


Introduction
Many outpatients with schizophrenia or schizoaffective disorder continue to suffer from persistent symptoms, or experience side effects associated with their current maintenance antipsychotic. Treatment strategies include dose adjustments, addition of adjunctive medications, or allowing the passage of time in the hopes of achieving further symptom reduction and the diminution of adverse effects. Another important treatment consideration is whether or not to change antipsychotic medications. The substitution of one antipsychotic agent by another is informally known as “switching.” Based on the above, it is not surprising that switching between antipsychotic medications commonly occurs in the routine treatment of schizophrenia, in an effort to find the optimal regimen for an individual patient.Reference Kane and Correll1–Reference Covell, Jackson, Evans and Essock3 Switching can be motivated by the desire to improve efficacy, tolerability, or both.Reference Weiden4, Reference Weiden and Buckley5 A number of switch techniques have been employed in evaluating optimal approaches to the initiation of the new antipsychotic and/or the discontinuation of the pre-switch agent.Reference Weiden, Aquila, Dalheim and Standard6–Reference Buckley and Correll9 Despite the appeal of switching antipsychotics, there are also potential concerns of complications from attempting a switch, such as symptom exacerbation, insufficient efficacy, and new tolerability problems emerging from the “post-switch” medication.
Lurasidone10 is a second-generation antipsychotic medication that received regulatory approval for the treatment of adults with schizophrenia in the U.S. in 201011 and in Canada in 2012.12, 13 Detailed systematic reviews of the overall efficacy, tolerability, safety, and place in therapy of lurasidone can be found elsewhere.Reference Citrome14, Reference Citrome15
This report is of a 6-month extension to a core 6-week study in which outpatients, who were previously treated with another first-line antipsychotic, were successfully switched to lurasidone monotherapy.Reference McEvoy, Citrome and Hernandez16 Details of the core study, where patients in a non-acute phase of illness were randomized to 1 of 3 lurasidone dosing regimens for the initial 2 weeks of the study and then flexibly dosed for the subsequent 4 weeks of the study, can be found elsewhere.Reference McEvoy, Citrome and Hernandez16 The primary objective of the extension study was to evaluate the long-term safety and tolerability of lurasidone as measured by the proportions of patients with AEs, serious AEs (SAEs), and discontinuations due to AEs or SAEs. One of the secondary objectives was to evaluate the long-term efficacy of lurasidone.
Method
This multicenter, open-label, 6-month study was conducted at 26 clinical sites in the United States (ClinicalTrials.gov identifier: NCT01143090). All patients had completed the 6-week core study, the details of which can be found elsewhere.Reference McEvoy, Citrome and Hernandez16 All patients received open-label lurasidone. The study was reviewed and approved by an institutional review board at each study center, and the trial was conducted in accordance with Good Clinical Practice as required by the International Conference on Harmonization guidelines. Compliance with these requirements also constitutes conformity with the ethical principles of the Declaration of Helsinki.
Participants
Key inclusion criteria for the 6-week lurasidone switch study that immediately preceded the extension study were that patients had to be adults with either schizophrenia or schizoaffective disorder who had at least a partial response to, and were stable on, a first-line antipsychotic at a dose consistent with product labeling. Patients who were unstable (eg, recent hospitalization) or known to be treatment-resistant (eg, prior treatment with clozapine) were excluded. Patients also had to have a clinically relevant basis to change antipsychotics such that clinically significant efficacy or tolerability concerns had to be present on their current medication despite efforts at optimization.
In order to enroll in the extension study, patients were required to have completed the 6-week core study, be judged suitable for participation in the extension trial and able to comply with the protocol in the opinion of the investigator, and to provide informed consent. The remaining study eligibility requirements were similar to that of the preceding core study.Reference McEvoy, Citrome and Hernandez16
Interventions
All patients started the extension study on treatment with the same dose of lurasidone taken at study endpoint of the 6-week core study; following this, lurasidone was flexibly dosed (40–120 mg/day), if clinically indicated, starting on Day 7 of the extension study. When made, dose adjustments occurred at weekly intervals and in increments/decrements of 40 mg/day, based on investigator judgment in order to optimize efficacy and tolerability. The study drug was taken once daily in the evening with food or within 30 minutes after eating. The study drug, packaged in blister cards, was dispensed by the designated site study staff. Patients took the study drug at home following the directions provided to them by the study site.
