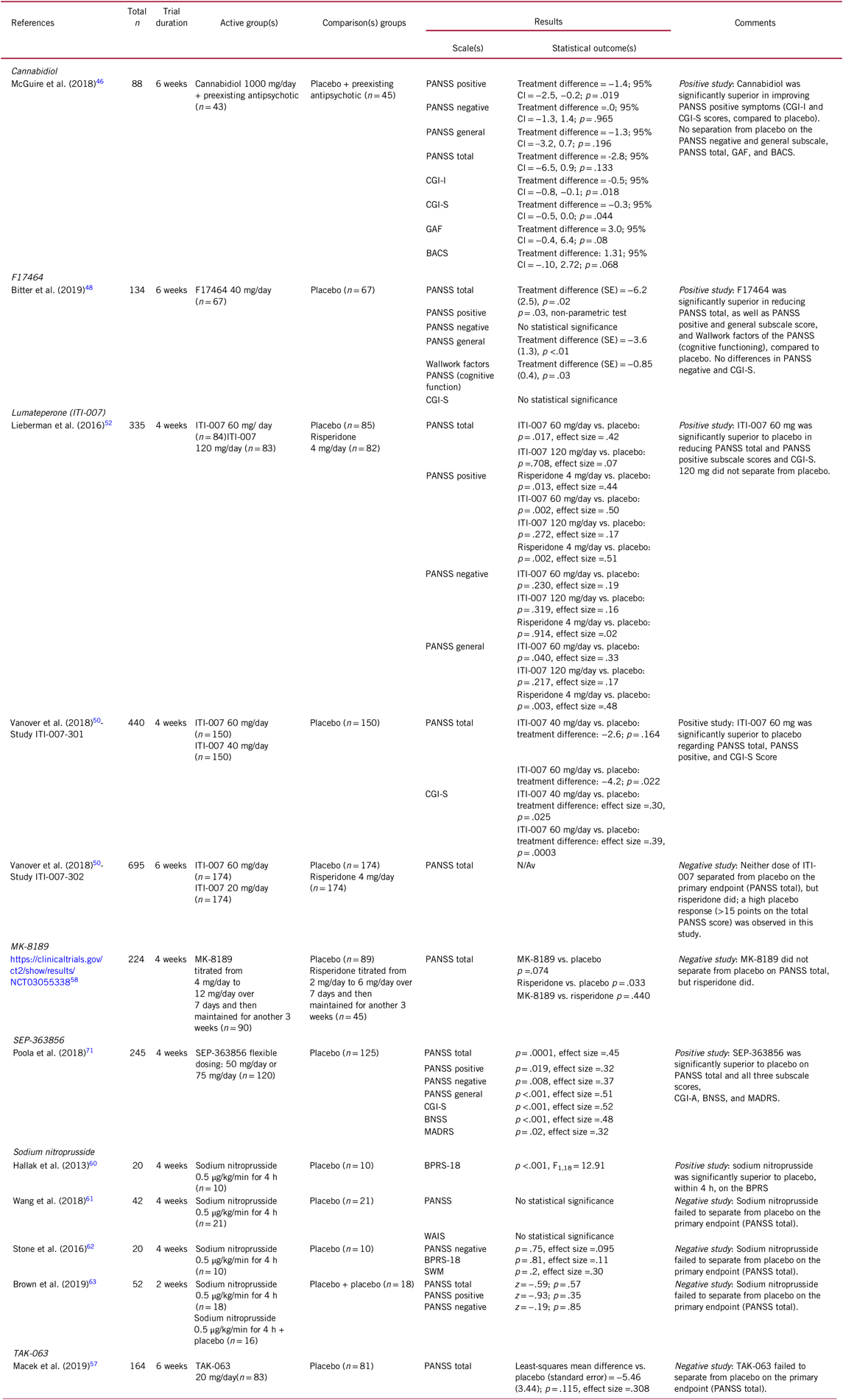
BACS, Brief Assessment of Cognition in Schizophrenia; BNSS, Brief Negative Symptom Scale; BPRS-18, 18-Item Brief Psychiatric Rating Scale; CBD, Cannabidiol; CGI-I, Clinical Global Impressions Scale – Improvement; CGI-S, Clinical Global Impressions Scale – Severity; CI, confidence interval; GAF, Global Assessment of Functioning; MADRS, Montgomery-Asberg Depression Rating Scale; OR, odds ratio; PANSS, Positive and Negative Syndrome Scale; SOC, standard of care; SWM, Spatial Working Memory; WAIS, Wechsler Adult Intelligence Scale.
F17464
F17464 is an oral, selective D3 antagonist/5-HT1A partial agonist that is developed for the treatment of schizophrenia.Reference Sokoloff and Le Foll47
In a phase-II, double-blind, randomized, placebo-controlled, parallel-group 6-week trial,Reference Bitter, Lieberman and Gaudoux48 patients (n = 134) with acute exacerbation of schizophrenia were randomized to 40 mg/day F17464 or placebo. The F17464 group was significantly superior to placebo in reducing the PANSS total score, PANSS positive and general subscale score, and of the PANSS Wallwork factors, which correspond to cognitive functioning. However, F17464 did not differentiate from placebo on the PANSS negative and CGI-S scores (Table 2).Reference Bitter, Lieberman and Gaudoux48
Adverse events were comparable to placebo. Overall, results of this phase-II study demonstrated therapeutic efficacy in improving total, positive, and PANSS-derived cognitive symptoms in acutely exacerbated schizophrenia patients, with a favorable safety profile.Reference Bitter, Lieberman and Gaudoux48
Lumateperone (ITI-007)
Lumateperone, also known as ITI-007, is an investigational agent with simultaneous modulation of serotonin, dopamine, and glutamate, which is being developed for the treatment of schizophrenia, bipolar depression, and other neuropsychiatric/ neurodegenerative diseases. The FDA has accepted the new drug application (NDA) for lumateperone for the treatment of acutely exacerbated schizophrenia in December 2018.
Lumateperone acts as a presynaptic partial agonist and postsynaptic antagonist at dopamine D2 receptors with functional mesolimbic/mesocortical selectivity.Reference Davis and Correll49 In addition, it has potent serotonin 5-HT2A receptor antagonism, serotonin transporter inhibition, and stimulates phosphorylation of glutamatergic N-methyl-D-aspartate (NMDA) GluN2B receptors, likely downstream of dopamine D1 receptor intracellular signaling.Reference Davis and Correll49 Recently, the additional and unique increase of AMPA currents via the mTOR protein pathway has been described.Reference Vanover, Dimitrienko and Glass50
At the therapeutic dose of 60 mg/day, lumateperone fully saturates 5-HT2A receptors, at modest levels of dopamine receptor occupancies (30–40%), at least partially due to its wider separation (60-fold) between its affinity for 5-HT2A receptors and D2 receptors, compared to other second-generation antipsychotics, including risperidone (12-fold) and aripiprazole (0.18-fold).Reference Snyder, Vanover and Zhu51
Lumateperone has been investigated in three placebo-controlled trials of acutely exacerbated schizophrenia, two of which included risperidone for assay sensitivity as an active comparator, and all conducted in the USA only.Reference Lieberman, Davis and Correll52 In a 4-week, phase-IIb clinical trial (n = 335; ITI-007-005), ITI-007 showed statistically significant efficacy vs. placebo in improving schizophrenia symptoms, measured with the PANSS total score and its subscale scores at a dose of 60 mg/day (but not at 120 mg/day), and at a similar effect size as risperidone.Reference Lieberman, Davis and Correll52 Compared to risperidone 4 mg/day, ITI-007 was superior at reducing negative symptoms, as well as on key safety and tolerability measures including prolactin, body weight, and glucose and lipid levels.Reference Lieberman, Davis and Correll52 In a first 4-week, phase-III trial (n = 440; ITI-007-301), lumateperone 60 mg/day separated from placebo regarding schizophrenia symptoms both in PANSS total and CGI-S.Reference Vanover, Dimitrienko and Glass50 Notably, although lumateperone 40 mg/day was not superior to placebo regarding PANSS total scores, the 40 mg/day group was significantly superior regarding the CGI-S and PANSS positive subscale score (although not formally tested as the primary outcome did reach statistical significance), like the 60 mg/day group (Table 2). In a second phase-III trial lasting 6 weeks (n = 695; ITI-007-302), however, lumateperone 60 mg/day as well as 20 mg/day did not separate from placebo regarding schizophrenia symptoms, while risperidone 4 mg/day did (Table 2).Reference Vanover, Dimitrienko and Glass50 Nevertheless, although risperidone separated from placebo, it also had the greatest dropout rate, with patients dropping out early being potentially the less responsive patient subgroup, and the placebo effect was much larger than in the other two trials, making the results less reliable than in the first two trials. In terms of adverse effects, lumateperone separated on few symptoms from placebo, including only somnolence, sedation, and fatigue (Table 1). However, these adverse effects were generally only mild or moderate, and lumateperone was associated with low and placebo-like discontinuation rates.Reference Vanover, Dimitrienko and Glass50
Finally, in phase 1 of a recently completed study (n = 302, ITI-007-303),Reference Vanover, O’Gorman and Glass53 investigating the safety of ITI-007 in an open-label setting in patients with stable schizophrenia switching from standard-of-care (SOC) antipsychotics to lumateperone, 6 weeks of treatment with lumateperone led to significant improvements in both schizophrenia symptoms and key cardiometabolic parameters, such as LDL-cholesterol and triglycerides as well as prolactin levels. After only 2 weeks of switching back to SOC antipsychotics, the safety variables that had significantly improved trended back to pre-lumateperone baseline and were not significantly improved anymore.Reference Vanover, Correll and Weingart54 Interestingly, in this study program, lumateperone was dosed in the evening, and the already-low adverse event rates seen in the acute studies in which AM dosing had been applied were further reduced.