Consistent with the preceding core study, medications used to treat movement disorders were not given prophylactically, but agents for emerging or worsening parkinsonism or akathisia were permitted. Concomitant use of agents for the treatment of anxiety symptoms, agitation, or insomnia was permitted during the study at the discretion of the investigator, as in the core study.Reference McEvoy, Citrome and Hernandez16 Patients may have initiated and/or continued treatment with concurrent lithium, divalproex, lamotrigine, or antidepressants during the course of the study. Patients could continue to participate in ongoing psychotherapeutic and psychosocial interventions during the course of this trial.
Compliance was monitored closely and determined at each visit. Patients were instructed to bring all used bottles and unused study drug with them to each visit. Patients who missed more than 25% of the scheduled doses or who took more than 125% of the scheduled doses were considered noncompliant.
Outcomes
The primary endpoints were the proportion of patients with adverse events (AEs), serious AEs (SAEs), or who discontinued due to AEs. AEs were defined as events with onset on or after the first study drug dose in the extension study until 7 days after the last dose in the extension study. AEs that occurred during the core study are reported elsewhere.Reference McEvoy, Citrome and Hernandez16 The secondary safety endpoints included the mean change from both core and extension study baselines in weight, lipids, and glycemic control, Simpson–Angus Scale (SAS),Reference Simpson and Angus17 Barnes Akathisia Rating Scale (BARS),Reference Barnes18 and Abnormal Involuntary Movement Scale (AIMS).Reference Munetz and Benjamin19 The secondary efficacy endpoints included the mean change from both core and extension study baselines in the Positive and Negative Syndrome Scale (PANSS)Reference Kay, Opler and Fiszbein20 total scores, Clinical Global Impression Severity of Illness (CGI-S)21 scores, and Calgary Depression Scale for Schizophrenia (CDSS)Reference Addington, Addington and Schissel22 scores. Additional assessments included were the Columbia-Suicide Severity Rating Scale (C-SSRS) (designed for the assessment of suicidal ideation and behavior in clinical and research settings),Reference Posner, Brown and Stanley23 Health Services Utilization Questionnaire (HSUQ) (designed to capture types and quantities of health and social services used by an individual),Reference Browne, Gafni, Roberts and Hoxby24 Personal Evaluation of Transitions in Treatment (PETiT) (designed to capture aspects of subjective responses to, and tolerability of, antipsychotic drugs, treatment adherence, and impact of antipsychotic drug therapy on the quality of life of individuals treated for schizophrenia),Reference Voruganti and Awad25 Short Form-12 Health Survey (SF-12) (self-rating scale designed to assess overall physical and emotional health and how well a person is able to accomplish his/her usual activities),Reference Ware, Kosinski and Keller26 and Medication Satisfaction Questionnaire (MSQ) (a single-item questionnaire that evaluates satisfaction with antipsychotic medication).Reference Vernon, Revicki and Awad27 In addition, physical examinations, ECGs, and routine fasting clinical laboratory measures were obtained. Regular study visits were scheduled at intervals of 1 month, with weekly telephone contacts between visits. A post-treatment follow-up visit occurred 1 week after the last dose of study drug to assess any post-treatment discontinuation AEs. At the follow-up visit, the patients were instructed to contact the study site if AEs were experienced in the 7 days following the last visit. If the patient was unable to make the visit in person, a telephone contact was made to assess any post-treatment discontinuation AEs.
Statistical analysis
The extension study baseline was defined as the last non-missing assessment during the treatment period of the core study. The “safety population” consisted of all patients who signed informed consent and received at least one dose of study drug in the extension study. The “intent-to-treat population” (ITT) consisted of all randomized patients who signed informed consent, received at least 1 dose of study drug in the core study, and had a core study baseline and at least 1 extension study efficacy measurement from the same scale. If a patient had only a core study baseline but no extension study values, all change-from-baseline values would be missing, and the patient would be effectively excluded from the ITT analysis.
No hypothesis testing was performed for the primary safety endpoints for this study; only descriptive statistics were compiled. Summaries of change from baseline in all continuous efficacy and safety measures included change from core study baseline and change from extension study baseline. For secondary endpoints, change from core study baseline and from extension study baseline in PANSS total scores, CGI-S, and CDSS were tested with a 1-sample t-test of the least squares (LS) means. Least squares means were provided from an analysis of covariance (ANCOVA) with treatment and pooled center as fixed factors and baseline value as a covariate. The same pooled sites created in the core study were used in this study.
Additional descriptive statistics were compiled for both time to discontinuation due to treatment failure and time to any discontinuation. “Treatment failure” was defined as any occurrence of discontinuation due to insufficient clinical response, exacerbation of underlying disease, or AE, and was the primary outcome measure of the core study.Reference McEvoy, Citrome and Hernandez16 Time to discontinuation due to treatment failure and time to any discontinuation were compiled starting from the first extension study dose. Kaplan–Meier (KM) plots were created for both time to discontinuation due to treatment failure and time to any discontinuation. Patients who did not discontinue were considered censored in the KM plots.
Results
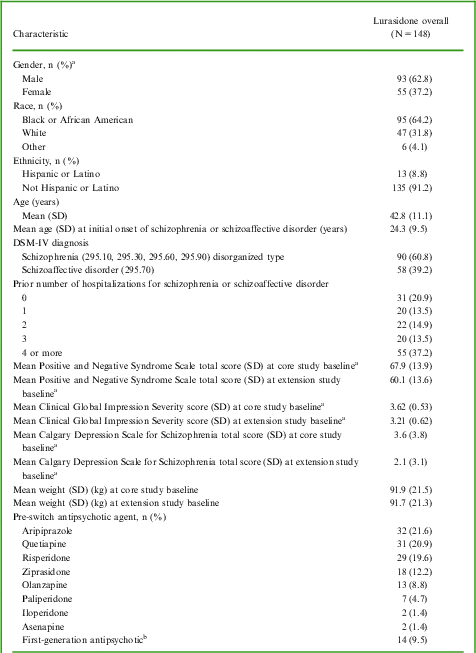
Of the 198 patients who completed the core 6-week study, 149 (75.3%) entered the extension study. The first patient was enrolled on August 10, 2010, and the last patient completed the study on November 28, 2011. A total of 98 patients (65.8%) completed the 6-month study. A total of 51 patients (34.2%) discontinued from the study: 18 (12.1%) withdrew consent, 17 (11.4%) discontinued due to AEs (of which 9 [6.0%] had AEs associated with exacerbation of underlying disease), 9 (6.0%) were lost to follow-up, 2 (1.3%) due to insufficient clinical response, 2 (1.3%) due to protocol violations, 1 (0.7%) failed to meet inclusion/exclusion criteria, 1 (0.7%) due to noncompliance with study drug, and 1 (0.7%) due to an administrative reason. Excluded from the safety and efficacy analyses was 1 patient who did not receive study medication in the extension study. See Table 1 for patient demographics and baseline clinical characteristics of the study population.
Table 1 Patient demographics and baseline clinical characteristics (safety population)

aIntent-to-treat population (N = 144).
bHaloperidol (n = 5), perphenazine (n = 4), chlorpromazine (n = 2), fluphenazine (n = 2), thiothixene (n = 1).
Abbreviations: DSM-IV = Diagnostic and Statistical Manual of Mental Disorders, fourth edition; SD = standard deviation.
Lurasidone 40, 80, and 120 mg were the modal daily doses for 19 (12.8%), 65 (43.9%), and 64 (43.2%) of patients, respectively. The overall mean (SD) daily lurasidone dose was 102.0 (77.1) mg, and the median daily dose was 89 mg.The mean (SD) extent of exposure was 134.4 (61.8) days, and the median extent of exposure was 170 days. Noncompliance with study dosing was reported for 27 (18.2%) of patients, with 3 (2.0%) missing >25% of scheduled doses and 24 (16.2%) classified as taking >125% of scheduled doses. Patients who did not return their blister cards at monthly study visits were conservatively regarded as taking >125% of scheduled doses; we note that some subjects may have had difficulty returning cards due to illness-related factors.