In phase 2 of the same study (n = 603, ITI-007-303)Reference Vanover, Correll and Weingart54 patients were enrolled for a planned treatment duration of up to 1 year. As the study is ongoing, data reflect an interim data cut that includes observed cases for those subjects who have had completed each visit (300 days, n = 184). After 300 days of treatment, lumateperone led to significant improvements in both schizophrenia symptoms (Table 2) and key cardiometabolic parameters, such as body weight and total cholesterol, LDL-cholesterol as well as prolactin levels.Reference Vanover, Correll and Weingart54 These results imply that patients with stable symptoms on other antipsychotics may further improve when switched to lumateperone.Reference Vanover, Correll and Weingart54
Overall, lumateperone represents a novel approach, with a unique mechanism of action, which seems to be promising in enhancing efficacy with a favorable safety profile.
Phosphodiesterase 10A (PDE10A) inhibitors
Phosphodiesterase 10A (PDE10A) inhibitors are expected to modulate via cAMP- and cGMP-dependent mechanisms both the dopamine D1-direct and D2-indirect striatal pathways and regulate the phosphorylation status of a panel of glutamate receptor subunits in the striatum.Reference Grauer, Pulito and Navarra55 Therefore, studies of PDE10 inhibitors are expected to target both dopaminergic and glutamatergic dysfunction in schizophrenia.
Several PDE10A inhibitors are currently investigated, including TAK-063 and MK-8189 for total schizophrenia symptoms.
TAK-063Reference Suzuki, Harada and Suzuki56 has shown mixed results. In a phase-II, 6-week, randomized, placebo-controlled study, subjects with an acute exacerbation of schizophrenia were randomized to TAK-063 20-mg/day (n = 83) or placebo (n = 81). Although in this sample, TAK-063 failed to separate from placebo on total PANSS at study endpoint, the effect size was small (0.308) (Table 2). Consistent with previous phase-I studies, TAK-063 was well tolerated, with higher rates of akathisia and dystonia vs. placebo.Reference Macek, McCue and Dong57
In addition, one recent negative study read out for MK-8189. In a placebo- and active-controlled, 4-week study of patients (n = 224) with an acute exacerbation of schizophrenia, MK-8189 (n = 90) was compared with placebo (n = 89) and with risperidone (n = 45). At study endpoint MK-8189 did not separate from placebo or risperidone on the PANSS total score. Conversely, risperidone significantly separated from placebo on the PANSS total score (Table 2). Furthermore, MK-8189 did not separate from placebo in other secondary outcomes (adverse events and study discontinuation).58
Sodium nitroprusside (nitric oxide donor)
Nitric oxide may be implicated in the pathophysiology of schizophrenia, as it is a gas that mediates the release of neurotransmitters, and seems to be involved in learning, memory, and neurodevelopment.Reference Crippa, Hallak and Abílio59 As a nitric oxide donor, sodium nitroprusside may thus improve symptoms of schizophrenia.
The original randomized, double-blind, placebo-controlled study of sodium nitroprusside, conducted in Brazil, examined the effectiveness and safety of a single intravenous administration of sodium nitroprusside (0.5 μg/kg/min for 4 hours) for the improvement of positive, negative, anxiety, and depressive symptoms in 20 patients with schizophrenia who were within the first 5 years of schizophrenia and who were taking antipsychotics.Reference Hallak, Maia-de-Oliveira and Abrao60 After the adjunctive infusion of sodium nitroprusside, a rapid improvement of symptoms was observed within 4 hours, with significant superiority of sodium nitroprusside vs. placebo on the 18-item Brief Psychiatric Rating Scale total score and subscale scores, which persisted for 4 weeks after infusion (Table 2).Reference Hallak, Maia-de-Oliveira and Abrao60
However, three recent studies were unable to replicate these original study results. In one study, 42 adults with schizophrenia were randomized to two placebo or sodium nitroprusside infusions (0.5 μg/kg per min for 4 h) during a 1-week interval.Reference Wang, Zhao and Hu61 The authors failed to find any significant effect of sodium nitroprusside vs. placebo on psychotic symptoms or cognitive functions, although sodium nitroprusside seemed relatively well tolerated. In a second study, 20 patients with a diagnosis of schizophrenia or schizoaffective disorder were randomized to an infusion of sodium nitroprusside (0.5 μg/kg per min for 4 h) or placebo and re-assessed for symptoms and cognitive performance immediately after the infusion, and 4 weeks later (Table 2).Reference Stone, Morrison and Koychev62 Similarly, the authors failed to observe any significant advantage of sodium nitroprusside vs. placebo for reducing psychotic symptoms or improving spatial working memory vs. placebo. Finally, in a third, 2-week acute treatment study,Reference Brown, Freudenreich and Fan63 patients (n = 52) were randomized to one of three treatment sequences (sodium nitroprusside + sodium nitroprusside; placebo + sodium nitroprusside; placebo + placebo), without separation of sodium nitroprusside from placebo in this study (Table 2). Consistent with the results of the two prior studies, no differences emerged in safety or tolerability measures.Reference Brown, Freudenreich and Fan63 However, as in the original, positive study patients were younger, in an earlier illness phase and more severely ill than in the negative trials, patient features that may enhance the efficacy of sodium nitroprusside may need to be considered in future trials.Reference Maia-de-Oliveira, Baker and Dursun64
Trace amine-associated receptor 1 (TAAR-1) agonists
Trace amine-associated receptor 1 (TAAR1) is a G-protein-coupled receptor that belongs to the TAAR family. Discovered in 2001, TAARs have been found in several tissues, including the central nervous system and olfactory epithelium.Reference Schwartz, Canales and Zucchi65 , Reference Revel, Moreau and Pouzet66 TAAR1 is activated by endogenous trace amines that are structurally related to monoaminergic neurotransmitters. TAAR1 agonists include amphetamine and methamphetamine, and TAAR1 seems to be most selective to dopamine, being less responsive to the endogenous tryptamine, norepinephrine, and serotonin, although glutamatergic transmission may also be modulated. In turn, these receptor features make TAAR1 agonists attractive targets for the treatment of schizophrenia.Reference Schwartz, Canales and Zucchi65 , Reference Revel, Moreau and Pouzet66 Currently two TAAR1 agonists are being evaluated for schizophrenia. These include SEP-36385667 , 68 and RO5263397Reference Revel, Moreau and Pouzet66 , Reference Espinoza, Leo and Sotnikova69 (for which wide variations in blood levels have been described based on genotype and ethnicity).Reference Fowler, Kletzl and Finel70
While for RO5263397, phase-II study results have to our knowledge not been publicly reported, results of a phase-IIb study of SEP-363856 have been presented as a poster.Reference Poola, Sunkaraneni and Galluppi71 In a phase-II, randomized, double-blind, placebo-controlled 4-week, flexible dose study, SEP-363856 50 mg/day or 75 mg/day (n = 120, modal dose=75 mg/day) was superior to placebo (n = 125) for PANSS total scores, PANSS positive, PANSS negative, and PANSS general psychopathology subscale score, CGI-S, Brief Negative Symptom Scale (BNSS), and the Montgomery-Asberg Depression Rating Scale (MADRS). All-cause discontinuation (approximately 20%) and intolerability-related discontinuation (<10%) were similar to placebo (Table 2). Moreover, changes in body weight, blood glucose, lipids, and prolactin levels were comparable to placebo. Adverse effects ≥2% and at a maximum 1–2.5% greater than with placebo included somnolence, nausea, diarrhea, and dyspepsia.Reference Poola, Sunkaraneni and Galluppi71
Negative symptoms
After a positive phase-II study,Reference Umbricht, Alberati and Martin-Facklam72 the bitopertin program of a GLYT-1 inhibitor was stopped due to following negative studies,Reference Bugarski-Kirola, Blaettler and Arango73 leaving again negative symptoms a highly desirable and sorely needed indication for the treatment of schizophrenia. Several different agents are currently investigated for this indication.
LuAF-11167
Since PDE10A inhibitors have not reliably separated from placebo in acutely exacerbated schizophrenia,Reference Macek, McCue and Dong57 , 58 LuAF-11167 is currently being developed for the treatment of negative symptoms.74 Since, possibly, dopamine blockade due to accompanying antipsychotic treatment in the adjunctive trial programs for acutely exacerbated schizophrenia patients may interfere with the optimized effects of a PDE10A inhibitor for negative symptoms, an ongoing study evaluates LuAF-11167 in monotherapy vs. placebo. Results are not available for this phase-II study.74
Pimavanserin
Pimavanserin is a 5-HT2A inverse agonist approved for the treatment of psychosis associated with Parkinson’s disease. In one prior study of schizophrenia patients,Reference Meltzer, Elkis and Vanover75 pimvanserin added to lower risperidone doses (2 mg/day) was equally efficacious as higher risperidone doses (6 mg/day); also pimavanserin augmentation seemed to reduce EPS. Currently, the ongoing ADVANCE study is a phase-II, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of adjunctive pimavanserin for the treatment of negative symptoms of schizophrenia.76 This 26-week study compares three doses of pimavanserin (34 mg, 20 mg, or 10 mg) plus background antipsychotic vs. placebo plus background antipsychotic, with the primary outcome measure being the Negative Symptom Scale-16 (NSA-16). Results are expected later in 2019.
Roluperidone (MIN-101)
Roluperidone, also known as MIN-101, is a cyclic amido derivative, with affinities for sigma-2, 5-HT2A, and alpha1-adrenergic receptors, developed to target negative symptoms and cognitive dysfunction in schizophrenia patients. MIN-101 has low affinity for dopaminergic, muscarinic, cholinergic, and histaminergic receptors. Recent studies have shown that MIN-101 is an antagonist at sigma-2 and 5-HT2A.Reference Keefe, Harvey and Khan77 , Reference Davidson, Saoud and Staner78
Reportedly, an unpublished phase-IIa studyReference Staner, Davidson and Noel79 was positive for negative symptoms in schizophrenia, which was replicated in a phase-IIb trial.Reference Davidson, Saoud and Staner78 In that 12-week study, 234 patients who had been symptomatically stable for at least ≥3 months and who had predominant negative symptoms (score ≥20 on the PANSS negative subscale, baseline mean = 26.8) were randomized to roluperidone 32 mg/day or 64 mg/day or placebo. At 12 weeks, MIN-101 32 mg/day and 64 mg/day were statistically significantly superior in the PANSS negative factor score (pentagonal structure model). In addition, positive results were also reported for PANSS negative symptom, total, and activation factor scores, CGI-S, and the BNSS (Table 3).Reference Davidson, Saoud and Staner78 In addition, roluperidone was safe, without clinically significant changes in vital signs, routine laboratory values, body weight, metabolic parameters, or neuromotor scale scores. Adverse events included headache, anxiety, insomnia, schizophrenia symptoms, asthenia, nausea, and somnolence.Reference Davidson, Saoud and Staner78
TABLE 3. Selected novel and emerging pharmacological treatments for schizophrenia targeting negative and cognitive symptoms

BACS, Brief Assessment of Cognition in Schizophrenia; CI, confidence interval; MATRICS, Measurement and Treatment Research to Improve Cognition in Schizophrenia; PANSS, Positive and Negative Syndrome Scale; SCoRS, Schizophrenia Cognition Rating Scale.
Results of secondary analyses from this trial also suggested that roluperidone may also have an enhancing effect on cognitive functioning.Reference Keefe, Harvey and Khan77 The BACS token motor, verbal fluency, and composite z scores improved significantly vs. placebo in the 32 mg roluperidone group, but not the 64 mg/day group, although the efficacy for negative symptoms was greater for the higher dose, and although negative correlations were seen for improvements in certain cognitive symptom domains and negative symptom reduction even in the 64 mg/day group (Table 3). Thus, it is important to note that dose-related effects of roluperidone diverged between negative and cognitive symptom improvement. Moreover, results do not yet indicate whether similar results would be observed for patients with a different range of schizophrenia symptoms.Reference Keefe, Harvey and Khan77 Nevertheless, roluperidone represents an innovative mechanism of action, which seems to be promising in improving the treatment of negative symptoms, as well as cognitive dysfunction while having a favorable safety profile. The ongoing phase-III trial program will have to confirm these initial results.80
TAK-831
D-serine is an endogenous ligand for the glycine modulatory binding site on the NR1 subunit of NMDA receptors in the brain, whose agonist functioning is thought to improve negative symptoms of schizophrenia.Reference Sacchi, Rosini and Pollegioni81 Since D-serine is degraded by the flavoenzyme DAAO, DAAO inhibitors may improve NMDA functioning and negative symptoms in schizophrenia.
TAK 831 is an oral, highly selective and potent DAAO inhibitor that is undergoing phase-II testing for the treatment of negative symptoms in schizophrenia.82 In this 12-week, placebo-controlled trial, three different doses (50 mg/day, 125 mg/day, 500 mg/day) are tested using the PANSS negative symptoms factor score as the primary outcome.
Cognitive impairment
Since the negative results for encenicline, an alpha-7 nicotinic acetylcholine receptor agonist for cognition, in schizophrenia,Reference Keefe, Meltzer and Dgetluck83 activity in this area has slowed down considerably. Since glycine is involved in gluta matergic NMDA transmission, which seems to be relevant for both negative and cognitive symptoms in schizophrenia,Reference Hashimoto, Malchow and Falkai84 GLYT-1 agonists may also improve cognitive dysfunction.