Safety and tolerability outcomes
A total of 98 patients (66.2%) had at least 1 AE. The incidences of the most commonly encountered AEs, as defined by frequency ≥ 2% among all patients, are noted in Table 2. The most commonly reported AEs were insomnia (13 patients [8.8%]), nausea (13 patients [8.8%]), akathisia (12 patients [8.1%]), and anxiety (9 patients [6.1%]). Most patients had only mild or moderate AEs. Severe AEs were uncommon; these events were reported by 10 patients (6.8%): 2 patients (1.4%) had severe dysphoria, and no other severe AE was reported by more than 1 subject. There were no deaths. Psychiatric hospitalizations occurred in 7 (4.7%) of patients.
Table 2 Adverse events with an incidence ≥ 2% (safety population)
The most common extrapyramidal symptom AEs were tremor (3 patients [2.0%]), dystonia (2 patients [1.4%]), and parkinsonism (2 patients [1.4%]). Drooling, extrapyramidal disorder, and trismus were each reported by 1 subject. The most common cardiovascular/metabolic AEs were hypertension (5 patients [3.4%]) and hyperlipidemia (3 patients [2.0%]). Three patients (2.0%) had a weight increase reported as an AE, and 2 patients (1.4%) had a weight decrease reported as an AE (we note that weight change reported as an AE by the investigator is complementary to the objectively collected quantitative weight data). Blood pressure increase and hypercholesterolemia were each reported by 1 subject. Sedation was reported by 4 patients (2.7%), and somnolence was reported by 1 patient (0.7%).
There were 8 patients (5.4%) with 11 treatment-emergent SAEs. Six patients had 1 SAE, 1 patient had 2 SAEs, and 1 patient had 3 SAEs. The most common treatment-emergent SAEs were exacerbations of schizoaffective disorder (3 patients [2.0%]) or schizophrenia (2 patients [1.4%]) and suicidal ideation (2 patients [1.4%]). Bipolar I disorder, paranoia, and homicidal ideation were each reported by 1 subject.
Discontinuation from the study because of an AE occurred in 16 patients (10.8%). The most common AEs leading to discontinuation were schizoaffective disorder (4 patients [2.7%]) and vomiting (2 patients [1.4%]). No other individual AE led to the discontinuation of more than 1 subject.
No clinically relevant changes were observed for changes from core study and extension study baselines to last observation carried forward (LOCF) endpoint in weight, body mass index, waist circumference, lipids, glucose, HbA1c, insulin, C-reactive protein, or prolactin. Figure 1 illustrates the median change in weight, total cholesterol, triglycerides, and glucose from core study baseline.
Figure 1 Median change from core study baseline to LOCF endpoint on metabolic variables. The number of patients available for assessment ranged from 117 for triglycerides and glucose to 144 for weight. Median changes from extension study baseline to LOCF endpoint were similarly small (weight −0.7 kg, total cholesterol −2.0 mg/dL, triglycerides 6.0 mg/dL, glucose 2.0 mg/dL).
A larger proportion of patients experienced ≥ 7% weight loss from baseline compared to ≥ 7% weight gain over the 6-month observation period (Table 3). The proportions differed depending on the antipsychotic received prior to receiving lurasidone in the core study. From core study baseline to extension LOCF endpoint, the percentage of subjects with ≥ 7% weight loss vs ≥ 7% weight gain were 3.2% vs 6.5% for subjects who had received aripiprazole, 23.1% vs 7.7% for olanzapine, 16.7% vs 13.3% for quetiapine, 22.2% vs 11.1% for risperidone, and 16.7% vs 11.1% for ziprasidone. There were absolute mean weight decreases from core study baseline to month 6 for all but the risperidone pre-switch antipsychotic medication subgroup (see Table 4). The largest mean decrease was −1.0 kg (ziprasidone subgroup). Change in lipid and glucose variables from the core study baseline to month 6 for patients who received quetiapine, olanzapine, aripiprazole, risperidone, and ziprasidone pre-switch are described in Table 4. From core study baseline to extension study LOCF endpoint, most patients had metabolic variables that did not shift categories (ie, stayed in same category: normal to normal, low to low, or high to high). Among the 72 patients with high LDL cholesterol values at core study baseline, 22 patients (30.6%) had normal LDL cholesterol values at extension study LOCF endpoint. Among the 11 patients with high glucose values at core study baseline, 6 patients (54.5%) had normal glucose values at extension study LOCF endpoint.