BI-425809
BI 425809 is a GLYT-1 inhibitor that was well tolerated in a phase-1 study in healthy male volunteers, displaying an adverse event profile suggestive of GLYT1-inhibiting effects.Reference Moschetti, Desch and Goetz85 BI 425809 is currently undergoing phase 2 testing for cognitive dysfunction in schizophrenia.86 In this 12-week, placebo-controlled study, four doses of once daily oral BI 425809 are evaluated for their effect on the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) Consensus Cognitive Battery (MCCB) total score, with everyday functional capacity, measured by Schizophrenia Cognition Rating Scale (SCoRS), being the key secondary outcome.86 Another 12-week, phase-II, placebo-controlled proof of concept study with the same outcomes is being planned that aims to explore the effect of oral once daily BI 425809 on cognition administration in patients with schizophrenia on stable antipsychotic treatment and adjunctive computerized cognitive training.87
Cannabidiol
Cannabidiol has also been investigated for the treatment of cognitive dysfunction in schizophrenia. In a 6-week, randomized, placebo-controlled, fixed-dose study targeting cognition, oral cannabidiol (600 mg/day) added on to background antipsychotics was not superior to placebo in 36 stable chronic schizophrenia patients on either the MATRICS CCB or the PANSS total score (Table 3).Reference Boggs, Surti and Gupta88 Similarly, in another 6-week study of 88 patients with schizophrenia focusing on total PANSS that separated on positive symptoms and CGI-I and CGI-S, there was no significant benefit of adjunctive cannabidiol (1000 mg/day) for the secondary outcome of cognition measured with the Brief Assessment of Cognition in Schizophrenia.Reference McGuire, Robson and Cubala46
Residual and treatment-resistant positive symptoms
Residual and refractory symptoms of schizophrenia remain a major unmet need in schizophrenia. Only clozapine is indicated for this indication, and few agents are being studied in this area.
LuAF-35700
LuAF-35700 is a D1-preferring D1 and D2 antagonistic investigational antipsychotic agent that is also an antagonist at serotonin 5-HT2A and 5-HT6 receptors with high affinity. Due to its relatively lower dopamine D2 occupancy, it is expected to have less antidopaminergic-related adverse effects, including EPS, prolactin elevation, dysphoria or anhedonia, and depressed mood, than most currently available antipsychotics. The FDA granted fast-track designation for LuAF-35700 in November 2015. Currently, this novel antipsychotic is in phase-III of its clinical development, evaluating two doses (10 mg/day and 20 mg/day) in the Daybreak89 and one dose (10 mg/day) in the ANEW90 study programs.
In the DAYBREAK study, 1098 patients who had failed at least one prior well-documented antipsychotic treatment trial without satisfactory clinical improvement were enrolled and underwent a single-blinded, prospective, 6-week treatment period with either risperidone (4–6 mg/day) or olanzapine (15–20 mg/day) to confirm antipsychotic treatment resistance. Those failing to adequately respond to prospective treatment with risperidone or olanzapine were randomized to either stay on this treatment or switch to LuAF-35700 for another 10 weeks. In a press release from on October 25, 2018, Lundbeck announced that in the DAYBREAK study LuAF-35700 was not superior, but as effective as olanzapine or risperidone for treatment-resistant patients (Table 4). The drug was well tolerated and safe at both doses.91
Table 4: Selected novel and emerging pharmacological treatments for schizophrenia targeting residual and treatment-resistant total symptoms
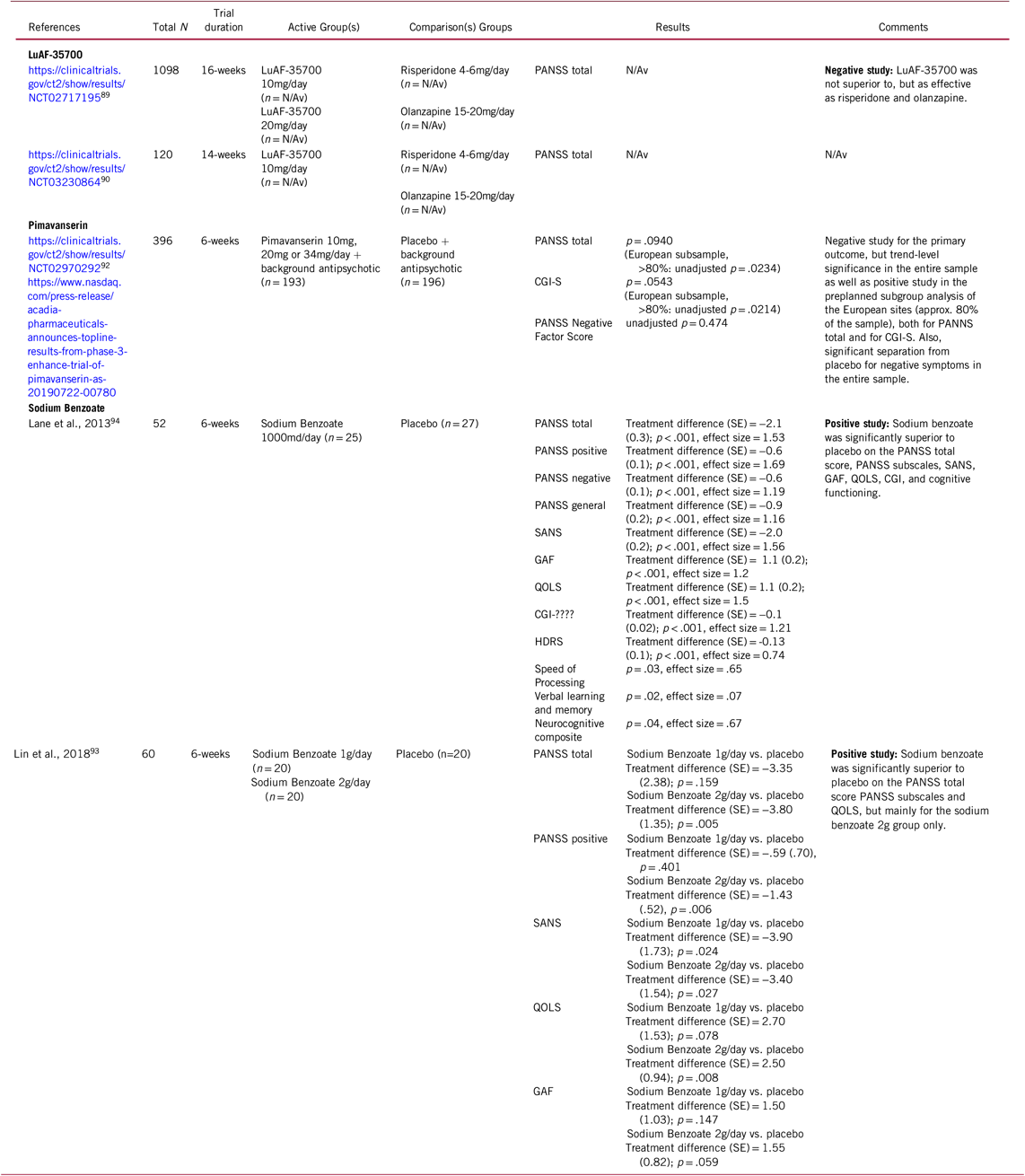
CGI = Clinical Global Impressions Scale; GAF = Global Assessment of Functioning; HDRS = Hamilton Depression Rating Scale; PANSS = Positive and Negative Syndrome Scale; N/Av = Not Available; QOLS = Quality of Life Scale; SANS = Scale for the Assessment of Negative Symptoms; SE = Standard Error
Due to these results, the ANEW was terminated that had a similar design, except for evaluating a single dose of 10 mg/day of LuAF-35700 vs. placebo and for stratifying randomization for treatment-resistant schizophrenia developing either within the first 5 years or after 10 years of the illness. Final results in a relatively small number of randomized patients are currently awaited.
Pimavanserin
Since a first augmentation study of risperidone with pimavanserin had shown that enhancing 5-HT2A receptor blockade by adding pimavanserin to a sub-effective risperidone dose provided faster onset of action, and at endpoint, equal efficacy and better safety, compared to standard dose risperidone, while reducing EPS,Reference Meltzer, Elkis and Vanover75 a program for schizophrenia patients with suboptimal positive symptom response was initiated.
The recently completed ENHANCE-1 study is a phase-III, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of adjunctive pimavanserin for the treatment of schizophrenia with residual positive symptoms.92 This international, 6-week study (n = 396, mean age = 37.2 years) compared pimavanserin (starting dose 20 mg, with adjustments possible to 34 mg or 10 mg) plus background antipsychotic vs placebo plus background antipsychotic (risperidone = 39.1%, olanzapine = 35.7%, aripiprazole = 21.3%).93 Despite numerical advantages, pimavanserin augmentation missed statistical separation from placebo for the primary outcome, PANSSS total score (p = 0.0940). A positive trend was also observed on the key secondary endpoint, the CGI-S score (p = 0.0543). In a pre-specified subgroup analysis by region, significant separation from placebo was seen in the European sites (>80% of the sample) both for the PANSS total score (unadjusted p = 0.0234), and the CGI-S score (unadjusted p = 0.0214). Furthermore, in the entire sample, pimavanserin reduced negative symptoms significantly more than placebo, both regarding the PANSS negative symptoms scale sub-score (unadjusted p = 0.0474) and the PANSS Marder negative factor score (unadjusted p = 0.0362).
Approximately 88% of pimavanserin and 96% of placebo patients completed the study. Additionally, pimavanserin was well-tolerated with low and similar rates of adverse events in the adjunctive pimavanserin (40.4%) and adjunctive placebo (36.9%) groups. Adverse events reported in at least 5% of patients in the pimavanserin group included headache, somnolence, and insomnia. Discontinuations due to adverse effects were low, being 2.5% for pimavanserin vs. 0% for placebo.93
Sodium benzoate
Not only dopaminergic hyperactivity but also NMDA receptor hypofunction has been implicated in the pathophysiology of schizophrenia, possibly especially in patients resistant to treatment with dopamine antagonizing agents.Reference Howes, McCutcheon and Agid32 In addition to NMDA partial or full agonists, NMDA functioning can be increased by raising d-amino acid levels via blockade of their metabolism. Sodium benzoate, a DAAO inhibitor and common food preservative, has this mechanism. In two placebo-controlled trials, sodium benzoate had beneficial efficacy and acceptable tolerability.93 , Reference Lin, Lin and Chang94
In the first randomized, double-blind, placebo-controlled trial,Reference Lin, Lin and Chang94 52 patients with chronic schizophrenia stabilized with antipsychotics for ≥3 months were randomized to 6 weeks of add-on treatment of 1 g/day of sodium benzoate vs. placebo. Sodium benzoate produced a 21% improvement in PANSS total score, and with large effect sizes (range = 1.16–1.69). Sodium benzoate was significantly superior to placebo in the PANSS total and subscales, Scales for the Assessment of Negative Symptoms-20 items, Global Assessment of Function, Quality of Life Scale and Clinical Global Impression. In addition, neurocognition subtests improved significantly, including the domains of processing speed and visual learning (Table 4). Sodium benzoate was well tolerated without significant adverse effects.
In the second randomized, double-blind, placebo-controlled trial,93 60 inpatients with schizophrenia stabilized with clozapine were randomized to 6 weeks of add-on treatment with 1 g/day sodium benzoate, 2 g/day sodium benzoate, or placebo. At 6 weeks, 2 g/day sodium benzoate was associated with significantly greater improvement than placebo in PANSS total score, PANSS positive subscore, and Quality of Life Scale. Furthermore, both the 1 mg/day and 2 mg/day sodium benzoate were associated with significantly greater improvement in the Scale for the Assessment of Negative Symptoms vs. placebo (Table 4). Sodium benzoate was well tolerated without relevant adverse effects. Furthermore, as an indication of the mechanism involved in the significant results, changes of the antioxidant catalase differed among the three groups and were correlated with the improvement of PANSS total score and PANSS positive score in the sodium benzoate group.
Agents targeting antipsychotic non-adherence
Non-adherence remains a serious issue with schizophrenia as with many other psychiatric and medical disorders.Reference Kane, Kishimoto and Correll36 Although several second-generation LAIs have been approved,Reference Lane, Lin and Green95 several have required oral cotreatment for several weeks until therapeutic antipsychotic levels have been reached. Moreover, historically, all LAIs were injected deep intramuscularly. Four innovative LAI formulations have been developed to avoid the need for loading or booster injections and/or oral cotreatment. Furthermore, two LAI formulations have received recent FDA approval, one being the first subcutaneously injected LAI.
Aripiprazole lauroxil NanoCrystal® Dispersion (ALNCD; Aristada Initio)
Aripiprazole lauroxil (AL) (Aristada®) is an extended-release, intramuscular long-acting injectable (LAI) aripiprazole prodrug with high dosage and considerable dosing interval flexibility (441, 662, and 882 mg monthly, 882 mg every 6 weeks, or 1064 mg every 8 weeks). However, due to being a prodrug and slow release, therapeutic drug levels are achieved only gradually, so that oral aripiprazole supplementation for 21 consecutive days is required after the first intramuscular injection.
In order to achieve faster efficacy and significantly shorten or eliminate oral supplementation during the initiation phase, a 1-day intramuscular initiation regimen for aripiprazole lauroxil (Aristada Initio™) was approved by the FDA on July 2, 2018.Reference Correll, Citrome and Haddad96 The single injection comprises the same prodrug used in aripiprazole lauroxil but with smaller particles, known as the nanocrystalline milled dispersion of aripiprazole lauroxil. The smaller nanoparticle size enables faster dissolution of aripiprazole lauroxil, resulting in faster release of aripiprazole into plasma.97 Simultaneous administration of the 1-day initiation regimen with all approved aripiprazole lauroxil dosing regimens is predicted to achieve the correct blood levels within 4 days.98
A recent phase-III, double-blind, placebo-controlled study compared a 1-day initiation regimen with the 21-day oral coverage regimen. Patients (n = 161) randomized to the 1-day initiation regimen received a 675 mg injection of aripiprazole lauroxil NanoCrystal® Dispersion (ALNCD), a single 30 mg oral aripiprazole tablet, and then an aripiprazole lauroxil injection (441 mg or 882 mg). Subjects randomized to the 21-day oral coverage regimen received 15 mg oral aripiprazole and an aripiprazole lauroxil injection (441 mg or 882 mg) on day 1, followed by 20 days of 15 mg oral aripiprazole. The 1-day regimen groups had comparable aripiprazole exposure to the corresponding 21-day groups (Table 5). The most common side effects were injection pain, headache, weight gain, insomnia, dyspepsia, and anxiety in both therapies.Reference Hard, Wehr and Sadler99