Table 3 Changes in weight ≥ 7% from core and extension study baselines to LOCF endpoint (N = 144)

Table 4 Metabolic variables – mean (standard deviation) change for weight and median change for lipids and glucose, from core study baseline at 6 months and to LOCF endpoint, by pre-switch antipsychotic medication
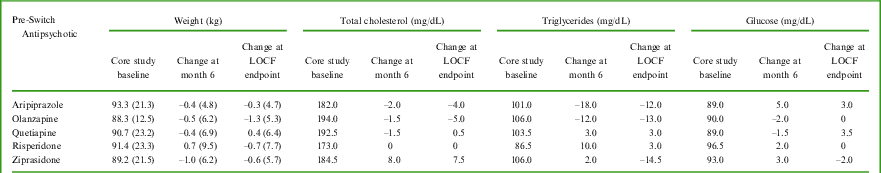
The number of patients available for assessment ranged from 22–30 for quetiapine, 8–13 for olanzapine, 21–31 for aripiprazole, 15–27 for risperidone, and 13–18 for ziprasidone.
The largest mean decreases in prolactin at month 6 were seen for patients previously treated with risperidone (−7.5 ng/mL [SD 17.0 ng/mL], median –5.8 ng/mL), and the largest mean increases in prolactin at month 6 were seen for patients previously treated with quetiapine (4.8 ng/mL [SD 13.7 ng/mL], median 2.0 ng/mL). Generally, larger median changes in prolactin were observed in females than males for each pre-switch antipsychotic medication. Overall, prolactin results for most patients did not shift categories from the core study baseline to extension study LOCF endpoint. Four patients (2.9%), 3 men and 1 woman, had normal to high shifts in prolactin. Other patients with increases in prolactin included 6 patients (4.3%) who shifted from low to normal and 2 patients (1.4%) who shifted from low to high. However, high to normal shifts were observed in 13 patients (9.4%), 7 women and 6 men.
There were no clinically meaningful changes at month 6 in any ECG or vital sign parameter, as assessed relative to core study baseline or extension study baseline. There were no patients with an ECG QTc (Fridericia) interval > 500 ms, or an increase from core study baseline in QTc > 60 ms, at any time during the study.
Median and mean SAS, BARS, and AIMS scores did not demonstrate meaningful changes from either core or extension study baselines (median changes were 0). BARS global scores remained unchanged between core study baseline and month 6 of the extension study for most of the patients (82.7%); a total of 12.2% of patients improved with respect to akathisia and 5.1% of patients worsened. Results were similar relative to extension study baseline.
Few patients had worsening in the C-SSRS relative to core study baseline. Four patients (2.8%) had worsening of their suicidal ideation (ie, more severe than at baseline). A total of 7 patients (4.9%) had emergence of suicidality (ideation and/or behavior). There were no suicide attempts/completions.
Efficacy outcomes
A summary of PANSS, CGI-S, and CDSS outcomes are presented in Table 5. The mean PANSS total score, mean CGI-S score, and mean CDSS score decreased consistently from core study baseline across extension visits, indicating an improvement in overall condition. PANSS total score results were generally similar across subgroups of gender, age group, race, and ethnicity; any differences were not considered clinically meaningful.
Table 5 Positive and Negative Syndrome Scale, Clinical Global Impressions–Severity, and Calgary Depression Scale for Schizophrenia – mean change (standard deviation) from core and extension study baselines to LOCF endpoint, LS mean (standard error), and within-group p-value
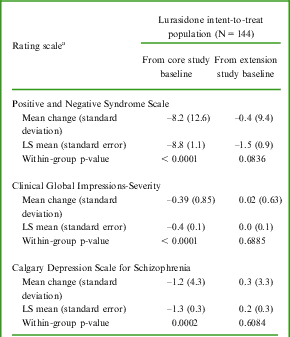
aSee Table 1 for baseline values.
The HSUQ responses indicated that in addition to the normal clinical visits, 17.4% of patients, on average, saw “a doctor or other healthcare provider for psychiatric problems or alcohol or drug use” per month (range: 11.0% to 20.5% across extension visits). Few patients visited an emergency room for an emotional/psychiatric problem (mean 0.8% per month [range 0% to 3.8%]) or for a medical problem (mean 2.1% per month [1.5% to 3.8%]). Few patients were in contact with the criminal justice system (mean: 1.8% per month [0% to 3.7%]).