TABLE 5. Selected novel and emerging pharmacological treatments for schizophrenia targeting the amelioration of antipsychotic non-adherence and body weight gain
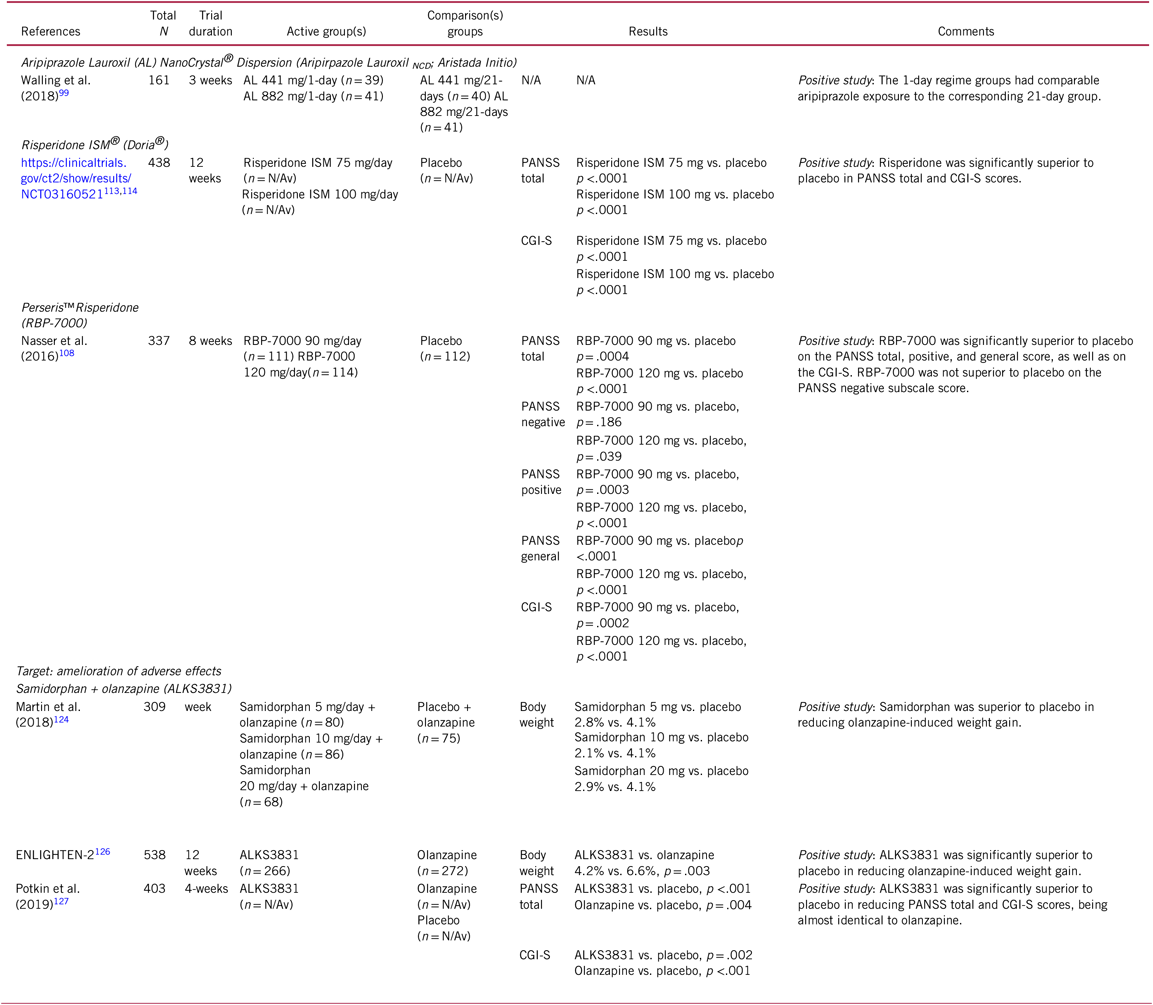
CGI-S, Clinical Global Impression Scale – severity; N/Av, not available; PANSS, Positive and Negative Syndrome Scale.
Therefore, aripiprazole lauroxilNCD Technology can offer an alternative aripiprazole lauroxil initiation regimen that ensures therapeutic aripiprazole levels during the first 21 days of treatmentReference Hard, Wehr and Sadler99 and has a comparably side effect profile, faster efficacy, and might positively affect adherence due to a very minimized (1 day) oral supplementation.
Paliperidone 6-monthly formulation
After developing a once-monthly and a 3-monthly intramuscular injectable formulation of paliperidone,Reference Walling, Hard and Wehr100 Janssen is currently conducting phase III trial for a 6-monthly formulation of paliperidone palmitate. Results are still outstanding.
Perseris™ risperidone for subcutaneous injection (RBP-7000)
Risperidone has been available since 1993 to treat schizophrenia and bipolar disorders. Risperidone blocks serotonergic 5-HT2A and dopaminergic D2 receptors with greater affinity to the 5-HT2A receptor. In addition, there is a much less affinity to other receptors such as alpha-1- and alpha-2-adrenergic receptors as well as H1-histaminergic receptors.Reference Mathews, Gopal and Nuamah101 Up until 2018, risperidone LAI was only available in a microsphere formulation that had to be injected deep intramuscularly every 2 weeks and that required a 21-day oral risperidone supplementation phase at the initiation of risperidone microsphere treatment.Reference Lane, Lin and Green95
RBP-7000 (Perseris™) is a recently (July 27, 2018) FDA-approved subcutaneous LAI formulation of risperidone102 that requires no oral supplementation and is the first antipsychotic available as a subcutaneously (intraabdominally) administered LAI.103 The once monthly injected depot formulation uses the ATRIGEL© administration system. After injection, the delivery system solidifies upon contact with bodily fluids, and the resulting biodegradable implant delivers the drug for a longer period depending on the dose strength and the injection volume.Reference Citrome104
Pharmacokinetic population analyses reported optimized occupancy of the dopamine D2 receptor for RBP-7000 after repeated doses of 90 mg/day and 120 mg/day and a potentially improved side effect profile compared to oral risperidone.Reference Patel105 , Reference Gomeni, Heidbreder and Fudala106 An 8-week, pivotal phase-III double-blind, placebo-controlled trial in acutely exacerbated patients with schizophrenia demonstrated the efficacy, safety, and tolerability of RBP-7000 (90 mg/day and 120 mg/day).Reference Laffont, Gomeni and Zheng107 –Reference Nasser, Henderson and Fava109 Common treatment-related side effects were injection site pain, headache, sedation/somnolence, and weight gain. Mean subject-reported injection site pain, as measured by the Visual Analog Scale scores (0 = no pain to 100 = unbearably painful), was similar in all treatment groups following both injections, with pain scores decreasing from a mean of 27 at 1 min after the first dose to a range of 3–7 at 30–60 min postdose. Moreover, de novo patients were recruited, and completers from the double-blind placebo-controlled study were transferred to a 1-year open-label study in which they received monthly injections of RBP-7000 (120 mg/day). In general, the safety achieved with 12 monthly injections of RBP-7000 was comparable to that of oral risperidone (Table 5). In addition, symptoms continued to improve in completers of the placebo-controlled trial who moved on to the long-term study.Reference Ivaturi, Gopalakrishnan and Gobburu110
RBP-7000 has fast efficacy without the need for loading or booster injections or oral cotreatment and the subcutaneous injection route, which is hoped to be associated with less injection pain improved convenience, acceptance, and adherence. However, like risperidone microspheres, RBP-7000 needs to be refrigerated; furthermore, it needs to be reconstituted, and a large bore needle (18 G 5/8-inch length needle) is required for the subcutaneous intrabdominal injection.103
Risperidone extended release for subcutaneous injection (TV-46000)
Currently, risperidone extended release (TV-46000) is being studied in different dose regimens administered subcutaneously in patients with schizophrenia aged 13–65 years. One placebo-controlled study evaluates the efficacy, safety, and tolerability of two dose regimens of TV-46000 as relapse prevention treatment of patients who are clinically stabilized and eligible for risperidone treatment.Reference Graham, Fava and Newcomer111 One TV-46000 dose-controlled study evaluates the long-term safety and tolerability of two dose regimens and will last up to 80 weeks (including a 4-week screening period, 12-week oral conversion/stabilization stage, 56-week double-blind maintenance stage, and 8-week follow-up period).112 Results are expected in the middle or end of 2020.
Risperidone ISM® or Doria®
Recently, positive topline results of the PRISMA-3 trial, a multicenter, randomized, placebo-controlled phase-III study of once-monthly intramuscular Doria® or Risperidone ISM®, were announced.113 ISM® is a technology for the release of drugs that is based on the in-situ formation of biodegradable matrices after the administration of a liquid carrier. Due to its special characteristics, therapeutic antipsychotic blood levels are achieved without requirement of initial oral antipsychotic coadministration or initial loading or booster injections. In the PRISMA-3 study114 of 438 acutely exacerbated patients with schizophrenia, once-monthly injections of 75 mg and 100 mg of Risperidone ISM® each showed statistically significant improvement (p < .0001) vs. placebo injections in the total score of the PANSS as well as in the CGI-Severity scale at 12 weeks, the pre-specified primary and key secondary efficacy endpoints (Table 5).113 A long-term, open label study that evaluates the safety, tolerability, and durability of the long-term effects of Risperidone ISM® is under way.