The PETiT inventory demonstrated a positive subjective response to treatment with a mean change from core study baseline to month 6 of 5.1 (SD 7.2) and 2.6 (SD 8.8) at extension study LOCF endpoint. Relative to the extension study baseline, however, there were decreases for the PETiT scores: −0.9 (SD 6.6) at month 6 and −1.9 (SD 7.2) at extension study LOCF endpoint.
The SF-12 scores showed numerical increases (improvement) relative to core study baseline for all measures (physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional, and mental health) at month 6. Overall changes were small as noted by a median change of 0 from either core study baseline or extension study baseline to extension LOCF for all the components of the SF-12.
The MSQ mean score at core study baseline was 4.2 (SD 1.5) (between MSQ questionnaire responses 4 = “neither dissatisfied nor satisfied” and 5 = “somewhat satisfied”). At month 6, the mean score had increased by 1.4 (SD 1.9) (between MSQ questionnaire responses 5 = “somewhat satisfied” and 6 = “very satisfied”). Relative to the extension study baseline, a slight decrease was observed for MSQ mean change; however, median change at extension LOCF was 0.
Effectiveness outcomes
Treatment failure (discontinuation due to insufficient clinical response, exacerbation of underlying disease, or AE) was observed for 19 patients (12.8%), of which 2 (1.4%) were for insufficient clinical response and 17 (11.5%) for AEs including exacerbation of underlying disease. Median time to treatment failure was 58 days (95% CI 22–86) (Figure 1). The discontinuation rate for any reason was 50/148 (33.8%), and median time to discontinuation was 62 days (95% CI 30–75) (Figure 2).
Figure 2a Time to treatment failure (Kaplan–Meier). Treatment failure is defined as discontinuation due to insufficient clinical response, exacerbation of underlying disease, or an adverse event.
Figure 2b Time to discontinuation for any cause (Kaplan–Meier).
Discussion
This was a 6-month, open-label extension to a study where non-acute patients who met Diagnostic and Statistical Manual of Mental Disorders, 4th edition, text revision (DSM-IV-TR) criteria for schizophrenia or schizoaffective disorder were switched to lurasidone due to insufficient efficacy and/or safety/tolerability concerns. The overall safety profile was consistent with findings from previous studies, and no new safety concerns were identified. The most commonly reported AEs were insomnia (8.8%), nausea (8.8%), akathisia (8.1%), and anxiety (6.1%). There were no signals to indicate clinically relevant alterations in metabolic variables associated with lurasidone treatment. Greater proportions of subjects experienced weight loss ≥ 7% from baseline than the equivalent amount of weight gain (Table 3). In terms of efficacy, there were consistent and statistically significant decreases in mean PANSS total score from the core study baseline to all extension visits, indicating consistent improvement at each month. Similar findings were observed on the CGI-S and CDSS. Positive subjective responses to treatment with lurasidone were generally observed in PETiT scores, SF-12, and MSQ changes relative to the core study baseline, denoting the acceptability of longer-term lurasidone treatment after switching from other agents. Improvement in these assessments, taken together, may also suggest the potential for enhanced adherence to treatment with lurasidone. Few patients required emergency room visits, or were in contact with the criminal justice system, as measured by the HSUQ. The overall incidence of discontinuation due to treatment failure in this study was low (12.8%), with a median time to treatment failure of 58 days.
Generalizability of the study results is enhanced by the inclusion of patients with schizoaffective disorder who comprised about 39% of the sample. The study was conducted entirely in the U.S., and the racial/ethnic distribution of subjects was similar to that in the Phase II schizophrenia trials of lurasidone, which were also conducted entirely in the U.S. In those studies, black/African Americans comprised the majority of study participants.Reference Ogasa, Kimura, Nakamura and Guarino28, Reference Nakamura, Ogasa and Guarino29
The current product label10 provides for a dosing range for lurasidone of 40–160 mg/d. In our study, investigators were free to dose flexibly within the range of 40–120 mg/day according to the perceived tolerability and efficacy of lurasidone experienced by the individual subject. In the extension study, almost equal numbers of patients received a modal dose of 80 or 120 mg/day (43.9% and 43.2% of patients, respectively), with only 12.8% receiving 40 mg/day. In the core study,Reference McEvoy, Citrome and Hernandez16 lurasidone 40, 80, and 120 mg/d were the modal daily doses for 21.3%, 49.6%, and 29.2% of subjects, respectively. At core study baseline, subjects were randomized to 3 different lurasidone dose initiation groups, with flexible dosing permitted from week 2 to the week 6 core study endpoint. In the current extension study, all subjects were flexibly dosed, starting at the dose achieved at core study endpoint. No comparisons among the initially randomized groups in the core study were performed in this extension study.