Amelioration of antipsychotic-related adverse effects
The recent FDA approval of the vesicular monoamine transporter-2 (VMAT-2) inhibitors valbenazine and deutetrabenazine for the effective amelioration of tardive dyskinesia associated with antidopaminergic medications in schizophrenia115 was the first indication of a novel mechanism molecule specifically for the management of an adverse effect of antipsychotic medications. Currently, ALKS3831 is under study to ameliorate weight gain associated with olanzapine treatment.
Olanzapine-related weight gain
Most currently available antipsychotics are associated with considerable weight gain and cardiometabolic adverse effects.Reference Solmi, Murru and Pacchiarotti22 , Reference Correll, Detraux and De Lepeleire23 , Reference Solmi, Pigato and Kane116 These adverse effects are relevant for cardiovascular morbidity and mortality,Reference Correll, Solmi and Veronese27 and are especially prominent early on in treatment and especially with clozapine and olanzapine.Reference Correll, Manu and Olshanskiy117 –Reference Correll, Lencz and Malhotra119 Since both clozapine and olanzapine have been associated with efficacy advantages,Reference Correll, Lencz and Malhotra119 , Reference Leucht, Cipriani and Spineli120 mitigating the weight gain and/or metabolic burden is of high clinical importance.
Fixed-dose combination of olanzapine and samidorphan (ALKS3831)
Samidorphan is an opioid antagonist, which originally has been investigated for the treatment of addiction and which is now examined for its mitigating or preventing effect of olanzapine-induced weight gain. Samidorphan (ALKS33) binds with high affinity to human µ-, k-, and ∂-opioid receptors and functionally acts as an µ-opioid receptor antagonist with low intrinsic activity at k- and ∂-opioid receptors.Reference Lieberman, Stroup and McEvoy121 By blocking opioid receptors involved in the brain reward pathway augmentation is decreased and cravings are reduced.Reference Wentland, Lou and Lu122 Results of a phase-I trial in healthy volunteers indicated that combining olanzapine with samidorphan (ALKS3831) did not affect the pharmacokinetic profile of either drug.Reference Silverman, Martin and Memisoglu123 Similarly, in a phase-I proof of concept study in healthy volunteers, samidorphan in combination with olanzapine was associated significantly with less weight gain than olanzapine alone.Reference Wentland, Lou and Lu122 In recent studies, the combination drug ALKS3831 has been investigated, comprising a flexible dose of olanzapine and a fixed dose (10 mg/day) of samidorphan.
Results of a 12-week, phase-II trial (ENLIGHTEN-1) suggested mitigating effects of ALKS3831 on olanzapine-induced weight gain in schizophrenia patients across all dose-ranges of olanzapine, with equivalent efficacy to olanzapine and with a similar safety profile, except for significantly less weight gain.Reference Sun, McDonnell and von Moltke124 This study had a 1-week open-label olanzapine lead-in period, which was followed by 12 weeks of double-blind treatment with olanzapine plus placebo (n = 75) or olanzapine plus 5 mg (n = 80), 10 mg (n = 86), or 20 mg (n = 68) of samidorphan. Treatment with ALKS3831 was associated with statistically significant lower weight gain (37% lower weight gain vs. olanzapine plus placebo) (Table 5). Adverse events reported at a frequency ≥5% in any of the ALSK3831 groups and occurring at least two times more frequently than with olanzapine plus placebo included somnolence, sedation, dizziness, and constipation. Since all patients received a 1-week olanzapine monotherapy run-in treatment and since olanzapine alters metabolic glucose and lipid parameters within a week, this study was not designed to assess metabolic effects of ALKS 3831.
These positive results have recently been replicated in a 6-month study (ENLIGHTEN-2) where ALKS-3831 was associated with statistically significant advantages over olanzapine plus placebo for both primary endpoints, percentage change in body weight at 6 months and proportion of patients gaining ≥10%, as well as for the key secondary endpoint, the proportion of patients gaining ≥7%.Reference Martin, Correll and Weiden125 At 6 months, weight increased with ALKS3831 (n=266) by 4.21% vs. 6.59% with olanzapine (n=272), translating into a 57% lower mean percent weight change (p=0.003). Similarly, weight gain ≥10% ALKS3831 occurred in 17.8% with ALKS 3831 vs. 29.8% for olanzapine (p = .003, number-needed-to-treat (NNT)=9). Finally, weight gain ≥7% ALKS3831 occurred in 27.5% with ALKS 3831 vs. 42.7% with olanzapine (Table 5). For weight gain ≥2%, ≥5%, and ≥15% at six months, similar results were observed, each favoring ALKS3831. In the ENLIGHTEN-2 study, there was no significant difference between ALKS3831 and olanzapine at the end of 6 months in any of the investigated glucose and lipid metabolism parameters. However, somewhat surprisingly, the mean increases in fasting glucose, insulin, HbA1C and lipids were relatively small, which may have reduced the power to differentiate ALKS 3831 from Olanzapine. The most common adverse events with ALKS3831 were weight gain, somnolence, and dry mouth; the most common adverse events with olanzapine were weight gain, somnolence, and increased appetite.Reference Correll, Kahn and Silverman126
Finally, in a 4-week, phase-III, randomized, double-blind active (olanzapine monotherapy) and placebo-controlled trial in patients experiencing acute exacerbation of schizophrenia, ALKS3831 was investigated to demonstrate that addition of the centrally active samidorphan to olanzapine in ALKS3831 does not reduce olanzapine’s efficacy. This study showed superior antipsychotic efficacy of ALKS3831 compared to placebo, with almost identical efficacy as olanzapine monotherapy (Table 5).127 ALKS3831 was generally well tolerated, with a safety profile similar to olanzapine. Adverse events leading to study discontinuation included weight gain, somnolence, dry mouth, anxiety, headache, and schizophrenia. In this short, 4-week study, body weight gain was not dissimilar between olanzapine and ALKS3831. Nevertheless, the results must be seen within the study’s limitations, including baseline difference between groups with regard to BMI, the acute nature of the study population, inpatient treatment, and a short duration of the trial, making it difficult to compare weight gain between the three groups, as ALKS3831 is known to exert its weight gain mitigating effect only after 3 weeks of treatment.127
Overall, ALKS 3831 has proven to be effective in the treatment of schizophrenia and has been associated with significantly less olanzapine-induced weight gain.127 Questions still remain regarding the potential of ALKS3831 to also mitigate olanzapine’s adverse effects on glucose and lipid parameters, and on oxidative stress and inflammation that are generally associated with significant body weight gain.
Summary and Conclusion
This targeted and selective review (i) differentiated the mechanisms of action of new and emerging antipsychotics and medications with novel mechanisms targeting total, positive, negative, and cognitive symptoms of schizophrenia, as well as residual and treatment-resistant positive symptoms, non-adherence, and olanzapine-related weight gain, and (ii) evaluated the latest clinical data on these agents inasmuch as these are already available. Successful phase-III and/or phase-II trial programs have included (i) new formulations of aripiprazole lauroxil LAI and risperidone LAI that have been able to avoid the need for oral cotreatment, as well as, in the case of the two risperidone LAI formulations, loading/booster injections in the initial phases of LAI treatment,Reference Hard, Wehr and Sadler99 , Reference Ivaturi, Gopalakrishnan and Gobburu110 , 113 leading to FDA approval of Aripiprazole Lauroxil NanoCrystal® Dispersion (ALNCD; Aristada Initio) and once-monthly subcutaneously injected Perseris™ Risperidone (RBP-7000); (ii) the treatment of acutely exacerbated schizophrenia with lumateperone, a novel antipsychotic modulating dopamine, serotonin and, via D1 agonism, glutamatergic transmissionReference Vanover, Dimitrienko and Glass50 that is currently being evaluated by the FDA for approval for acutely exacerbated schizophrenia, and (iii) the combination of samidorphan plus olanzapine in one pill for the reduction of body weight gain with ALKS3831 at 3 and 6 months,Reference Wentland, Lou and Lu122 , Reference Sun, McDonnell and von Moltke124 , Reference Martin, Correll and Weiden125 while maintaining olanzapine’s efficacy.Reference Sun, McDonnell and von Moltke124 , 127 , Reference Potkin, Kunovac and Silverman128
The other selectively reviewed agents have had either suggestive or mixed or, even, negative findings, or are at different stages of phase-II or phase-III development for different symptom domains of schizophrenia. Suggestive and encouraging results exist for monotherapy with roluperidone (MIN-101) for negative symptoms,Reference Davidson, Saoud and Staner78 and the TAAR1 agonist SEP-363856,Reference Poola, Sunkaraneni and Galluppi71 D3 antagonist/5HT1A partial agonist F17464, cannabidiol, and sodium benzoate for total psychotic symptoms. Negative findings have been reported for total symptom reduction with PDE10A inhibitors (e.g., MK-8189 and TAK-063) and sodium nitroprusside given adjunctively with background antipsychotics in acutely/severely ill patients, for cannabidiol targeting cognitive dysfunction in schizophrenia, and for monotherapy with the D1-preferring D1/D2-antagonist LuAF-35700 for treatment-resistant schizophrenia. The pimavanserin trial for residual positive symptoms was negative for the primary and key secondary outcome in the overall sample, but showed significant separation from placebo in the prespecified regional analysis of the European sites that had enrolled >80% of the sample, and showed significant separation from placebo for negative symptoms in the overall analysis set.93 Relevant data are expected soon for pimavanserin augmentation of antipsychotics for predominant negative symptoms. Trials are under way, but in the early stages for the DAAO inhibitor TAK-831 given adjunctively with antipsychotics and the phosphodiesterase 10A inhibitor LuAF-11167 in monotherapy for negative symptoms, as well as the GLYT-1 inhibitor BI-425809 targeting cognitive dysfunction in schizophrenia.
Of note, the findings of lumateperone showing that the lower 40 mg dose was significantly superior to placebo for positive symptoms and CGI-S, but not for total symptoms,Reference Vanover, Dimitrienko and Glass50 suggest that in future studies of novel agents that have minimal adverse effects, the broad-spectrum endpoint of total PANSS may be better exchanged for the more specific PANSS positive subscale when targeting the reduction of psychotic symptoms.
Interestingly, since dopamine blockade of background antipsychotic treatment may possibly interfere with the efficacy of novel, non-dopamine receptor targeting agents that focus on the improvement of negative symptoms, two trial programs attempt the testing of monotherapy of a non-dopaminergic mechanism molecular entity. These include the phase-II - PDE-10 inhibitor LuAF-11167 trial program74 and the 5HT2A/sigma 2 antagonist roluperidone (MIN-1010).Reference Davidson, Saoud and Staner78 While data from the ongoing phase-II PDE10A inhibitor LuAF-11167 trial program are not available, roluperidone has already shown in one phase-II trial superiority vs. placebo for negative symptoms.Reference Davidson, Saoud and Staner78 Although this superiority did not seem to have been accompanied by too many psychotic exacerbations (surprisingly both in the roluperidone and in the placebo arm), despite withdrawal of the antipsychotic for the 3-month study period (plus an up to 4-week washout period), concern remains as to the safety of both roluperidone and LuAF-11167 that may not be sufficiently antipsychotic in and of themselves to protect patients from a psychotic relapse. It will be important to observe longer-term data with each of these agents and to identify characteristics of patients with relevant levels of negative symptoms who could possibly be sufficiently stable to be switched safely to a non-dopamine targeting novel agent. These studies will need to prove that, in fact, such non-dopamine modulating agents can improve negative symptoms while maintaining positive symptom stability, despite the removal of the prior dopamine modulating antipsychotic agent that at one point was needed to reduce schizophrenia symptoms.
Even 66 years after the serendipitous discovery of chlorpromazine as the first “antipsychotic” agent, the most challenging issue in the pharmacological management of this complex and multifactorial disorder is the need for increased efficacy, reduced side effects, and exploring novel approaches to effectively treat negative symptoms, cognitive dysfunction, and residual/treatment-resistant positive symptoms, which currently remain the biggest gaps in the management of schizophrenia.129
Although emerging data exist, results from important trial programs are still outstanding and further high-quality trials are required to evaluate the efficacy and tolerability of novel pharmacological approaches to the management of different domains of schizophrenia. Unfortunately, to date, only limited progress has been made in the exploration of novel psychopharmacological treatment targets for schizophrenia that go beyond the D2 hypothesis. Therefore, the need for novel treatment approaches, which target the entire range of complex symptoms in schizophrenia, remains urgent. Nevertheless, part of the limited success for the search of novel treatments for schizophrenia remains the limited understanding of the pathophysiology of schizophrenia.Reference Kahn, Sommer and Murray1 Therefore, a better understanding of pathophysiological mechanism and biomarkers is indispensable to ensure improved drug development for schizophrenia.
Acknowledgements
We thank Jonathan M. Meyer, MD, clinical professor in the Department of Psychiatry at the University of California, San Diego School of Medicine in La Jolla, CA, for sharing a presentation given at NEI congress 2018 that contained some of the reviewed material.
Funding
No funding declared.
Disclosure
Ms. Krogmann, Peters, von Hardenberg, Dr. Bödeker, and Mr. Nöhles have nothing to disclose.
Dr. Correll has been a consultant and/or advisor to or has received honoraria from: Alkermes, Allergan, Angelini, Boehringer-Ingelheim, Gedeon Richter, Gerson Lehrman Group, Indivior, IntraCellular Therapies, Janssen/J&J, LB Pharma, Lundbeck, MedAvante-ProPhase, Medscape, Merck, Neurocrine, Noven, Otsuka, Pfizer, Recordati, Rovi, Servier, Sumitomo Dainippon, Sunovion, Supernus, Takeda, and Teva. He has provided expert testimony for Bristol-Myers Squibb, Janssen, and Otsuka. He served on a Data Safety Monitoring Board for Boehringer-Ingelheim, Lundbeck, Rovi, Supernus, and Teva. He received royalties from UpToDate and grant support from Janssen and Takeda. He is also a shareholder of LB Pharma.
Optional Posttest and CME Certificate
CME Credit Expires: May 15, 2022
Posttest Study Guide
The posttest can only be submitted online. The below posttest questions have been provided solely as a study tool to prepare for your online submission. Faxed/mailed copies of the posttest cannot be processed and will be returned to the sender. If you do not have access to a computer, contact NEI customer service at 888-535-5600.
Phosphodiesterase 10A (PDE10) inhibitors currently being investigated, such as TAK-063 and MK-8189, are expected to target both ___________ and ___________ dysfunction in schizophrenia.
A. Cholinergic, serotoninergic
B. Dopaminergic, glutamatergic
C. Dopaminergic, serotoninergic
D. Histaminergic, glutamatergic
Which of the following investigational agent being tested as a monotherapy does not have a non-dopaminergic mechanism
A. ALKS3831
B. LuAF-11167
C. MIN-1010
Lumateperone (ITI-007), an investigational agent for the treatment of schizophrenia, simultaneously modulates which of the following:
A. Dopamine
B. Glutamate
C. Serotonin
D. A and C
E. B and C
F. A, B, and C
RBP-7000 (Perseris), a recently FDA-approved long-acting injectable (LAI) formulation of risperidone, requires an oral risperidone supplementation after the first subcutaneous injection.
A. True
B. False
The investigational agent sodium benzoate is a … .
A. d-amino acid oxidase (DAAO) inhibitor
B. glycine transporter-1 (GlyT-1) inhibitor
C. phosphodiesterase 10A (PDE10) inhibitor
Optional Online Posttest and CME Certificate Instructions
Read the article
Complete the posttest, available only online at www.neiglobal.com/CME (under “CNS Spectrums”)
Print your certificate (passing score = 70% or higher)
Questions? call 888-535-5600, or email [email protected]






 Open access
Open access