The safety and tolerability results reported here are consistent with data from other open-label lurasidone extension studies with durations of 6 monthsReference Stahl, Cucchiaro and Simonelli30 and 22 months,Reference Silva, Correll and Simonelli31 and from 12-month double-blind studies comparing lurasidone with quetiapine extended-releaseReference Loebel, Cucchiaro and Xu32 and lurasidone with risperidone.Reference Citrome, Cucchiaro and Sarma33 A common finding that arises is lurasidone's relatively benign weightReference Leucht, Cipriani and Spineli34, Reference De Hert, Yu and Detraux35 and metabolicReference De Hert, Yu and Detraux35 profile, including the potential for normalization of weight and lipid parameters.Reference Stahl, Cucchiaro and Simonelli30 This has long-term implications for morbidity and mortality when treating patients with schizophrenia, and can make the choice of lurasidone compelling.
Several study limitations should be noted. Although the extension study was of 6 months duration, it may still have been too short to fully characterize lurasidone's long-term safety profile. Moreover, the open-label design and lack of a parallel control group limits interpretability. As with any extension study, there may be a decrease in apparent incidence of some AEs if these AEs had led to study discontinuation from the core study or if they had been transient or successfully treated.
Conclusions
In this 6-month, open-label study, treatment with lurasidone continued to be generally well-tolerated in patients with schizophrenia or schizoaffective disorder who had switched to lurasidone from a broad range of antipsychotic agents. The most commonly reported AEs were insomnia, nausea, akathisia, and anxiety. There was no signal for clinically relevant adverse changes in body weight, lipids, glucose, insulin, or prolactin. Patients were able to maintain clinical improvement (as assessed by PANSS, CGI-S, and CDSS) over 6 months of follow up. Positive subjective responses to treatment with lurasidone were also observed (as assessed by PETiT scores, SF-12, and MSQ).
Disclosures
In the past 12 months, L. Citrome was a consultant for, has received honoraria from, or has conducted clinical research supported by the following: Alexza, Alkermes, AstraZeneca, Bristol-Myers-Squibb, Eli Lilly, Envivo, Forest, Genentech, Janssen, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer, Reckitt Benckiser, Reviva, Shire, Sunovion, and Takeda; consultant, consulting fees for Lundbeck; and a small number of shares, common stock for Johnson & Johnson. P. J. Weiden was a consultant for, has received honoraria from, or has conducted clinical research supported by the following: Delpor, Genentech/Roche, Johnson & Johnson (Janssen), Lundbeck, Neurocrine, Otsuka USA, Merck, Novartis, Sunovion. J. McEvoy was a consultant for, has received honoraria from, or has conducted clinical research supported by the following: Alkermes, Eli Lilly, Merck, Psychogenics, Roche, and Sunovion; consultant, consulting fees for Otsuka; and grants, consultant, research support and consulting fees for Ameritox. C. U. Correll was a consultant for, has received honoraria from, or has conducted clinical research supported by the following: Actelion, Alexza, Bristol-Myers-Squibb, Cephalon, Eli Lilly, Genentech, Gerson Lehrman Group, IntraCellular Therapies, Lundbeck, Medavante, Medscape, Merck, Janssen/J&J, Otsuka, Pfizer, ProPhase, Roche, Sunovion, Takeda, Teva, and Vanda. J. Cucchiaro, J. Hsu, and A. Loebel are full-time employees of Sunovion Pharmaceuticals.
This study was supported by Sunovion Pharmaceuticals Inc., Marlborough, MA, and Fort Lee, NJ.






 Open access
Open access